Putative azithromycin resistance mutations in Chlamydia trachomatis are globally distributed but arose before azithromycin was discovered
Parul Sharma, Deborah Dean, Timothy D. Read

TL;DR
This study finds that mutations linked to azithromycin resistance in Chlamydia trachomatis existed before the drug was used clinically and are common in certain infection types.
Contribution
The study reveals that putative azithromycin resistance mutations in C. trachomatis predate the drug's clinical use and are lineage-specific.
Findings
Mutations in the rplV gene are common in urogenital and anorectal infection lineages but absent in ocular lineages.
Time-scaled phylogenetic analysis shows these mutations existed before azithromycin was introduced clinically.
No consistent resistance patterns were found in 23S rRNA or rplD genes.
Abstract
Azithromycin is widely used to treat Chlamydia trachomatis infections, yet the extent of resistance to the drug across the species has not been addressed. We surveyed mutations and substitutions linked to putative azithromycin resistance across 1,349 high-quality C. trachomatis genomes. Mutations in the rplV gene encoding three non-synonymous substitutions, compared with the canonical C. trachomatis reference strain D//TW-3/Cx sequence, were found to be common but largely conserved within phylogenetic lineages causing prevalent urogenital and anorectal infections and lymphogranuloma venereum. However, no mutations were identified in the ocular lineage. Time-scaled phylogenetic analysis suggested that these mutations predate the clinical introduction of azithromycin. In contrast, no consistent resistance-associated patterns were observed in 23S rRNA or rplD genes. This large-scale…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
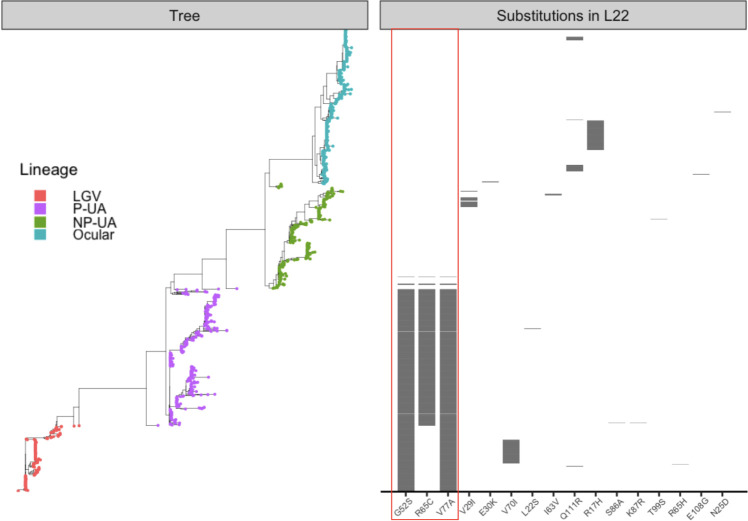 Fig 1
Fig 1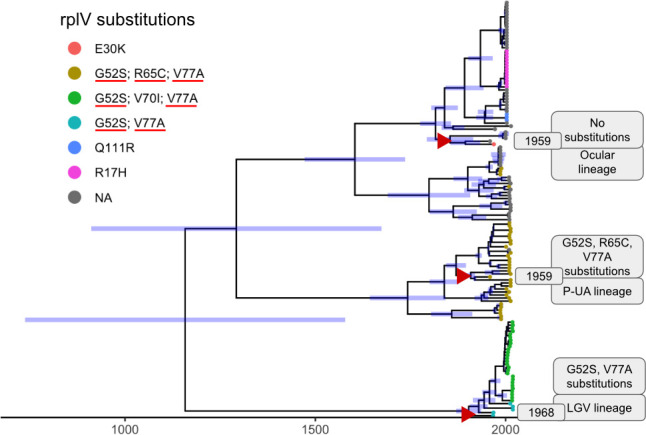 Fig 2
Fig 2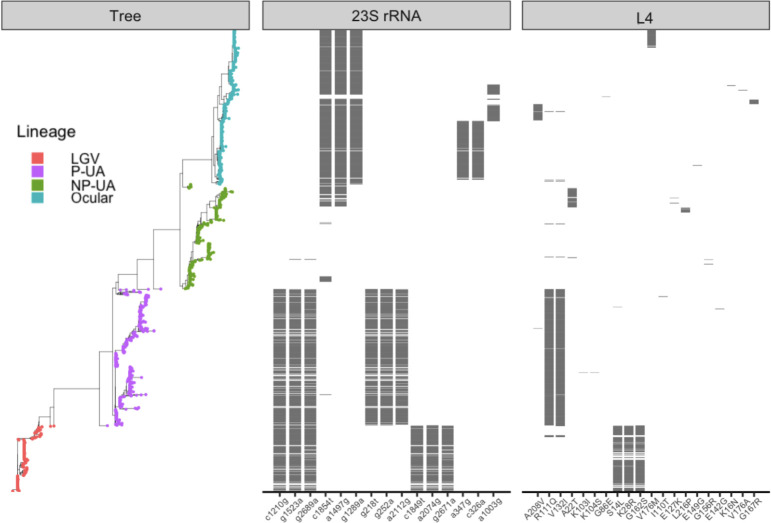 Fig 3
Fig 3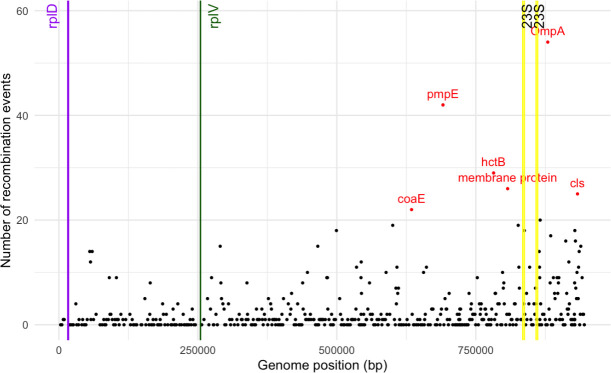 Fig 4
Fig 4| Source | Allele | Reference | Reference species | CT-numbering | Detected |
|---|---|---|---|---|---|
| NCBI | a2074c/a2074g/a2074t |
| a1879c/a1879g/a1879t | Yes (a->g) | |
