Regioselective Hydrosilylation Catalysis with Supported Well-Defined Pt(0) Complexes: Effects of Surface Anions and Phosphenium Ligands
Damien B. Culver, Conor Neill, Frédéric A. Perras, Mita Halder, Angela Chartouni

TL;DR
Scientists created stable, selective catalysts using platinum and surface-bound ligands, achieving high performance in chemical reactions.
Contribution
The study introduces a new method for creating selective single-atom catalysts using surface-bound phosphenium ligands and Pt(0) complexes.
Findings
Bulky aromatic ligands improve regioselectivity in hydrosilylation reactions.
Stronger-coordinating anions enhance the selectivity of the precatalysts.
Surface-bound Pt(0) complexes perform comparably to molecular Pt catalysts.
Abstract
Achieving stable, selective single-atom catalysts is challenging because localsurface-site structures are difficult to control. Surface organometallic chemistry and organic–inorganic hybrid materials offer partial solutions, but applications to supporting zero-valent metals are limited. We demonstrate that well-defined, ionically bound N-heterocyclic phosphenium ([NHP]+) ligands can be generated on silylium-functionalized sulfated zirconia ([iPr3Si][SZO]). These surface-bound [NHP][SZO] ligands coordinate Pt(0) centers, forming [(NHP)Pt(0)L][SZO] precatalysts that are highly active for alkyne hydrosilylation. Systematic studies reveal that sterically bulky aromatic ligands enhance regioselectivity, achieving performances comparable to molecular Pt catalysts. Further, more-coordinating anions support more-regioselective precatalysts; therefore, SZO supports more-selective species…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
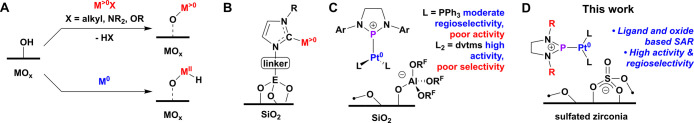 Figure 1
Figure 1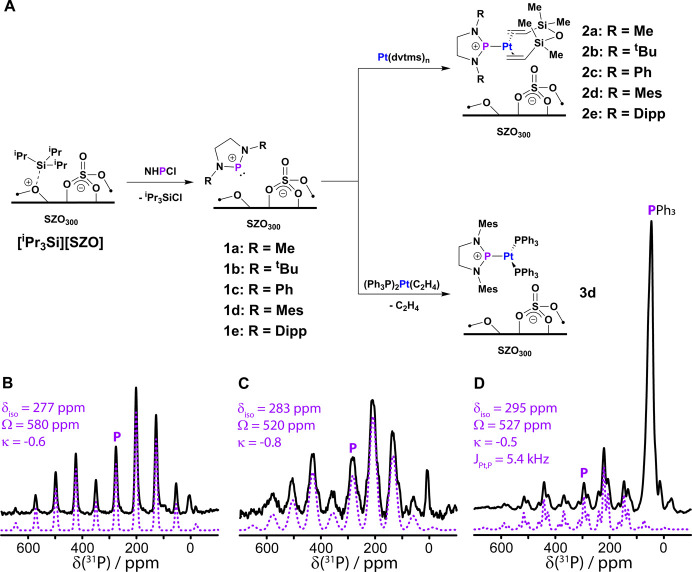 Figure 2
Figure 2 Figure 3
Figure 3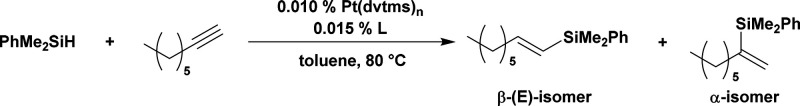 Figure 4
Figure 4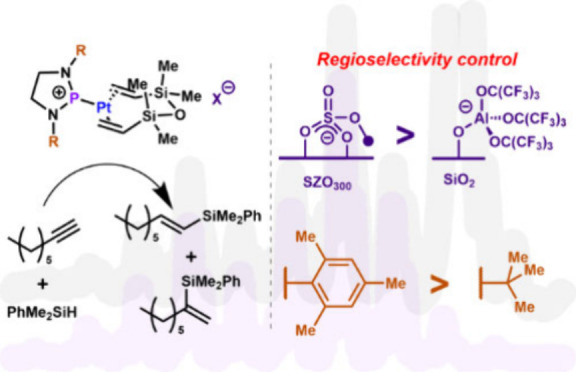 Figure 5
Figure 5- —Workforce Development for Teachers and Scientists10.13039/100006210
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganoboron and organosilicon chemistry · Catalytic Cross-Coupling Reactions · Catalytic C–H Functionalization Methods
The formation of selective heterogeneous single-atom catalysts (SACs) remains a significant challenge, particularly for low-valent transition metals, which are generally unstable on support surfaces. ?−? ? ? ? ? ? A key example is Pt(0) SACs, which are difficult to stabilize for reactions such as regioselective alkyne hydrosilylation. ?−? ? ? ? Conventional oxide-supported Pt particle or single-atom hydrosilylation catalysts typically require high metal loadings and fail to match the selectivity of molecular catalysts. This is partially attributable to the plurality of active sites in these systems, which prevents the structural control required to develop and leverage structure–activity relationships (SARs).
