Proton-Initiated Reversible Chalcogen-Vertex Extrusion in Macropolyhedral Chalcogenaboranes
Jonathan Bould, Miroslava Litecká, William Clegg, Marcel Ehn, John D. Kennedy, Michael G. S. Londesborough

TL;DR
This paper describes a reversible chemical transformation in chalcogenaborane clusters triggered by protonation and deprotonation.
Contribution
The study reveals a proton-induced reversible structural rearrangement in macropolyhedral chalcogenaborane clusters.
Findings
Protonation converts [E2B17H18]− into metastable E2B17H19 with distinct subcluster architectures.
Deprotonation regenerates the original anions through boronotropic dehydrogenation.
Metastable compounds eliminate dihydrogen or selenium to form known neutral products.
Abstract
Protonation of the macropolyhedral chalcogenaborane anions [E2B17H18]− (E = S, 1a – Se, 1b – ) induces a radical cluster rearrangement, yielding the new metastable isoelectronic neutral species E2B17H19 (2a and 2b). This transformation converts the original structure comprising two identical “arachno” 10-vertex subclusters into a new architecture with two distinct “nido” 10-vertex subclusters. Notably, one of the chalcogen atoms, which originally was a triconnected cluster vertex in anions 1 – , is extruded, forming a bridging μ2-{EH} unit. Remarkably, deprotonation of compounds 2 facilitates a boronotropic dehydrogenation, giving reintegration of the extruded {EH} unit and regeneration of the original [E2B17H18]− anions 1 – . Compound 2a is characterized by a single-crystal X-ray diffraction (SCXRD) study, and by agreement between experimental and density functional theory…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1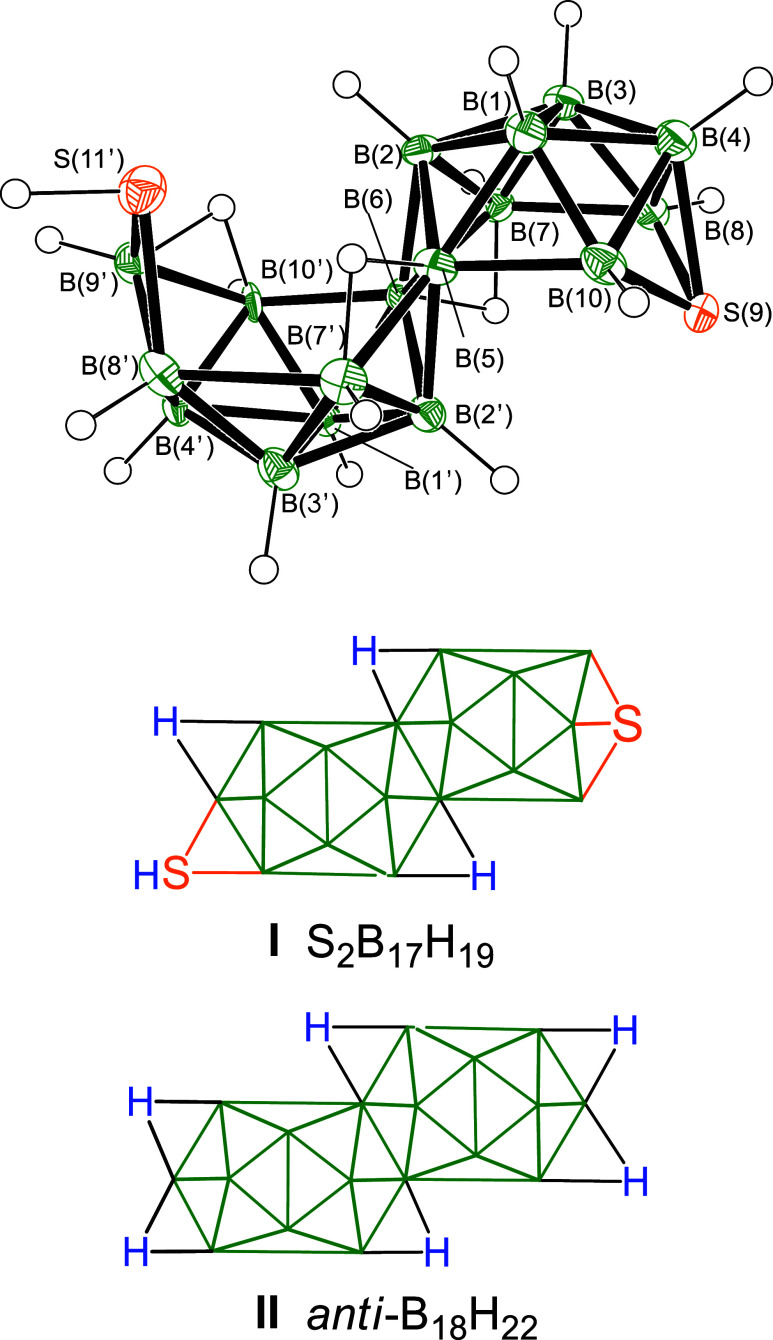 1