| a2075g |
| a1880g | |||
| a2059g |
|
| a2036g | Yes | |
| a2058g / a2058t |
|
| a2038g | ||
| a2114g |
|
| a2449g | ||
| a2115g |
|
| a2450g | ||
| c2627a |
| c2592a | |||
| c2611t |
| c2592t / c2592g | Yes (c->t) | ||
| g2032t |
|
| g2012t | ||
| c2630a/c2630g |
|
| na | ||
| t2609c |
|
| t2590c | ||
| t754a |
|
| t740a | Yes (t->g) | |
| CARD | c196a |
|
| c196a | |
| a2107c/a2107g |
|
| a2107c / a2107g | ||
| a2109g |
|
| a2109g | ||
| a2663c / t2663c |
|
| a2663c / t2663c |
| Source | Number of genomes |
|---|---|
| NCBI Assembly | 87 |
| AllTheBacteria | 1,083 |
| Reference genomes | 20 |
| NCBI SRA | 159 |
| Total | 1,349 |
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsReproductive tract infections research · Urinary Tract Infections Management · Syphilis Diagnosis and Treatment
INTRODUCTION
Chlamydia trachomatis is an obligate intracellular pathogen and the most common cause of bacterial sexually transmitted infections (STIs) worldwide. Ocular strains of C. trachomatis cause trachoma, the leading cause of infectious blindness, particularly among women due to their increased exposure to children who are reservoirs of infection in trachoma-endemic regions (1). In STIs, treatment commonly involves antibiotics, such as doxycycline, administered twice a day over 7 days or azithromycin as a single dose (2). Recent clinical evidence indicates that doxycycline is more effective than azithromycin for treating rectal chlamydial infections (3). For trachoma, topical tetracycline ointment or oral azithromycin has historically been the preferred option (4). Azithromycin is the antibiotic of choice in many mass drug administration (MDA) campaigns implemented under the World Health Organization (WHO) Surgery, Antibiotics, Facial cleanliness, and Environmental improvement (SAFE) strategy, aimed at eliminating blinding trachoma (5). Despite large-scale efforts, elimination targets have not been fully achieved, and several countries, particularly in sub-Saharan Africa, such as Ethiopia and Sudan, remain endemic. In recognition of these challenges, the WHO extended its global elimination goal for blinding trachoma from 2020 to 2030 (6, 7).
