High-quality genome assembly and linkage map for a rapidly evolving plant species: Silene uniflora
Owen G Osborne, Daniel P Wood, Mariya P Dobreva, Luke T Dunning, Rachel Tucker, Sarah E R Coates, Jaume Pellicer, Jon Holmberg, Adam C Algar, Greta Bocedi, Cecile Gubry-Rangin, Leonel Herrera-Alsina, Berry Juliandi, Lesley T Lancaster, Pascal Touzet, Justin M J Travis

TL;DR
This paper provides a high-quality genome assembly and linkage map for Silene uniflora, a plant species useful for studying rapid adaptation and evolution.
Contribution
The study presents a chromosome-scale genome assembly and linkage map for Silene uniflora, revealing widespread genome rearrangements in the genus.
Findings
A 1,268 Mb genome assembly with a scaffold N50 of 40.72 Mb was generated for Silene uniflora.
The genome contains 41,603 protein-coding genes with a BUSCO completeness score of 91%.
Genome comparisons reveal widespread rearrangements in the Silene genus, suggesting a role in evolutionary processes.
Abstract
The genus Silene is an important model system for fields as diverse as sex chromosome evolution, speciation, and disease ecology. However, genomic resources remain scarce in the genus. Here, we present a near chromosome-scale genome assembly and high-density linkage map for S. uniflora, a hermaphroditic/gynodioecious species which is an important model for rapid adaptation to anthropogenic disturbance and the role of phenotypic plasticity in adaptive evolution. Using a combination of long-read and Hi-C sequencing technologies, we generated a 1,268 Mb genome assembly with a scaffold N50 of 40.72 Mb and 682 Mb assembled into 12 chromosomes. We annotated the genome using evidence from transcriptome and protein mapping in combination with ab initio gene prediction, resulting in 41,603 protein-coding genes and a BUSCO completeness score of 91%. We also present a linkage map which we used to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
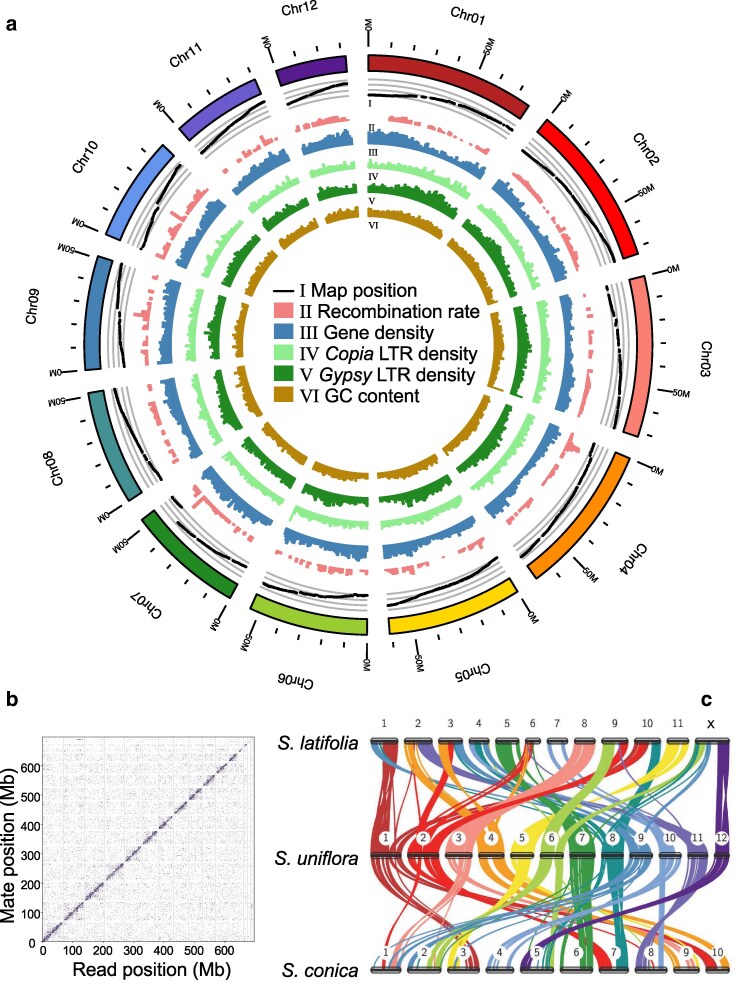 Fig. 1
Fig. 1- —UK Natural Environment Research Council10.13039/501100019151
- —ASTP10.13039/100029350
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Chromosomal and Genetic Variations · Genetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities
Introduction
Sea campion, Silene uniflora, is a perennial herb native to coastal Northern and Western Europe (Davy et al. 2024). Morphological and genetic studies of S. uniflora and its close relative S. vulgaris in the mid-20th century were fundamental to the development of the field of experimental taxonomy (Marsden-Jones and Turrill 1957; Davy et al. 2024). Subsequently, it became a model species for the study of castrating sexually transmitted fungal diseases (Chung et al. 2012; Abbate et al. 2018). It has also attracted attention for its rapid adaptation to the extreme environments resulting from non-ferrous metal mining (Baker 1974, 1978; Baker and Dalby 1980). More recently, the species has become an emerging model system for research into the genetic basis of parallel evolution (Papadopulos et al. 2021) and the role of plasticity and gene expression during rapid adaptation (Wood et al. 2023; Coates et al. 2025). More broadly, Silene has been the focus of intense research to understand sex chromosome evolution (Papadopulos et al. 2015; Yue et al. 2023), cytonuclear incompatibilities (Garraud et al. 2011; Postel et al. 2022), the evolution of mitochondrial genomes (Sloan et al. 2012; Wu et al. 2015), biological invasions (Keller and Taylor 2010; Jenkins and Keller 2011; Castillo et al. 2014), adaptation, and speciation (Bratteler et al. 2006; Muir et al. 2012; Favre et al. 2017; Zemp et al. 2018). Nevertheless, the first chromosome-scale assembly was published only recently (Fields et al. 2023).