1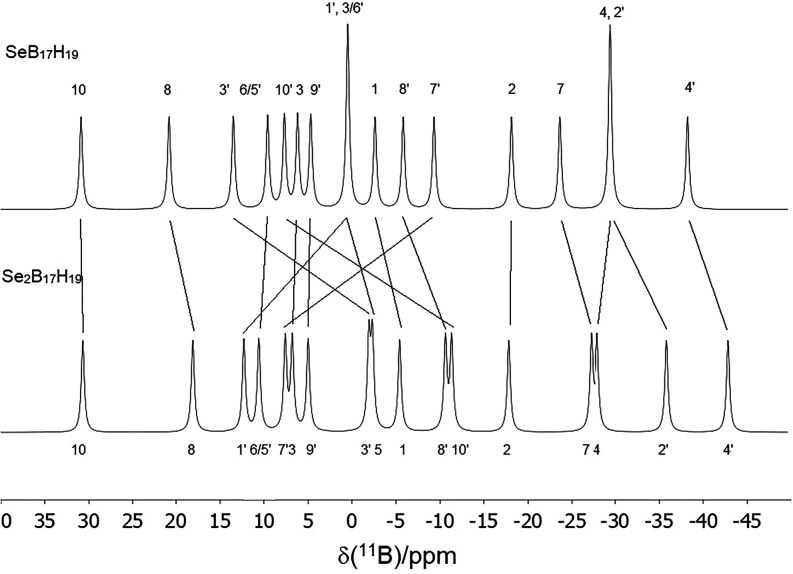 2
2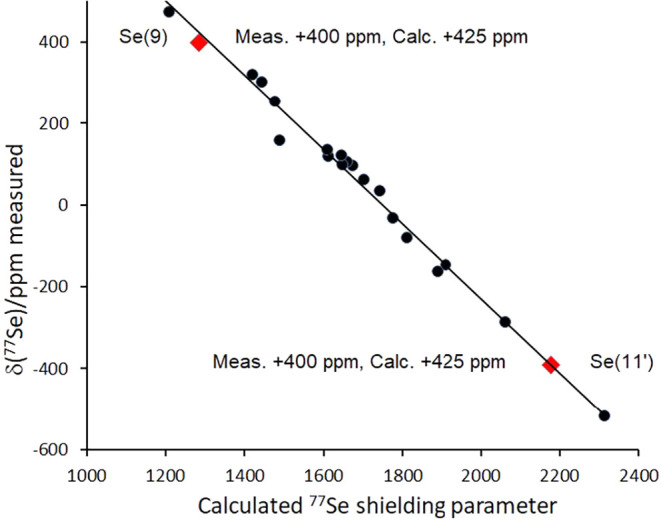 3
3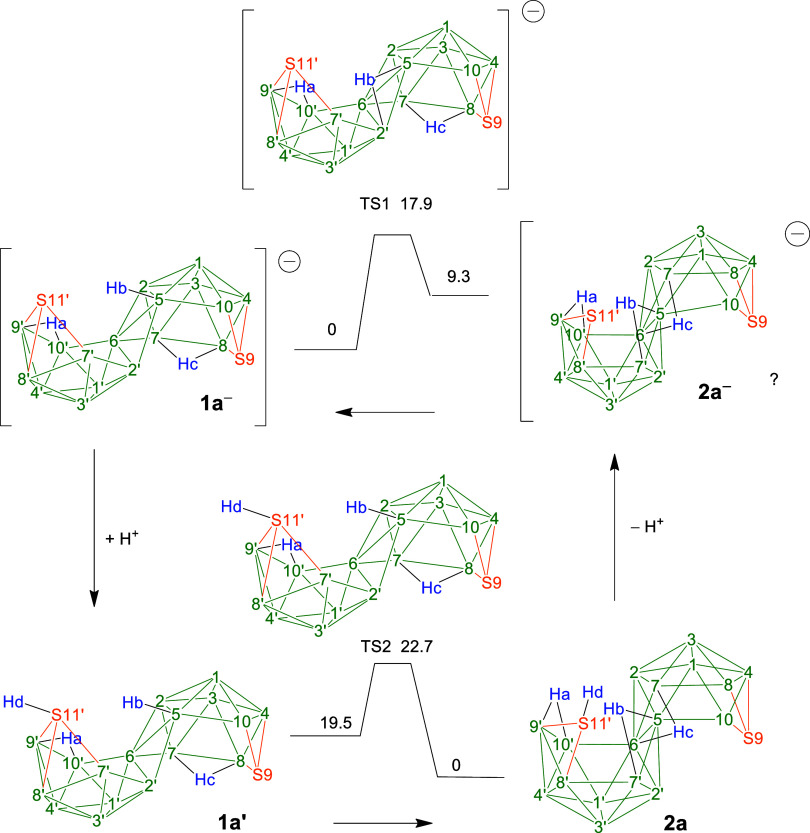 2
2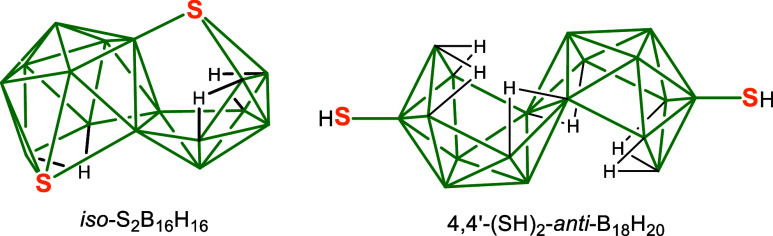 3
3- —Grantov? Agentura Cesk? Republiky10.13039/501100001824
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBoron Compounds in Chemistry · Organoboron and organosilicon chemistry · Boron and Carbon Nanomaterials Research
Introduction
Polyhedral boron hydride chemistry occupies a distinctive position within inorganic chemistry, offering rare insights into electron-delocalized cluster architectures, unusual bonding motifs, and a capacity for structural adaptability that rivals the diversity seen in organic frameworks.? Among these compounds, macropolyhedral chalcogenaboranes have emerged as particularly intriguing systems, not least because the incorporation of chalcogens perturbs the delicate balance between electron count, three-dimensional (3D) architecture, reactivity, and photophysical properties of the borane cluster core.? The anions [E_2_B_17_H_18_]^−^ (E = S, Se) exemplify this complexity. ?,? They consist of two equivalent arachno 10-vertex subclusters linked by two common boron atoms (see Scheme), situating them at the intersection of classical Wade–Mingos electron-counting concepts? and the more flexible behavior exhibited by large, maropolyhedral metallaboranes, categorized by Jemmis et al.?