Azithromycin is an azalide, a sub-class of macrolide antibiotics (8). It was discovered in 1980, patented in 1981, and its widespread use began after 1991 when it was launched by Pfizer (9). Since then, it has become one of the most widely prescribed antibiotics for bacterial infections, including C. trachomatis, due to its broad-spectrum activity, tissue penetration and long half-life, and patient compliance advantages (10, 11). However, its extensive use has also raised concerns about the potential for emerging resistance.
Several STI studies have reported C. trachomatis treatment failures with azithromycin (12–14). These failures are often attributed to either reinfection of another C. trachomatis strain after clearance of the initial infection or persistence of the initial infection. Persistent infections have often been attributed to the development of C. trachomatis antibiotic resistance (14, 15). Several studies have identified point mutations in 23S rRNA, rplV, and rplD genes as potentially linked to macrolide resistance in C. trachomatis (16–18). In rplV, encoding the L22 protein, mutations resulting in G52S, R65C, and V77A substitutions were detected in isolates from patients with azithromycin treatment failure (19, 20), although the minimum inhibitory concentrations (MICs) were within the sensitive range. The same variants were observed in Russian urogenital isolates in both macrolide-resistant and sensitive strains (21), and among endocervical, vaginal, and rectal samples in Fiji (22). For RplD, the L4 protein, studies reported P109L, P151A (20), and Q66K substitutions (16), while other studies (19, 23) did not detect rplD mutations in clinical isolates. 23S rRNA gene mutations A2057G, A2058C, A2059G, and T2611C have been associated with resistance, although their presence and role remain inconsistent across studies (20, 21, 24, 25). Notably, all the above 23S rRNA mutations are also listed in the NCBI Reference Gene Catalog (26), a public database that compiles known antimicrobial resistance genes and associated mutations across diverse pathogens. Similarly, the Comprehensive Antibiotic Resistance Database (CARD; 27) serves as another curated resource, detailing resistance determinants, mechanisms, and mutation-based associations. The latest version of CARD (accessed October 2025) specifically documents 23S rRNA mutations linked to azithromycin resistance in C. trachomatis, while the NCBI catalog lists rplD, rplV, and 23S rRNA mutations reported across multiple bacterial species.
In this study, we analyze the patterns of DNA mutations in the 23S rRNA gene and DNA mutations and amino acid substitutions in L22 and L4 associated with azithromycin resistance across a globally distributed collection of 1,349 C. trachomatis genomes from public databases. Our aim was to characterize the prevalence, lineage specificity, and evolutionary patterns of putative azithromycin resistance variants.
MATERIALS AND METHODS
Compiling and pre-processing genomic data
C. trachomatis genomes were compiled from multiple sources, including the NCBI Assembly database (accessed March 2025), AllTheBacteria database (version 0.2 with the incremental release from 08 to 2024) (28), published reference genomes (29), and available NCBI SRA samples. For NCBI SRA samples, raw reads were down-sampled to achieve even coverage using the bbnorm.sh script from BBMap (v39.01) (30), followed by assembly with SPAdes (v4.0.0) (31) using default parameters. Quality filtration was applied across the complete data set of genomes from all sources using checkM (v1.2.3) (32) retaining only genomes with more than 98% completeness, less than 6% contamination, and less than 2,000 contigs. Metadata for genomes in this study in Table S1.
Identifying azithromycin resistance-associated mutations
A list of point mutations and substitutions linked to macrolide resistance was obtained from the NCBI Reference Gene Catalog (accessed March 2025) (26) and CARD (27). Additional candidate mutations reported in the literature for azithromycin resistance in C. trachomatis were also compiled (Table 1). To ensure comparability, all mutations were converted to C. trachomatis numbering by alignment to C. trachomatis reference strain D/UW-3/Cx (NCBI Accession ID: NC_000117.1) (33).
All genomes in the data set were re-annotated using Bakta (v1.9.2) (34) to minimize annotation heterogeneity, and sequences corresponding to the 23S rRNA, rplV, and rplD genes were extracted. Each gene set was aligned to the reference sequence using MAFFT (v7.526) (35), and variants were identified relative to the reference coordinates. Mutations were detected and summarized using custom python scripts (available on GitHub: https://github.com/parul-sharma/CT-AMR).