There are currently only high-quality reference genomes available for 2 Silene species—S. latifolia and S. conica (Fields et al. 2023; Yue et al. 2023). S. latifolia is dioecious and has been the focus of intense research into the evolution of young, homomorphic sex chromosomes. The majority of Silene species are diploid with 12 pairs of chromosomes (2n = 24, including S. latifolia and S. uniflora; (Bari 1973). S. conica is purely hermaphroditic and is unusual as it only possesses ten chromosome pairs (2n = 20; (Fields et al. 2023). S. latifolia and S. conica also sit at the extreme ends of genome size for Silene species—S. latifolia has one of the largest diploid genome sizes (2.8 Gb; (Pellicer and Leitch 2020), whereas S. conica is one of the smallest (0.9 Gb; (Williams et al. 2021). Populations of S. uniflora can exclusively contain hermaphrodites or be gynodioecious (Davy et al. 2024). Its genome size is intermediate for the genus (1.2 Gb), it has 12 pairs of chromosomes (Williams et al. 2021) and B chromosomes have also been observed in karyotyping studies (Cobon and Murray 1983). High-quality genomic resources for the species have the potential to accelerate research into the genomic and epigenetic mechanisms that underlie rapid adaptation but also contribute to wider research in the genus by adding additional resources for studies of sex chromosomes and mating systems. Here, we report a near chromosome-level genome assembly and a detailed genetic map for S. uniflora, providing a comprehensive resource for future eco-evolutionary research.
Materials and methods
Plant material and sequence data generation
Cuttings from a single coastal individual were collected in Tresaith (West Wales, UK) and propagated and self-fertilized as part of a previous study (Papadopulos et al. 2021). A single individual inbred F1 (SUTF1P; draft genome ASM1898310v1) was selfed and the F2 individual sequenced here was grown under controlled conditions at the Henfaes Research Centre, Bangor University, UK. High molecular weight DNA was extracted for Pacific Biosciences (PacBio) and Oxford Nanopore (ONT) sequencing using a Qiagen DNAeasy Maxi plant kit. PacBio library preparation and sequencing was completed at the Genomics Laboratory, University of Sheffield, UK (Supplementary note 1). Liquid nitrogen frozen leaf tissue was sent to Dovetail Genomics, LLC for Hi-C, Chicago and Illumina Truseq library preparation and sequencing on a HiSeqX sequencer (Illumina).
RNA extraction was conducted on root, flower, and leaf tissue for the genome plant using the RNeasy Plant Mini Kit (Qiagen), including a DNase digestion step, and RNA extracts were sent to the Beijing Genomics Institute (Hong Kong) for library preparation and sequencing. 100 bp paired end RNA-seq libraries were prepared according to the BGISEQ-500 RNA-Seq Library Preparation Protocol and libraries were sequenced on a BGISEQ500 sequencer (Beijing Genomics Institute).