Overall Reaction Pathways
Comparison with carborane chemistry provides further context. Carboranes, particularly closo-C_2_B_10_H_12_ and its derivatives, are renowned for their structural resilience and for the rich, position-specific substitution chemistry made possible by their rigid icosahedral scaffolds.? Large-scale topological rearrangements are rare in carboranes, reflecting the exceptional stability of the polyhedral cage. In contrast, the heteroboranes considered here display a striking structural plasticity: as we shall show, a single proton triggers a profound rearrangement that reshapes the macropolyhedral topology. This divergence illustrates how chalcogen incorporation unlocks new modes of reactivity inaccessible to purely boron–carbon clusters.
An understanding of the structure and behavior of metastable intermediates is crucial for elucidating mechanistic pathways in such complex reorganizations. Although transient species are widely recognized as key actors in organic and organometallic chemistry, their roles in boron macropolyhedral cluster transformations remain comparatively underexplored. The acid-induced structural dynamism of the [E_2_B_17_H_18_]^−^ anions, where E refers to sulfur (a) or selenium (b) as appropriate, provides a compelling case study and is the subject of our work here. The [S_2_B_17_H_18_]^−^ anion (1a ^ – ^), first described in 1994,? was reported then to seemingly undergo immediate conversion to S_2_B_17_H_17_ (3a) upon protonation, accompanied by a notable ∼33° rotation in the relative orientation of the two arachno subclusters. We later showed that the selenium analogue [Se_2_B_17_H_18_]^−^ (1b ^ – ^) displays similar reactivity.? In both cases, however, our contemporary work, which benefits from the possibilities afforded by modern NMR spectroscopic studies of the reaction, revealed the fleeting formation of metastable 19-vertex intermediates, now identified as E_2_B_17_H_19_ (2a E = S; 2b E = Se), bridging the transformation of 1 into 3. Despite repeated observation of these species in our previous NMR studies, the intrinsic instability of these species has, until now, prevented their definitive structural characterization.
Results and Discussion
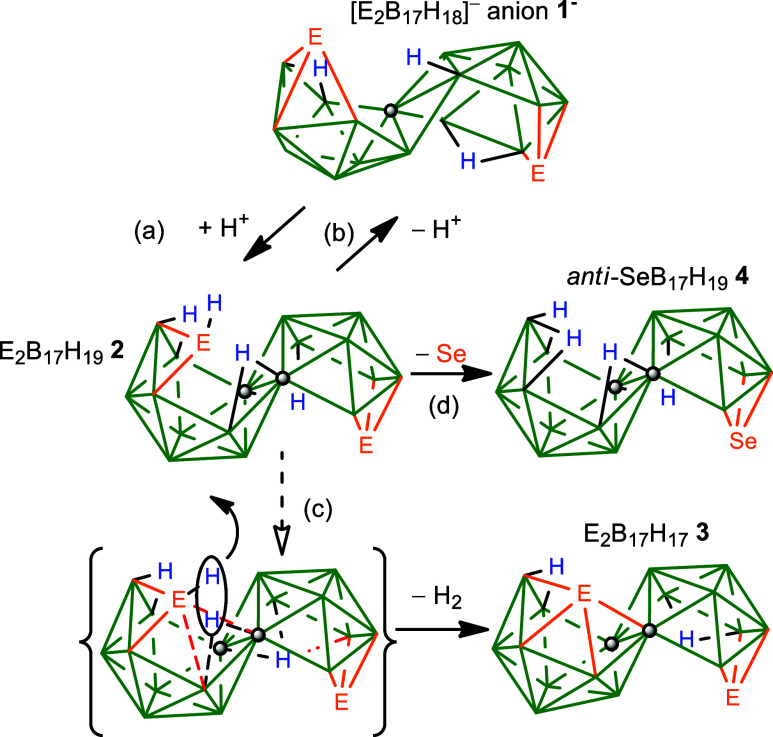
Acidification of dichloromethane solutions of the [E_2_B_17_H_18_]^−^ anions 1 with concentrated H_2_SO_4_ yields neutral E_2_B_17_H_19_ compounds 2, which, over several hours, undergo spontaneous loss of dihydrogen to give neutral E_2_B_17_H_17_ 3 and, in the case of selenium, a small amount of EB_17_H_19_ 4 (Scheme). From a single-crystal X-ray diffraction study (SCXRD) the molecular structure of 2a (Figure) is seen to exhibit a {μ_2_-SH}-SB_17_H_18_ unit. The configuration is analogous to that of the long-known anti-B_18_H_22_, but with an {SH} unit bridging an open-face B–B link in a position occupied by a bridging hydrogen atom in *anti-*B_18_H_22_ (Schematic structures I and II in Figure). Bridging thiol groups are rare in polyhedral borane chemistry, with only S_2_B_16_H_18_ ? and mercaptoboranes such as μ_2_-HS(B_2_H_5_)? as known exemplars. In a further analogy with anti-B_18_H_22_, the structure of 2a can be viewed as two “nido” 10-vertex subclusters that are fused with two boron atoms held in common.