Phylogenetic trees and time-scaled analysis
Core-genome genes were identified for all annotated genomes in the data set using PIRATE (v.1.0.5) (36) under a single workflow. The resulting core gene alignment, composed of 853 genes 0.83Mb in length, was used as input for IQ-TREE2 (version 2.3.0) (37) to construct a maximum-likelihood phylogenetic tree with automated model selection. A similar phylogenetic tree was obtained for the subset of genomes (138 out of 1,354) with associated dates of isolation. For these dated strains, dates of isolation were added using the “–date” parameter to supply temporal metadata to the IQ-TREE run. The resulting tree was subsequently used as input for BEAST (v2.7.7) (38) to perform time-scaled phylogenetic inference.
Using BEAST, we evaluated multiple models for phylogenetic inference. All models used a strict molecular clock but differed in population models: (i) Bayesian Skyline; (ii) Bayesian Skyline with HKY nucleotide substitution; (iii) exponential growth; (iv) constant population size; and (v) extended Bayesian Skyline with invariant sites. All models produced broadly similar topologies and divergence times (Supplementary Data). For clarity, the results presented here are based on the strict molecular clock with an exponential growth population model. Random Local Clock models were not attempted, as the low overall divergence of C. trachomatis sequences would provide insufficient signal to reliably estimate lineage-specific rate variation.
Recombination and visualizations
Recombination analysis was performed on the core genome alignment (described above) using ClonalFrameML (v1.13) (39) with default parameters. The resulting importation status file was processed with a custom Python script (available on GitHub https://github.com/parul-sharma/CT-AMR) to identify recombination regions. Briefly, the genome was divided into 1,000 bp windows, the total number of recombination events within each window was counted, and windows with elevated recombination activity (more than 20 recombination events) were designated as recombination “hotspots.”
To investigate whether resistance-associated loci showed phylogenetic patterns distinct from the core genome, we constructed individual gene trees for the 23S, rplV, and rplD sequences. These were then compared with the core-genome phylogeny using tanglegrams, allowing us to check congruence in tree structure and evaluate potential evidence of recombination or lineage-specific inheritance at these loci. All visualizations were generated in R, with trees constructed using ggtree (v3.13.0) (40), time-scaled trees in combination with treeio (v1.28.0) (41), and tanglegram comparisons performed using phytools (v2.4-4) (42). Supporting R scripts are available in the GitHub repository.
RESULTS
Putative-resistance mutations in the rplV gene are lineage specific
We examined resistance-associated mutations in our data set of 1,349 high-quality C. trachomatis genomes (Table S1). A core-genome phylogeny divided the genomes into four major lineages, consistent with prior studies: lymphogranuloma venereum (LGV), ocular, prevalent urogenital/anorectal (P-UA), and non-prevalent urogenital/anorectal (NP-UA), reflecting their disease tropisms (43–45). All substitutions were called in relation to the canonical reference D/UW-3/Cx sequence (NC_000117.1), an NP-UA genome (33).
We first focused on the three amino acid substitutions in L22: G52S, R65C, and V77A that have been associated with azithromycin resistance in C. trachomatis (19, 21). In each case, we found that the substitution was caused by a single mutation (g151a, c190t, and t227c, respectively). These substitutions showed strong lineage specificity (Fig. 1). All LGV genomes (191/191, 100%) carried G52S and V77A, while nearly all P-UA genomes (399/404, 98.8%) harbored the complete set of triple substitutions (G52S, R65C, and V77A). In contrast, these putative resistance-associated substitutions were completely absent from ocular genomes (0/450) and were rare in NP-UA genomes (4/304, 1.3%). In the rare NP-UA genomes with the G52S, R65C, and V77A substitutions, the sequence was identical to the P-UA allele, suggesting exchange by homologous recombination. A similar pattern in reverse was seen in the rare P-UA genomes missing these mutations (Table S2).
Distribution of L22 substitutions across genomes. Genomes are ordered according to core-gene phylogeny (left) with tips colored by C. trachomatis lineage (LGV, ocular, P-UA, NP-UA). The panel on the right shows a heatmap of the 16 L22 substitutions, marked along the x-axis, with gray tiles indicating presence and empty spaces indicating absence across samples. The three azithromycin-associated substitutions are marked with a red box.