For linkage mapping, 2 plants were grown from seed collected from populations (i) WWA-C and (ii) WWA-M (Papadopulos et al. 2021) and were subject to a controlled cross. One of the offspring of this cross was selfed and the resulting seeds (F2s) collected (Supplementary note 2). When the plants were 8 mo old, leaf punches were taken for DNA extraction and sequencing using LGC Genomics GmbH SeqSNP service. LGC Genomics constructed SeqSNP sequencing libraries for F2 progeny using 25,000 custom probes (Supplementary Table 1). These libraries were sequenced on an Illumina NextSeq 500.
Genome size estimation
The nuclear DNA content of Silene uniflora was determined by flow cytometry following the 1-step procedure described in Doležel et al. (2007); see Supplementary note 3.
Genome assembly
An initial draft hybrid assembly was constructed from raw Illumina short read data and PacBio longread data using the MaSuRCA pipeline (kmer size = 99; PacBio N50 = 6,149 bp; PacBio mean sequencing depth = 29.21x; Supplementary Table 2). Contigs from this draft assembly were then arranged into scaffolds using the Hi-C and Chicago data using the Dovetail HiRise pipeline. To further improve the assembly, we utilized Oxford Nanopore MinION (ONT) long reads. ONT data (N50 = 11,254 bp; mean sequencing depth = 12.55x; Supplementary Table 2) were first corrected using the short reads produced from the Hi-C scaffolding (Supplementary note 4). Corrected long reads were first used to perform additional scaffolding of the assembly before being used to close assembly gaps. Scaffolding was conducted using SLR v1.0.0 (Luo et al. 2019) with a minimum alignment length of 300 nucleotides. Gap closing was conducted using TGS-GapCloser v0.56 (Xu et al. 2020). Contamination was assessed using the NCBI Foreign Contamination Screen pipeline (Astashyn et al. 2024) and any adaptor and non-target organism sequences were masked from the final assembly. For each step in assembly improvement, QUAST (Gurevich et al. 2013) was used to assess assembly size and contiguity. To further assess assembly contiguity, we calculated the LTR Assembly Index (LAI) using LTR retriever v2.9.0 (Ou et al. 2018; Ou and Jiang 2018).
Genome annotation
Prior to gene prediction, we masked repeat regions using RepeatMasker (v4.1.0; http://www.repeatmasker.org/) against the MIPS plant repeat database (v.9.3; (Nussbaumer et al. 2013). For gene annotation, we employed RNA-seq mapping, assembled transcript mapping, protein mapping and ab initio approaches, which were then combined into a single non-redundant gene model (Supplementary note 5). We used eggnog-mapper v2.0.1 with database version 2.0 (Cantalapiedra et al. 2021) to functionally annotate predicted proteins. We used diamond as a mapper, a minimum query and subject coverage of 70%, an e-value cutoff of 0.0001, a taxonomic scope of Viridiplantae and defaults for all other options. To produce a final set of high-quality gene annotations, we filtered out annotations with non-canonical splice sites or in-frame stop codons, those annotated as transposable elements in eggnog-mapper, those without start or terminal stop codons, and those without either RNA evidence or matches to know ortholog groups in eggnog-mapper. Annotation completeness was assessed using BUSCO v4 (Simão et al. 2015) in protein mode with the Viridiplantae, Embryophyta, and Eudicot gene sets. To estimate the position of centromeres in the 12 largest scaffolds, we used CentIER v2.0 (Xu et al. 2024) with our final gene annotation and default settings. To identify telomeres within the 12 largest scaffolds, we used quartet v1.2.5 (Lin et al. 2023).
Linkage map construction and analysis
Raw reads from the SeqSNP data were trimmed and mapped to the genome (Supplementary note 6). Linkage maps were constructed using LepMap3 v0.4 (Rastas 2017); see Supplementary note 7. The linkage map was used to provide validation of the genome assembly, indicate the position of unassigned scaffolds, and estimate local recombination rate. The mapping parents originated from different populations to the reference individual, so structural variation between populations could introduce misassemblies. Therefore, the linkage map was not used for scaffolding. Recombination rate was estimated using a LOESS local regression (span 0.1 Mb), with recombination rate estimated as the slope in non-overlapping 1 Mb windows. To avoid inaccurate recombination rate estimates caused by error or rearrangements between the plants used for genome assembly and linkage mapping, we did not calculate recombination rate for windows containing no markers or where the map window contained markers from other scaffolds.