Surface organometallic chemistry (SOMC) combines molecular and surface strategies to create well-defined heterogeneous catalysts, primarily for non-zero-valent metals. ?−? ? ? ? ? ? ? While SOMC has enabled selective catalysis, including limited examples of alkyne and alkene hydrosilylation, ?−? ? ? ? SOMC typically relies on chemisorption of M-X species via protonolysis, halide abstraction, ?,? or more recently addition of zero-valent metals through oxidative grafting (FigureA). ?−? ? ? ? All of these result in metals in oxidation states greater than zero. Supporting zero-valent metals remains challenging, as reducing conditions often yield nanoparticles. ?−? ? ? ? Molecular ligands stabilize zero-valent metals; functionalizing supports with these ligands should enable anchoring such species and studying structure–activity relationships in SACs. Ligands tethered to polymers, graphene, or oxides form organic–inorganic hybrid materials (OIHMs), such as N-heterocyclic carbenes (NHCs) on silica, which coordinate metals for heterogeneous catalysis (FigureB). ?−? ? However, these syntheses are costly, labor-intensive, and limited to non-zero-valent metals.
We showed that N-heterocyclic phosphenium ([NHP]^+^) ligands ?−? ? ? can be generated on Lewis-acid-functionalized silica (ASO) without a tether, enabling the first well-defined Pt(0) complexes for alkyne hydrosilylation (FigureC).? These precatalysts are highly active but poorly regioselective at low loadings compared to molecular NHC-Pt analogues, likely due to limited stability and electronic differences.? We hypothesized that alternative [NHP]^+^ ligands could improve the regioselectivity and strengthen the P–Pt bond. Because functionalized silica decomposes at elevated temperatures, ?,? we replaced it with thermally stable sulfated zirconia (SZO), which also enabled the study of support effects (FigureD). ?,?,?
[NHP][SZO] ligands were synthesized by chloride abstraction from NHPCl ?−? ? ? precursors using [^i^Pr_3_Si][SZO] ?,? (FigureA), following the approach for [DippNHP][ASO].? ICP-OES and ^i^Pr_3_SiCl quantification confirmed P loadings of 0.067–0.136 mmol/g, consistent with ∼1 [^i^Pr_3_Si] site reacting per P (Table S1). Compounds 1a–e were characterized by ^1^H, ^13^C, and ^31^P solid-state (SS)NMR spectroscopy in addition to 2D ^1^H homonuclear double-quantum/single-quantum (DQ/SQ) correlation. The ^31^P spectrum of 1d (FigureB) and others in the Supporting Information (SI) contain signals at 252–277 ppm, comparable to those of [DippNHP][ASO] (^31^P δ = 272 ppm) and molecular analogues. ?,?
^1^H and ^13^C SSNMR spectra further support the proposed structures.