We next broadened our analysis to identify additional rplV mutations that resulted in amino acid substitutions relative to the reference D/UW-3/Cx protein sequence (accession ID: NC_000117.1) across the data set. Thirteen other substitutions were detected, most at low frequencies and restricted to specific lineages, with the only exception being the Q111R substitution present in few of both Ocular (28/450, 6.2%) and LGV (2/191, 1.0%) genomes (Fig. 1). In the P-UA lineage, S86A and K87R co-occurred in a single genome, while L22S was detected in another. Among ocular genomes, R17H was the most common secondary variant (86/450, 19.1%), followed by Q111R (28/450, 6.2%), with several others present at very low frequencies (N25D, E30K, and E108G, each in ≤2 genomes). In NP-UA genomes, V29I was observed in 26/304 (8.5%), I63V in 4/304 (1.3%), and T99S in a single genome. Within LGV genomes, Q111R was present in 2/191 (1.0%), while V70I was more frequent (68/191, 35.8%); R65H occurred only once.
Together, these findings demonstrated that the putative azithromycin-associated rplV mutations were tightly clustered in P-UA and LGV lineages. The absence of these variants in ocular genomes likely reflects their long-standing evolutionary separation from P-UA and LGV lineages. There were other substitutions, but aside from Q111R, there was no evidence of homoplasy. There was also evidence of gene conversion events in the P-UA and NP-UA lineage strains.
L22 resistance-associated substitutions predate the widespread clinical use of azithromycin
For a subset of 138 (10.2%) of the 1,349 genomes, metadata on the year of isolation were available (Table S1). The most recent samples in our data set were from 2019. Genome SAMEA767935 (P-UA lineage, E/Bour ompA type), collected in 1959 (46), carried all three canonical L22 resistance-associated substitutions (G52S, R65C, and V77A). Another 1959 genome, SAMEA1973344 (Ocular lineage, C/TW-3/OT ompA type), had no substitutions in L22. The second-oldest genome, SAMEA767923 (LGV lineage, L3/404 ompA type), collected in 1967, carried the double mutations (G52S and V77A). The presence of resistance-associated rplV mutations in genomes dating from 1959 and 1967 indicated that these mutations arose prior to the widespread clinical use of this antibiotic after 1991. Time-scaled phylogenetic analysis (Fig. 2) further supported this observation. Using the strict molecular clock with an exponential growth population model, we estimated a 95% likelihood that genomes carrying the triple mutations diverged between 1,016 and 1,779, while those with the double mutations diverged between 850 and 1,695.
Time-scaled phylogeny constructed using BEAST. Branch lengths are proportional to time (in years), and the 95% highest posterior density (HPD) intervals of node heights are indicated as blue bars. Tip labels annotated by color representing rplV substitutions as per the legend with azithromycin-associated substitutions marked with red underline. The three oldest genomes (highlighted with red triangles) are annotated with metadata, including year of isolation, lineage, and presence of rplV substitutions.
Patterns of mutations in rplD and 23S rRNA
The three L4 protein substitutions previously reported as azithromycin resistance-associated (P109L, P151A [20], and Q66K [16]) were not found across the 1,354 C. trachomatis genomes in this study. Due to the high sequence divergence of the rplD gene across bacterial species, none of the putative resistance-associated substitutions listed in the NCBI Reference Gene Catalog could be identified in our data set, as protein sequences from other species did not align with the C. trachomatis L4 protein (Table S3). A small number of substitutions were observed within the protein, most of which were lineage-specific (Fig. 3). Notably, two homoplasic substitutions—R111Q and V132I—were predominant in P-UA genomes (399/404, 98.8%), though they also appeared in a few NP-UA (n = 2), Ocular (n = 3), and LGV (n = 6) genomes. These variants were absent in five P-UA genomes and partially missing in three others, missing R111Q in two and V132I in one (Table S4).
Distribution of mutations in 23S rRNA and substitutions in L4 across genomes. Genomes are ordered by core-gene phylogeny (left), with tree tips colored by genome lineage. The first panel shows the 15 most frequent 23S rRNA mutations; the second panel shows all L4 substitutions. In both panels, the x-axis lists individual mutations or substitutions.