Above: Crystallographically determined structure of the 19-vertex dithiaborane S2B17H19 2a with 50% probability ellipsoids for non-hydrogen atoms. Below: schematic diagram comparing projections of the basic cluster architectures of S2B17H19 2a and anti-B18H22. The selenium analogue 2b is isostructural.
The results from the SCXRD study are substantiated by the close agreement between the experimental ^11^B NMR data for 2a and the values predicted via density functional theory (DFT)/GIAO calculations for the molecular structure and boron nuclear shieldings (Tables S1 and S2). Comparison of the calculated and measured ^11^B NMR spectra for 2a is in Figures S1 and S2. Further substantiation derives from comparing the ^11^B spectra of the now substantiated selenium analogue 2b with SeB_17_H_19_ 4 (Figure), which show that the effective replacement of a bridging hydrogen atom in 4 by {SeH} results in the largest changes in resonance positions for those vertices associated with the bridging {SeH} unit. Further, the measured ^77^Se NMR spectrum of 2b (Figure S4) exhibits two resonances at +400 and −390 ppm, which correspond to Se(9) and Se(11′) respectively in Figure. To support the characterization of 2b, we employed published correlations between measured ^77^Se NMR chemical shifts and DFT/mPW1PW91-calculated selenium nuclear shieldings for a range of selenaborane species.? These correlations have proven valuable in substantiating measured ^77^Se chemical shifts where direct structural data are unavailable for new species, and thence allowing structural conclusions. For 2b, the SCXRD data were suboptimal because of twinning and disorder and their refinement involved the support of the DFT structural model, particularly with regard to the positions of bridging and selenol hydrogen atoms. The calculated ^77^Se chemical shifts for the proposed structure of the selenium analogue 2b (Figure) are thence in agreement with the experimental values, thereby providing strong extra support for the structure. The assignments of the selenium and boron resonances might, in principle, be further strengthened by the observation of ^77^Se satellites on the boron resonances and correlating these with their assignments from DFT calculation. However, the line widths for ^11^B and ^77^Se are of the order of 80 and 150–300 Hz respectively, and any selenium satellites are very weak, 3.5% either side of the resonances, and are too broad to observe.
An illustration (with arbitrary peak widths) of the relative positions of the measured 11B-{1H} resonances in SeB17H19 4 and Se2B17H19 2b emphasizing the changes induced by the bridging {μ-SeH} moiety in 2b. It may be noted that there is only a small change in the chemical shift of the B(4′) position, which is trans to the {μ2-SeH} moiety, in going from SeB17H19 to Se2B17H19. The substitution of the bridging hydrogen atom in SeB17H19 4 with the {μ-SeH} moiety might be expected to produce a more significant effect. The sulfur analogue of SeB17H19 4 does not yet exist and so a direct comparison is not possible. However, the calculated relative chemical shifts for the 4′ positions in SB17H19 and S2B17H19 pair also show the same 5 ppm upfield shift. This suggests that the B(4′) chemical shift is relatively insensitive to the identity of E.
Correlation of the experimentally determined 77Se NMR chemical shifts and the mPW1PW91–calculated selenium magnetic nuclear shieldings for a range of macropolyhedral and single-cluster selenaboranes. The 19 selenium centers (black circles) range over 1000 ppm. , The points for the two selenium vertices in now-substantiated Se2B17H19 2b are shown as orange diamonds. The trend line is calculated from the published data , and does not include compound 2b. The linear regression analysis gives δcalc = A × δexp + B 0.906x + 21.4, R 2 = 0.989. The measured highly shielded Se–H chemical shift at −390 ppm compares to that in SeH2 of −345 ppm, which might support its characterization as a “pseudo bridging-hydrogen”.