Treatment of 1a–e with Karstedt’s catalyst? (FigureA) generates well-defined [(NHP)Pt(dvtms)][SZO] sites analogous to previously reported ASO-supported species (see SI). ICP-OES analysis confirmed Pt coordination (0.038–0.081 mmol/g) and revealed a slight decrease in phosphorus content, likely due to the partial re-formation of NHPCl from reactions with physisorbed ^i^Pr_3_SiCl not fully removed during toluene washings. The P:Pt ratios are 0.9–1.9, with some samples containing unreacted [NHP][SZO] (see Table S1 and SSNMR in SI), and the dvtms:Pt ratios range from 0.6 to 1.3. Samples 2a and 2b are deficient in dvtms, which may result from bis-coordination of [NHP]^+^ ligands forming minor [(NHP)_2_Pt][SZO] sites, consistent with their high phosphorus loadings (>0.6 P/nm^2^). MesNHPOTf? reacts with Karstedt’s catalyst to form [(NHP)(NHP(OTf))Pt][OTf] in solution (see SI), supporting that bis-coordination is possible. In contrast, 2c–e exhibit a dvtms:Pt ratio near 1.0, supporting the stoichiometry in FigureA.
2a–e were characterized by SSNMR spectroscopy, and the ^31^P SSNMR spectrum of 2d is provided in FigureC as an example (see SI). In all cases, the ^31^P SSNMR spectra confirm the successful formation of the [(NHP)Pt(0)(dvtms)]^+^ species. The ^31^P SSNMR resonances are broadened by the interaction with Pt, and they all shift to higher ppm (277–292 ppm), making them comparable to those of other [(NHP)Pt(0)L_n_] cations. ?,?,? The broadening of the ^31^P NMR signal is likely due to a distribution in the strength of the Pt–P bonding or from the dynamics of the complexes. For instance, we see the appearance of millisecond-order dynamics in 2a and 2c, which prevents the uniform cross-polarization of the spinning sideband pattern, even at 100 K. As a result, our attempts at ^1^H-detecting the ^195^Pt NMR signals from these species using fast-MAS were unsuccessful. ?−? ? Indeed, even the ^1^H–^31^P dipolar interactions are undetectable at room temperature. The ^1^H SSNMR spectra acquired for 2a–e (see SI) displayed resonances unique to the [NHP]^+^ and dvtms ligands, suggesting the structure is similar to (NHC)Pt(dvtms) complexes, as proposed (FigureA),? although most clear double-quantum correlations were intraligand. We also synthesized [(MesNHP)Pt(0)(PPh_3_)2][SZO] (3d), which contains 0.045 mmol/g of Pt and 3.2 equiv of P (FigureA). The ^31^P SSNMR of 3d (FigureD) is similar to that of [(DippNHP)Pt(0)(PPh_3_)2][ASO] and exhibits a resolvable J Pt–P coupling of 5.4 kHz, comparable to spectra of molecular Pt(0) phosphenium complexes, confirming coordination. ?,?
A major challenge in heterogeneous catalysis is achieving control over SARs. Precatalysts 2a–e and 3d exhibit tunable steric and electronic properties, offering a valuable platform for studying SARs. Precatalysts 2a–e demonstrate high activity in the hydrosilylation of 1-octyne, achieving full conversion at loadings of 0.005–0.05 mol% Pt within 8 h (Table). Notably, activity and regioselectivity vary systematically with precatalyst structure. Complexes bearing bulky ligands, 2b, 2d, and 2e, exhibit high turnover frequencies (TOFs > 400 min^–1^ at 15 min), whereas less-bulky 2a and 2c are less than half as active. This trend suggests that reductive elimination is rate-limiting, paralleling molecular systems.?
Notably, regioselectivity correlates with ligand electronics and steric bulk. Electron-withdrawing aryl R groups in 2c–e yielded the highest selectivities, with 2c being slightly more selective than 2a and 2b, and 2d being the most selective. 2b shows selectivity similar to that of 2a and lower than that of 2c despite containing sterically larger ^t^Bu groups, highlighting the importance of bulky and electron-withdrawing ligands. Interestingly, 2e is more selective than the ASO analogue (6.2–7),? suggesting there may be an effect from the anionic support.