Comparative genomics
Synteny between the S. uniflora genome and the previously published assemblies of S. conica and S. latifolia was examined using the MCscan algorithm implemented in the JCVI toolkit v.1.3.8 (Wang et al. 2012; Tang et al. 2024). Previously identified sex-linked transcripts from S. otites, S. pseudotites, and S. latifolia (Chibalina and Filatov 2011; Zemp et al. 2016; Martin et al. 2019) were mapped to the genome using minimap2 v.2.24-r1122 (Supplementary note 8).
Results and discussion
The mean estimated genome size from our flow-cytometry analysis was 1C = 1.28 pg, corresponding to a haploid genome size of 1251.48 Mb (Supplementary Table 3). The final genome assembly was 1269.36 Mb long, closely matching the estimated genome size. The assembly contained 12,573 scaffolds, had a scaffold N50 of 40.72 Mb and a scaffold L50 of 11. Approximately 53.73% of the assembly was contained within the 12 largest scaffolds which were between 32.17 and 76.86 Mb in size (henceforth chromosomes Chr01–Chr12), corresponding to the haploid chromosome number of S. uniflora. Additionally, 5 further scaffolds were over 1 Mb in length. Contamination screening identified no foreign organism sequences and 5 putative adaptor sequences (totaling 134 bp; Supplementary Table 4) which were masked from subsequent analysis. Starting from an initial assembly with an N50 of 34 Kb, each step in the genome assembly significantly improved genome contiguity (N50 values: Chicago: 169 Kb; Dovetail Hi-C: 27.01 Mb; ONT scaffolding: 34.97 Mb, ONT gap-filling: 40.72 Mb; Supplementary Table 5). The genome contains ∼29% repetitive sequences, with LTR retrotransposons contributing the largest fraction (∼25%; Supplementary Table 6). The assembly had a raw LAI score of 10.29 and a corrected LAI score of 4.66. The final gene annotation contained 41,603 protein coding genes encoding 48,596 putative transcripts. Of these, 98.89% were complete, including both start and stop codons and 32.26% also contained annotations for 5′ and 3′ untranslated regions (Supplementary Fig. 1). BUSCO analysis suggested that the annotation was largely complete (86% to 91% BUSCO completeness depending on the gene set used; Supplementary Fig. 2).
The linkage map contained 8,372 markers on 1,357 scaffolds which were assigned to 12 linkage groups (LGs; Supplementary Table 7). The linkage map was used to validate the assembly of the chromosomes and estimate the correct placement of other large scaffolds.
LGs were broadly concordant with assembled chromosomes. Between 87.2 and 97.5% (mean = 95.6) of markers from each chromosome were assigned to a single LG, and the LG differed for each chromosome (Supplementary Table 8). The assembled sequence for ten of the chromosomes covered over 90% of the map length of its corresponding LG (Supplementary Table 8). This value was lower for Chr11 and Chr12 (58 and 79% respectively; Supplementary Table 8), and the map suggested that 3 of the 5 unplaced scaffolds >1 Mb were linked to Chr12 in LG.4 (Supplementary Fig. 3) and one to Chr11 in LG.1 (Supplementary Fig. 4). The final unplaced scaffold >1 Mb was linked to Chr02 in LG.3 and embedded within Chr02 in the map (Supplementary Fig. 5). One putative centromere location was identified for all chromosomes except Chr01, for which 2 were identified. We did not identify telomeres in any chromosomes. Regions of high repeat density and low gene density were found toward the center of several chromosomes (Fig. 1; Supplementary Fig. 6), and these corresponded to centromere locations estimated by CentIER for these chromosomes (Supplementary Fig. 7). Therefore, evidence from linkage map-genome concordance and gene and repeat distribution indicate that, while this is not a telomere-to-telomere assembly, the majority of the assembled chromosomes are largely complete, with the exceptions of Chr11 and Chr12.
Genome assembly of Silene uniflora and comparison to other Silene genomes. The S. uniflora chromosomes (ie the twelve largest scaffolds) are shown as a circular plot (a) with GC content (%), gypsy and copia LTR retrotransposon density, gene density (genes per 1 Mb), and estimated recombination rate (cM/Mb) shown in non-overlapping 1 Mb windows. The relationship between physical and genetic distance is shown as a “Marey map” (Chakravarti 1991), with each point representing one genome-anchored marker and the y-axis representing its position in our linkage map. The Hi-C scaffolding is shown as a link density histogram (b) with the genomic position of each read and its mate shown on the x and y axes, respectively and color intensity representing number of read-pairs per bin. Comparison of the S. uniflora genome with previously published Silene genomes shows widespread rearrangements between the species (c). Links between the chromosomes of each species represent syntenic blocks shared between the species, colored by S. uniflora chromosome as in panel A.