The mechanistic implications of the four transformations–viz. the processes (a) to (d) in Scheme–that ultimately lead to the effective 33° cluster subcluster reorientations, as previously noted,? in the overall conversions of species of type 1 ^ – ^ to type 3, may now be rationalized. In this context, the structural elucidation of the intermediate species 2 allows the following proposals:
First, regarding the transition of anions 1 ^ – ^ to compounds 2, process (a).
- 1.That 2a features an {H–S} unit suggests that the most probable site for the initial protonation of compounds 1 ^ – ^ is the E atom.
- 2.For the sulfur species 1a ^ – ^, its initially protonated entity, neutral 1a′, is calculated to be 19.5 kcal/mol higher in energy than the resulting metastable isomer 2a′, providing a considerable driving force for the transformation process (Scheme).
Schematic Structures to Show the Proposed Relative Positions of the Vertices and Bridging Hydrogen Atoms, with Energies as Calculated for the Sulfur System as Exemplars
Thus, protonation of one of the chalcogen atoms in 1 ^–^ to give compounds 1′ triggers an extrusion of this vertex from its original three-connected, cluster-integrated position to a more exposed {μ-EH}-bridging unit in compounds 2. This displacement of one E atom as {EH} reduces the formal cluster bonding electron count by three, assuming that the three-connected E vertices in compounds 1 ^ – ^ act as 4-electron donors, while the bridging {EH} units behave as one-electron, pseudo-hydrogen bridges: the nature of bridging heteroatoms in boranes as bridging or as vertices has been discussed in detail elsewhere.? The rearrangement involves cleavage of the S(11)–B(7) connectivity 1a ^ – ^ (in Scheme) and formation of a new B(7)–B(5) linkage in 2a. The substantial reorganization of the framework prompts the exo-hydrogen atom BH(5) (Hb in 1a ^ – ^) to shift position, now to function as a bridging μ-H(7′,5) hydrogen atom across the newly established B–B linkage. In doing so, the delocalized negative charges present in anions 1 ^ – ^ become localized as μ-H(7,5) bridges, increasing the total number of bridging hydrogen atoms from two in anions 1 ^ – ^ to three in compounds 2. Accordingly, process (a) represents a formal oxidative cluster transformation, effecting a geometrical shift from arachno to nido in both subclusters of compounds 2. It also results in the migration of the μ-H(7,8) bridge (Hc in anions 1 ^ – ^) to a μ-H(6,7) position in both compounds 2 (Scheme).
The extrusion of the heteroatom cluster vertex to a bridging position is unusual enough to warrant a brief discussion. Thiolated derivatives of the single-cluster species B_10_H_14_ and of the isomers of macropolyhedral B_18_H_22_ are known.? These compounds feature thiol groups that have replaced, by Friedel–Crafts–type electrophilic substitution, exo-polyhedral hydrogen atoms on the periphery of an unchanged boron skeleton. This is in contrast to the current case where the thiol group arises from an internal migration and partial ejection from the cluster. A bridging sulfur has been observed? in the 18-vertex species iso-S_2_B_16_H_16_ in which the sulfur may be regarded as replacing both the bridging hydrogen atom in one subcluster and the exo-terminal hydrogen atom in the adjacent subcluster (Scheme). Here the bonding could be described as comprising one 2-electron 3-center bond and one 2-electron 2-center bond. Architecturally, the sulfur atom in iso-S_2_B_16_H_16_ could be regarded as resembling a thioether.? However, the thiol motif in 2 is not fully comparable to either a thioether or a thiiriane as the latter is an S–C–C 3-membered ring with the sulfur bonded to the carbon atoms via two strong 2-center, 2-electron bonds. Also, in thiiranes, the S bridges a C–C 2-electron σ-bonded linkage whereas in 2a the B8′-B9′ connectivity at 2.058(10) Å (1.92 Å in iso-S_2_B_16_H_16_) is looser than a direct B–B sigma linkage which would be expected to be ca. 1.6 Å, and the bridged two boron atoms are part of a diffuse multicenter bonding matrix.
Schematic Structures of iso-S2B16H16 and 4,4-(HS)2-anti-B18H20
Second, regarding the reversion of 2a into 1a ^ – ^, DFT calculations show, for the sulfur exemplar:
- 1.Deprotonation at the {H–S} position in 2a produces an anion, [S_2_B_17_H_18_]^ – ^ (2a ^ – ^), which is 14.6 kcal/mol lower in energy than for the deprotonation of any of the cluster bridging hydrogen atoms in 2a (see below), and thus is the most likely candidate for deprotonation in compounds 2.
- 2.This [S_2_B_17_H_18_]^ – ^ anion 2a ^ – ^, from deprotonation of μ-{H–S}, is 9.3 kcal/mol higher in energy than isomeric [S_2_B_17_H_18_]^ – ^ anion 1a ^ – ^ into which it rearranges via transition state 1 (TS1).