Similarly, examination of the 23S rRNA sequence showed no obvious correlation with previously reported resistance mutations. Of the 10 resistance-associated positions previously described in NCBI AMR and CARD databases (Table 2), four were present in all strains, including the reference strain used for alignment. This suggests that these variants represent fixed or highly conserved positions rather than markers of acquired resistance. Across the data set, a total of 104 additional mutations were detected, ranging from one to eight mutations per genome, with 77.8% (81 out of 104) present in fewer than 1% of genomes. Additionally, since C. trachomatis carries two copies of the 23S rRNA gene, we examined whether any mutations were heterozygous. Only 11 of the 89 complete genome assemblies exhibited heterozygous mutations, all of which were rare (present in less than 1% of genomes). To better understand lineage-specific patterns, we therefore visualized the 15 most frequent mutations. The 15 most prevalent mutations were non-homoplasious and largely lineage-specific, with genomes from the NP-UA lineage displaying the most rare mutations. However, the observed frequencies of mutations were influenced by the use of the NP-UA type strain (NC_000117.1, D/UW-3/Cx) as the reference for mutation calling, meaning that all differences were computed relative to this sequence.
Collectively, these observations indicate that, similar to rplV, variation in rplD and 23S rRNA is largely shaped by lineage and genomic background, and there is no strong evidence for recent selection for azithromycin resistance.
rplV, rplD, and 23S rRNA are not in genomic recombination hotspots
To investigate whether resistance-associated mutations in rplV, rplD, or 23S rRNA arose via intra-specific recombination, we compared gene-specific phylogenies with the core-genome phylogeny for a random subset of genomes. Across all comparisons, rplV vs core tree, rplD vs core tree, and 23S rRNA vs core tree (Supplementary Data), we observed strong congruence, with no evidence of anomalous branching patterns indicative of horizontal transfer or recombination. The observation was further supported by genome-wide recombination analyses. None of the sequences in these ribosomal genes were located within identified recombination hotspots (Fig. 4). In contrast, recombination events were predominantly observed in expected loci, such as genes encoding membrane proteins, with ompA exhibiting the highest density of recombination sites.
Recombination hotspots across C. trachomatis genome. Frequency of recombination events mapped to the C. trachomatis reference strain D/UW-3/Cx (NCBI Accession: NC_000117.1). The vertical lines indicate the genomic positions of the rplD (purple), rplV (green), and two copies of the 23S rRNA (yellow) genes. Regions with more than 20 recombination events are shown as red dots, representing recombination hotspots across the C. trachomatis genome.
DISCUSSION
Many methods of inference of resistance from genome sequences require catalogs of mutations linked to phenotypes (47). Here, using 1,354 available C. trachomatis genomes, we investigated the phylogenetic patterns of previously characterized mutations that were purported to be linked to azithromycin resistance but varied in the degree of susceptibility or resistance based on MICs. The increasing availability of large public genomic data sets now provides an opportunity to systematically explore resistance-associated mutations across diverse populations with more comprehensive data sets. This large-scale genomic approach provides a framework for better understanding resistance dynamics in C. trachomatis and lays the foundation for improved surveillance and clinical management strategies.
Our study provides evidence that putative azithromycin resistance alleles were already present in C. trachomatis lineages before the widespread clinical use of azithromycin that occurred after 1991. The high lineage specificity of these alleles suggests that their persistence more likely reflects neutral evolution rather than resistance acquired after azithromycin use. These findings align with a growing body of work demonstrating that the genetic foundations of antimicrobial resistance often predate human antibiotic use (48), with ancient selective forces shaping the genomic background upon which modern resistance emerges (49). However, the mutations may not be directly linked to elevated resistance to azithromycin. Possibly, the putative resistance phenotype linked to rplV found by other groups (19, 21) is linked to additional factors such as epistatic mutations (50). Nonetheless, these findings underscore the importance of considering historical and lineage-specific variation when interpreting the possible emergence of antibiotic resistance in C. trachomatis.
Pinpointing azithromycin resistance in C. trachomatis is complicated by several factors. Although treatment failures with azithromycin have been repeatedly reported, these events are multifactorial in nature. Reinfection from an untreated partner, inadequate drug treatment, or noncompliance with medication are common confounders that can mimic resistance-associated persistence (13, 51). Moreover, while genotypic evidence of resistance alleles provides valuable insights, it cannot be equated directly to a resistance phenotype. Several older studies highlighted this disconnect, where treatment failures were observed without clear links to resistance-associated mutations, underscoring the complexity of resistance as a trait (52). Such observations reinforce the idea that resistance in C. trachomatis is a multifactorial trait, with potential contributions from epistatic mutations, epigenetic regulation, host-pathogen interactions, or other yet-uncharacterized mechanisms.