Some inconsistencies between the map and assembly may represent rearrangements between the populations that the genome and mapping plants originated from. For example, plots of physical vs map position highlight a putative intrachromosomal translocation on chromosome Chr07, and putative inversions on Chr01, Chr03, Chr06, Chr09, and Chr10 (Supplementary Fig. 8). Frequent chromosomal rearrangements may have important implications in S. uniflora, since these have the potential to contribute to reproductive isolation between locally adapted populations (Kirkpatrick and Barton 2006).
Mean local recombination rate varied between chromosomes (ranging from 1.35–2.12 cM/Mb; Supplementary Fig. 9) and was negatively correlated with chromosome length (Spearman's rank correlation: ρ = −0.87; P = 0.0004; Supplementary Fig. 10), a common finding across eukaryote species (Haenel et al. 2018). Within chromosomes, and on a genome-wide scale, local recombination rate was significantly positively associated with gene density, but not GC content, despite gene density and GC content being correlated with each other (Supplementary Fig. 11). Gypsy and Copia LTR retrotransposon density were both significantly negatively associated with gene density (Supplementary Fig. 10).
Comparison to the S. latifolia and S. conica genomes revealed widespread rearrangements between the species. Notably, the X chromosome of S. latifolia was linked to 4 S. uniflora chromosomes. It has previously been suggested that chromosomal translocations have played a causative role in sex chromosome formation in S. latifolia and its sister species S. dioica (Bačovský et al. 2020). However, our synteny analysis suggests that most large-scale rearrangements in the S. latifolia X chromosome relative to S. uniflora are shared by S. conica—a hermaphrodite lacking sex chromosomes—suggesting these rearrangements occurred prior to the evolution of dioecy (Fig. 1c). Sex chromosomes have evolved independently in multiple Silene lineages. We found that previously identified sex-linked transcripts from S. latifolia, S. otites and S. pseudotites all mapped to different chromosomes in the S. uniflora genome (Supplementary Fig. 12), confirming previous evidence that sex chromosomes have evolved from different autosomes in each of these 3 species (Martin et al. 2019).
Here, we present a high-quality genome assembly and linkage map for S. uniflora, which we use to infer the recombination landscape of S. uniflora and investigate genome rearrangements between Silene species. This will be an important resource for studies using Silene as a model organism. S. uniflora populations have repeatedly and rapidly evolved tolerance to heavy metal contamination. A high-quality genome assembly will allow the role of genomic rearrangements in this process to be examined. Furthermore, as one of only 3 Silene species with a high-quality genome assembly, this resource is likely to be important for studies of other species and Caryophyllaceae more broadly.
Supplementary Material
jkag002_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abbate JL et al 2018. Co-occurrence among three divergent plant-castrating fungi in the same Silene host species. Mol Ecol. 27:3357–3370. 10.1111/mec.14805.PMC 634078730030861 · doi ↗ · pubmed ↗
- 2Astashyn A et al 2024. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol. 25:60. 10.1186/s 13059-024-03198-7.38409096 PMC 10898089 · doi ↗ · pubmed ↗
- 3Bačovský V et al 2020. The formation of sex chromosomes in Silene latifolia and S. dioica was accompanied by multiple chromosomal rearrangements. Front Plant Sci. 11:205. 10.3389/fpls.2020.00205.32180787 PMC 7059608 · doi ↗ · pubmed ↗
- 4Baker AJM . 1974. Heavy metal tolerance and population differentiation in Silene maritima with. University of London.
- 5Baker AJM . 1978. Ecophysiological aspects of zinc tolerance in Silene maritima with. New Phytol. 80:635–642. 10.1111/j.1469-8137.1978.tb 01596.x. · doi ↗
- 6Baker AJM, Dalby DH. 1980. Morphological variation between some isolated populations of Silene maritima with. In the British isles with particular reference to inland populations on metalliferous soils. New Phytol. 84:123–138. 10.1111/j.1469-8137.1980.tb 00755.x. · doi ↗
- 7Bari EA . 1973. Cytological studies in the genus Silene L. New Phytol. 72:833–838. 10.1111/j.1469-8137.1983.tb 02697.x. · doi ↗
- 8Bratteler M, Lexer C, Widmer A. 2006. Genetic architecture of traits associated with serpentine adaptation of Silene vulgaris. J Evol Biol. 19:1149–1156. 10.1111/j.1420-9101.2006.01090.x.16780515 · doi ↗ · pubmed ↗