The hydrogen atoms bridging to the commo boron atoms in *anti-*B_18_H_22_ are known to be highly acidic,? and compounds 2, as analogues of *anti-*B_18_H_22_, likewise each hold three acidic bridging hydrogen atoms. Among the two possible bridging hydrogen atoms, DFT calculations reveal that deprotonation of H(6,7′) on 2 (Hc on 2a in Scheme) yields the lowest-energy structure. However, rather than deprotonation at a bridging site, deprotonation at the {SH} site (Hd in 2a) leads to formation of the [S_2_B_17_H_18_]^ – ^ anion 2a ^ – ^, with a bare bridging sulfur atom, which, as summarized above, is 14.6 kcal/mol lower in energy than the H(6,7′)-deprotonated anion (Hc). Thence, the anion 1a ^ – ^ is 9.3 kcal/mol lower in energy than the isomeric 2a ^ – ^ from which it rearranges. This rearrangement involves the breaking of the B(7′)-B(5) connectivity and reformation of the S(11′**)–B(**7′) linkage. The increased integration of the chalcogen atom into the cluster bonding framework and the resultant injection of electron density reflect in the open-face hydrogen-atom reorganization.
Third, regarding the transformation of compounds 2 into compounds 3, process (c) in Scheme:
The bridging {EH} unit can subsequently reinsert into a more intimate cluster environment upon loss of dihydrogen, ultimately forming the 19-vertex E_2_B_17_H_17_ 3 (Scheme). This reinsertion involves formation of new E(11′)–B(7′) and E(11′)–B(5) connectivities. Intuitively, the eliminated H_2_ molecule derives from the μ-H(7′,5) proton and the {EH} hydride. Indeed, protonation of anion 1a ^ – ^ with D_2_SO_4_ ultimately produces S_2_B_17_H_17_ 3a in which all exo-terminal and bridging atoms are hydrogen, i.e., the deuterium on sulfur is lost as HD. The required structural transition would involve the exo-{E–H} thiol/selenol unit (shown in Figure) adopting an inwardly pointing endo-{E–H} polyhedral vector, thereby enabling a configuration that facilitates dihydrogen loss to afford compounds 3 as illustrated with the encircled hydrogen atoms in Scheme(c). DFT calculations show that the prospective species with endo **-**polyhedral orientation for S_2_B_17_H_19_ 2a are only 3 kcal/mol higher in energy than the exo-terminal configuration; the low endo-to-exo barrier of 11.2 kcal/mol for S_2_B_17_H_19_ indicates a feasible transition for this postulated first step. It may be noted here, with regard to the refinement of the SCXRD data and the derived positions of the hydrogen atoms, that calculated selenium magnetic nuclear shielding data for the hypothetical 2b species featuring an endo-polyhedral {Se–H} hydrogen yields a δ(^77^Se) value of −500 ppm, approximately 100 ppm more shielded than the experimental and calculated values for 2b(exo); this therefore eliminates the endo possibility in the solid state structural model. This emphasizes the value of accurate chemical shielding predictions in selenaborane systems for confirming structural assignments.?
Finally, and uniquely in the case of selenium, a competing process emerges –process (d) –in which the chalcogen atom can be eliminated altogether, with retention of its associated {SeH} hydrogen atom, leading to the 18-vertex SeB_17_H_19_ 4, as we have previously reported.? This competing pathway does not occur to any observable extent in the corresponding sulfur system.
Conclusion
The isolation and structural characterization of the metastable 19-vertex S_2_B_17_H_19_ 2a provides the identity of the intermediate in the acid-mediated transformation of anions [E_2_B_17_H_18_]^−^ 1 ^ – ^ to the known neutral chalcogenaboranes E_2_B_17_H_17_ 3, ?,? the remarkable luminescent properties of which have only recently been described.? This rearrangement involves an unusual, and unusually reversible, extrusion of a chalcogen vertex, accompanied by significant topological reorganizations of the boron framework from “arachno” to “nido” character in both subclusters. The ability of 2a and 2b to undergo this reversible structural reorganizationincluding reintegration of the extruded {EH} unit via dehydrogenationunderscores the dynamic and versatile nature of chalcogenaborane clusters. The reversibility of this rearrangement positions this chemistry within a broader trend of boronotropic processes, wherein proton transfer events mediate skeletal reintegration and molecular “self-repair”. This behavior contrasts with many classical borane rearrangements, such as the protonation of the polyhedral anion [closo-B_10_H_10_]^2–^ under superacidic conditions,? which are typically irreversible once the cluster has relaxed into a lower-energy polyhedron.
To provide further context, there are a number of base-induced extrusion and reinsertion of vertices in metallaborane clusters that have previously been observed on the addition or removal of certain Lewis bases (L = PMe_2_Ph or PPh_3_). For example, [(PPh_3_)2_CO-nido-B_5_H_9] affords [(PPh_3_)CO-exo-arachno-B_4_H_8_-(BH_2_•L)] on addition of phosphine.? Similarly, Lewis base induced vertex isomerizations have been reported in 10-vertex nido-metalladecaboranyl species such as 8-[(C_2_H_5_)3_N(CH_2)4_O]-6-(CO)3-6-(CO)3-6-MnB_9_H_12 and [6,6,6-(PPh_3_)2_CO-nido-6-B_9_H_13] which were rationalized by intramolecular vertex “shifts”? or “swings”? around the cluster and, in the case of [9-(C_5_Me_5_)-nido-6,9-NRhB_8_H_11_], a thermally induced rearrangement via a proposed transition state involving the extrusion of one cluster vertex. More pertinent to the work here, cluster isomerizations induced by protonation, and reversed by deprotonation have been observed in the dicarbadecaboranyl species X-nido-5,6-C_2_B_8_H_11_ (X = Cl, Br, I), with the proposed mechanism supported by DFT calculations.? Here the authors proposed intramolecular rearrangements to rationalize the products and they excluded the vertex swing or extrusion mechanisms.