Our analysis also underscores the limitations of transferring resistance markers across species. Variants annotated in public resistance databases, often curated based on other pathogens, do not consistently predict resistance in C. trachomatis. For example, macrolide resistance mutations in 23S rRNA (e.g., A2059G, A2074G, C2611T), which are well established in Neisseria gonorrhoeae, Campylobacter coli, and Escherichia coli, do not show consistent associations with resistance in C. trachomatis. In fact, we found these mutations in nearly all genomes in our data set, indicating that they likely reflect natural variation rather than resistance. This lack of correlation highlights the non-transferability of resistance markers between species. Instead, 23S variation in C. trachomatis appears to track more closely with phylogenetic background than with resistance, underscoring the need for organism-specific surveillance.
A major challenge in advancing C. trachomatis antimicrobial resistance research lies in the lack of phenotypic confirmation. Due to its obligate intracellular replication, traditional culture-based MIC testing is technically challenging, labor-intensive, and rarely performed in routine laboratories (53). This reliance on limited phenotypic data has constrained our ability to validate resistance mutations at scale. Nonetheless, in-depth genomic analyses, such as ours, are crucial for prioritizing candidate alleles and selecting samples for functional studies. By refining the pool of targets, these studies provide a framework to design phenotypic assays more efficiently, ultimately bridging the gap between genotypic surveillance and clinical relevance.
Taken together, our findings reinforce several key messages. First, resistance-associated alleles in C. trachomatis are shaped by lineage-specific evolution and likely predate clinical use of azithromycin. Second, mutations annotated as resistance markers in public databases derived from other pathogens cannot be assumed to apply to C. trachomatis. Third, while large-scale genomic surveys provide valuable insights into the evolutionary context of putative resistance mutations, they cannot on their own establish clinical relevance. Functional validation through phenotypic MIC testing and experimental work remains essential. Moving forward, integrating genomic surveillance with phenotypic evidence will be key to resolving the complex relationship between genetic variation and antimicrobial resistance in C. trachomatis, ensuring that resistance monitoring and treatment strategies are both accurate and tailored to this pathogen’s unique biology.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dean D. 2010. Chapter 77, Pathogenesis of chlamydial ocular infections, p 678–702. In Tasman W, Jaeger EA (ed), Duane’s foundations of clinical ophthalmology. Vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA.
- 2World Health Organization. Chlamydia. Available from: https://www.who.int/news-room/fact-sheets/detail/chlamydia. Retrieved 12 Sep 2025.
- 3Dombrowski JC, Wierzbicki MR, Newman LM, Powell JA, Miller A, Dithmer D, Soge OO, Mayer KH. 2021. Doxycycline versus azithromycin for the treatment of rectal Chlamydia in men who have sex with men: a randomized controlled trial. Clin Infect Dis 73:824–831. doi:10.1093/cid/ciab 15333606009 PMC 8571563 · doi ↗ · pubmed ↗
- 4Dawson CR, Schachter J, Sallam S, Sheta A, Rubinstein RA, Washton H. 1997. A comparison of oral azithromycin with topical oxytetracycline/polymyxin for the treatment of trachoma in children. Clin Infect Dis 24:363–368. doi:10.1093/clinids/24.3.3639114186 · doi ↗ · pubmed ↗
- 5World Health Organization. 2020. Executive summary. WHO guideline on mass drug administration of azithromycin to children under five years of age to promote child survival. World Health Organization, Geneva, Switzerland. https://www.ncbi.nlm.nih.gov/books/NBK 561639/.
- 6O’Brien KS, Emerson P, Hooper PJ, Reingold AL, Dennis EG, Keenan JD, Lietman TM, Oldenburg CE. 2019. Antimicrobial resistance following mass azithromycin distribution for trachoma: a systematic review. Lancet Infect Dis 19:e 14–e 25. doi:10.1016/S 1473-3099(18)30444-430292480 · doi ↗ · pubmed ↗
- 7Ageed A, Khan M. 2024. Eliminating trachoma in Africa: the importance of environmental interventions. Cureus 16:e 52358. doi:10.7759/cureus.5235838234389 PMC 10792353 · doi ↗ · pubmed ↗
- 8Bakheit AHH, Al-Hadiya BMH, Abd-Elgalil AA. 2014. Azithromycin. Profiles Drug Subst Excip Relat Methodol 39:1–40. doi:10.1016/B 978-0-12-800173-8.00001-524794904 · doi ↗ · pubmed ↗