We observed a positive correlation between the regioselectivity and precatalyst loading for 2d and 2e. Doubling the loadings from 0.005 to 0.01 mol% increases the β-(E)/α from 9.8 and 8.9 to 12.9 and 9.8 for 2d and 2e, respectively. Further increasing the 2d loading to 0.05 mol% raises the β-(E)/α to 17.5. The absence of similar increases for 2b suggests the higher selectivity of 2d and 2e may be due to kinetic effects involving alkyne orientation by bulky aryl ligands; similar effects have been observed in molecular complexes.? 2d and 2e are the most active and regioselective solid-state Pt alkyne hydrosilylation precatalysts reported to date ?,?,?,? and are comparable to the best molecular catalysts. ?,?,?−? ? ?
Similarly, 3d produced a regioselectivity of 22.5 at 0.45 mol% Pt, double the ratio of [(DippNHP)Pt(PPh_3_)2][ASO].? 3d is also more active at lower Pt loadings, completing the reaction in 2 days at 0.045 mol% Pt, whereas [(DippNHP)Pt(PPh_3_)2][ASO] achieved only 22% yield under comparable conditions, showcasing 3d’s improved stability. The regioselectivity, nevertheless, dropped over time from an initial selectivity of 9.9 to a final ratio of 7.2 (see SI), unlike 2a–e. A similar observation was made at 0.0045 mol% Pt. This likely reflects PPh_3_-induced leaching that forms poorly selective Pt particles. ?,?
A significant challenge in the stabilization of single-atom Pt(0) and other low-valent metals is leach prevention. ?,?,?,? Low-coordinate Pt with small ligands, including the precursor Karstedt’s catalyst, produces β-(E)/α ratios of ∼5–6,? so Pt released from [NHP]^+^ in 2a–e would lower selectivity. Hot-filtration tests (0.05 mol% Pt) after partial silane consumption (<54%) at 80 °C showed continued and comparable product formation in filtrates, indicating the formation of soluble species (see SI). ICP-OES of the 2d filtrate detected small amounts of Pt and Zr, and SEM images of SZO? revealed <1 μm agglomerates, supporting that [NHP-Pt]^+^-bearing SZO particles leach into solution.
Further tests with 2d (0.005 mol% Pt) confirmed that most particles are <0.1 μm, as Celite and fine frit filtration only modestly reduced filtrate activity. Heating 2d with 1-octyne or PhMe_2_SiH before filtration and then adding the other substrate cut filtrate-catalyzed product yields to <50%, with the 1-octyne-contacted precatalyst filtrate being least active. This suggests that heat and silane disrupt SZO particle-particle cohesion, and silane may stabilize particles by reacting with pyrosulfates? or residual hydroxyls. Catalyst reuse (0.08 mol% Pt) after partial silane conversion (44%) and hot-filtration showed ∼75% yield loss after one cycle and a gradual decline over four cycles, while regioselectivity dropped slightly (14→12). Despite leaching, residual activity and maintained regioselectivity indicate that the local structure persists and Pt clusters do not form.
The difference in regioselectivity between products formed by 2e and [(DippNHP)Pt(0)(dvtms)][ASO] suggests that the support plays a significant role in catalytic performance, beyond acting as a passive counterion. Prior studies showed that SZO surfaces coordinate cations more strongly than ASO.? Similar support effects have been observed for SOMC catalysts supported on sulfated oxides. ?,?−? ? To evaluate the influence of the anion on catalysis, we generated molecular precatalyst analogues by combining Karstedt’s catalyst with [MesNHP]^+^ with a non- ([B(C_6_F_5_)4]^−^) or weakly (OTf^–^) coordinating anion (see SI)? and compared their regioselectivities (Table). The OTf^–^ supported a more regioselective catalyst than [B(C_6_F_5_)4]^−^, comparable to precatalyst 2d, indicating that more-coordinating anions enhance regioselectivity. We further established that the ligand structural trend observed in 2a–e applies to the molecular analogues, with smaller MeNHPOTf and PhNHPOTf generating less regioselective catalysts than MesNHPOTf. Combinations of the ligand and Karstedt’s catalyst confirm coordination to form [NHP-Pt]^+^ species by NMR (see SI). SSNMR of 1d and 1e showed no evidence of surface interaction with the phosphorus cation, but it is plausible that sulfates coordinate to Pt or P during catalysis. ?,?,? These findings suggest that the surface sulfates may contribute to regioselectivity by acting as hemilabile ligands.