Thus, the E_2_B_17_H_19_ 2 species are examples that combine aspects of both features, the extrusion of a vertex induced by protonation and deprotonation. Taken together, these results demonstrate that chalcogen substitution enables a previously inaccessible regime of reversible cluster editing, one that challenges traditional assumptions about borane rigidity. They demonstrate how the addition or removal of a single proton can propagate structural change across an entire macropolyhedral framework, offering new mechanistic insight into the dynamic behavior of electron-deficient clusters and suggesting future opportunities in the design of responsive molecular architectures. The structural and behavioral versatility compared to binary borane macropolyhedra is caused by the valence flexibility of sulfur and selenium as compared to the first-row element boron.
Experimental Section
The preparation of S_2_B_17_H_19_ and Se_2_B_17_H_19_ essentially followed the published method for the preparation of E_2_B_17_H_17_ 3 and SeB_17_H_19_ 4.? In an NMR tube a concentrated dichloromethane solution of the N,N′-dimethyl-naphthalenediamine (tmnd, Proton Sponge) salt of the [S_2_B_17_H_18_]^−^ anion (60 mg, 0.13 mmol in 0.4 mL CH_2_Cl_2_) was immersed in a cold bath (−20 to −30 °C) and concentrated H_2_SO_4_ (ca. 0.2 mL of 95%, VWR Chemicals BDH) was quickly added and the mixture then agitated. The boron spectrum was measured at −20 °C and the sample ejected from the spectrometer and warmed slightly in the hand and then reinserted. This was repeated until all the starting material had reacted to give the S_2_B_17_H_19_ metastable intermediate 2a and before significant amounts of S_2_B_17_H_17_ 3a had formed. The initial formation of 2a is quantitative by NMR but the subsequent rate of 3a formation increases at room temperature and the isolation of large amounts of pure 2a is not feasible. Solutions of [tmnd][Se_2_B_17_H_18_] 1b were treated similarly but this system is much more difficult to handle and to obtain reasonably impurity-free product 2b. Many attempts were required for both 2a and 2b and no reliable isolated yields could be established. Single crystals for X-ray diffraction analysis were obtained by holding a highly concentrated dichloromethane solution of S_2_B_17_H_19_ 2a in an NMR tube in a freezer at −25 °C for ca 2 weeks. A mass of yellow crystalline and semicrystalline material formed. The compound is very soluble in dichloromethane and so, while still cold, the NMR tube was broken open to drain away the solvent as rapidly as possible and before it could redissolve. A suitable crystal was discovered in the isolated mass of material.
Notes on Acid
Used
Sulfuric acid was found to be the most useful acid in practice. This formed a 2-phase mixture, which allowed the very simple decanting of the dichloromethane layer. Indeed, when cold, the acid is immobile and it allows to pour off the dichloromethane while the acid remains in the NMR tube. CF_3_COOH does protonate the anion but, as a liquid, it requires the removal of the solvent and the acid together by evaporation. This leads to deprotonation of compounds 2 by the Proton Sponge and reformation of 1.
NMR Spectroscopy
NMR spectra were recorded on a JEOL ECZ 600 R (14.1 T) spectrometer, using ^77^Se, ^11^B, ^11^B{^1^H}, ^1^H, ^1^H{^11^B(broadband)}, ^1^H{^11^B(selective)}, and HMQC (Heteronuclear Multiple-Quantum Correlation) techniques. NMR spectra of all compounds were measured in CDCl_3_ solution. ^11^B chemical shifts are given relative to BF_3_·OEt_2_, δ^11^B = 0.0 ppm for Ξ(^11^B) = 32,083,971 Hz. ^77^Se chemical shifts are reported relative to SeMe_2_, δ(^77^Se) = 0.0 ppm for Ξ (^77^Se) = 19,071,523 Hz.
^77^Se NMR spectra were measured using a standard single-pulse sequence (available from the spectrometer library) with a 90° pulse length and relaxation delay of 0.1–0.2 s. The line widths of the ^77^Se resonances were in the range 200–300 Hz and the spectra were recorded with an FID resolution of 3–4 Hz.
Computational
Details
Calculations were performed using the Gaussian16 package.? The DFT/B3LYP methodology was employed for the calculation of boron and hydrogen nuclear shieldings with the 6-31+G(d,p) basis sets for B and H and the Binning and Curtiss 962+d polarization basis set for Se taken from BasisSetExchange.? The selenium nuclear shielding calculations were carried using the same basis sets and the mPW1PW91 methodology. The Polarizable Continuum Model was implemented with CH_2_Cl_2_ solvation. Frequency calculations at the appropriate level confirmed the theoretical geometries as energy minima. Transition states TS1 and TS2 were calculated using the Synchronous Transit-guided Quasi-Newton method,? QST3, and the B3LYP/6-31+G(d,p) method and basis sets and confirmed by Intrinsic Reaction Coordinate (IRC) calculations. See Supporting Information for TS1 and TS2 animations.