In summary, sulfated zirconium oxide (SZO) supports well-defined [NHP]^+^ OIHM ligands that coordinate the Pt(0) centers for alkyne hydrosilylation. SAR studies reveal that sterically bulky ligands enhance catalytic activity, while aromatic ligands yield higher regioselectivity than alkyl counterparts. Precatalysts 2d and 2e exhibit activities and selectivities comparable to those of molecular systems, positioning them as promising heterogeneous alternatives. Preliminary molecular studies suggest that the anion significantly influences regioselectivity. These insights provide a foundation for designing next-generation OIHM catalysts. Ongoing work is focused on further exploring the role of the supporting anion and elucidating the mechanism of this emerging class of supported Pt(0) complexes.
Safety Statements
Caution!
-
Extreme care should be taken in both the handling of the cryogenic liquid nitrogen and its use in the Schlenk line trap to avoid the condensation of oxygen from air.
Caution!
- The GHS Category 1 corrosive and toxic sulfuric acid (also oxidizing), hydrofluoric acid, nitric acid (also oxidizing), hydrochloric acid, and phosphorus trichloride constitute significant safety hazards and must be handled with extreme care.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ji S.Chen Y.Wang X.Zhang Z.Wang D.Li Y.Chemical Synthesis of Single Atomic Site Catalysts Chem. Rev.202012021119001195510.1021/acs.chemrev.9b 0081832242408 · doi ↗ · pubmed ↗
- 2Kaiser S. K.Chen Z.Faust Akl D.Mitchell S.Pérez-Ramírez J.Single-Atom Catalysts across the Periodic Table Chem. Rev.202012021117031180910.1021/acs.chemrev.0c 0057633085890 · doi ↗ · pubmed ↗
- 3Li J.Stephanopoulos M. F.Xia Y.Introduction: Heterogeneous Single-Atom Catalysis Chem. Rev.202012021116991170210.1021/acs.chemrev.0c 0109733172277 · doi ↗ · pubmed ↗
- 4Mitchell S.Pérez-Ramírez J.Single Atom Catalysis: A Decade of Stunning Progress and the Promise for a Bright Future Nat. Commun.2020111430210.1038/s 41467-020-18182-532855411 PMC 7453014 · doi ↗ · pubmed ↗
- 5Zhang Z.Li H.Wu D.Zhang L.Li J.Xu J.Lin S.Datye A. K.Xiong H.Coordination Structure at Work: Atomically Dispersed Heterogeneous Catalysts Coord. Chem. Rev.202246021446910.1016/j.ccr.2022.214469 · doi ↗
- 6Endo K.Saruyama M.Teranishi T.Location-Selective Immobilisation of Single-Atom Catalysts on the Surface or within the Interior of Ionic Nanocrystals Using Coordination Chemistry Nat. Commun.2023141424110.1038/s 41467-023-40003-837454144 PMC 10349889 · doi ↗ · pubmed ↗
- 7Fang J.Chen Q.Li Z.Mao J.Li Y.The Synthesis of Single-Atom Catalysts for Heterogeneous Catalysis Chem. Commun.202359202854286810.1039/D 2CC 06406 E 36752217 · doi ↗ · pubmed ↗
- 8Troegel D.Stohrer J.Recent Advances and Actual Challenges in Late Transition Metal Catalyzed Hydrosilylation of Olefins from an Industrial Point of View Coord. Chem. Rev.2011255131440145910.1016/j.ccr.2010.12.025 · doi ↗