X-ray Crystallography
X-ray diffraction analysis was performed with an XtaLAB Synergy, Dualflex, HyPix diffractometer. The crystal was kept at 100 K during data collection. CrysAlisPro software was used for data collection and cell refinement, data reduction and absorption correction.? Data were corrected for absorption effects using a semiempirical absorption correction (spherical harmonics), implemented in the SCALE3 ABSPACK scaling algorithm; neither a numerical absorption correction based on Gaussian integration over a multifaceted crystal model? nor an analytical absorption correction made any significant difference to the data. ?,? The structure was solved with the ShelXT structure solution program? using an iterative dual-space method and refined with the ShelXL? refinement package using least-squares minimization, native and implemented in Olex2.? Anisotropic displacement parameters were refined for all non-H atoms. The hydrogen atoms were calculated to idealized positions based on the DFT-calculated structure.
Crystallographic data for structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 2452393. Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge CB2 1EY, U.K. (fax: + 44–1223–336033; Email: [email protected] or http://www.ccdc.cam.ac.uk).
Crystal Data for S_2_B_17_H1_19_ 8 (M = 267.04 g/mol): monoclinic, space group P2_1_/n (no. 14), a = 21.9921(12) Å, b = 6.7157(2) Å, c = 22.1404(13) Å, β = 117.789(7)°, V = 2892.8(3) Å^3^, Z = 8, T = 100 K, μ(Cu–Kα) = 2.947 mm^–1^, D calc = 1.226 g/cm^3^, 8398 reflections measured (7.756° ≤ 2θ ≤ 154.206°, 5811 unique, R int = 0.1681, R sigma = 0.0203) which were used unmerged in all calculations because of the twinning. The final R 1 was 0.1190 (I > 2σ(I)) and wR 2 was 0.3789 (all data). Twinning and whole-molecule disorder of both independent molecules in the asymmetric unit of this structure hampered solution, which was eventually achieved by recognizing the probably correct framework after fitting appropriate idealized rigid cluster fragments to selected observed electron density peaks; theoretical calculations optimizing the geometry of this S_2_B_17_ framework and identifying H atom positions, satisfactorily matching the observed NMR spectra for this and the Se analogue, provided a starting rigid-geometry model for fitting the major and minor disorder components, which was then relaxed with appropriate restraints on geometry and displacement parameters and constrained riding H atoms to give the final refinement. Relatively high crystallographic R factors and residual electron density indicate imperfect modeling of the disorder and/or twinning.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1b Housecroft, C. E. Boranes and Metallaboranes: Structure, Bonding and Reactivity; Halsted Press, 1994; Vol. 1994.
- 2a Bould J.Londesborough M. G. S.LiteckáM.Macías R.Shea S. L.Mc Grath T. D.Clegg W.Kennedy J. D.Macropolyhedral Chalcogenaboranes: Insertion of Selenium into the Isomers of B 18H 22 Inorg. Chem.20226141899191710.1021/acs.inorgchem.1c 0301835049289 · doi ↗ · pubmed ↗
- 3a Jelínek T.Kennedy J. D.Štíbr B.Thornton-Pett M.Macropolyhedral Boron-Containing Cluster Chemistry: Isolation and Characterization of the First Macropolyhedral Thiaborane, the arachno-Type [9,9′-S 2B 17H 18]− Ion Angew. Chem., Int. Ed. Engl.19943315–161599160110.1002/anie.199415991 · doi ↗
- 4Jemmis E. D.Balakrishnarajan M. M.Pancharatna P. D.A unifying electron-counting rule for macropolyhedral boranes, metallaboranes, and metallocenes J. Am. Chem. Soc.2001123184313432310.1021/ja 003233 z 11457198 · doi ↗ · pubmed ↗
- 5Grimes, R. N. Carboranes, 3rd ed.; Elsevier: Oxford, UK, 2016.
- 6Bould J.Tok O.LiteckáM.Londesborough M. G. S.Ehn M.Two New Macropolyhedral Chalcogenaboranes: S 2B 16H 18 and Se B 16H 18 Inorg. Chim. Acta 2022542112114810.1016/j.ica.2022.121148 · doi ↗
- 7a Binder H.Ziegler A.Ahlrichs R.Schiffer H.μ4-S(B 2H 5)2, (H 2BSH)2, 1,2-(HS)2B 2H 4: Neue Thiaborane aus der Reaktion zwischen Diboran und Schwefelwasserstoff Theoretische Untersuchung der Molekülstrukturen Chem. Ber.198712091545155210.1002/cber.19871200913 · doi ↗
- 8Bould J.Londesborough M. G. S.Tok O.Experimental and Computational 77Se NMR Spectroscopic Study on Selenaborane Cluster Compounds Inorg. Chem.202463161861619310.1021/acs.inorgchem.4c 0189039160773 PMC 11372749 · doi ↗ · pubmed ↗