Intermittent White Urine With Nephrotic-Range Proteinuria and Preserved Renal Function: A Diagnostic Dilemma Between Chyluria, Pseudochyluria, and Amyloidosis
Firdaus Jabeen, Vaibhav Shukla, Salil Vallecha, Monis Khan, Saboor Mateen

TL;DR
A woman with white urine and kidney issues had a difficult diagnosis between chyluria, pseudochyluria, and amyloidosis.
Contribution
The case highlights diagnostic challenges in distinguishing chyluria from pseudochyluria and amyloidosis with limited tissue samples.
Findings
The patient had nephrotic-range proteinuria with preserved kidney function and intermittent white urine.
Tests for chyluria, tuberculosis, and amyloidosis were negative despite suggestive symptoms.
The case illustrates diagnostic uncertainty when tissue samples are non-representative.
Abstract
Milky or “white” urine is usually attributed to chyluria but may also result from lipid-rich proteinuric urine or crystals (“pseudochyluria”). We describe a 47-year-old woman with a 6-month history of intermittent white urine, bilateral edema, nephrotic-range proteinuria (20.3 g/24 h), preserved renal function, near-normal serum albumin, and chronic bronchiectasis. Evaluation for parasitic and non-parasitic chyluria, tuberculosis, and systemic amyloidosis was repeatedly negative. A fasting urine sample showed markedly raised triglycerides but negative tests for chyle. The kidney biopsy was non-representative, and the abdominal fat-pad biopsy was Congo-red-negative. Serum anti-PLA2R antibodies and free light chains were normal. The case illustrates the gray zone between chyluria and pseudochyluria and the challenge of defining the underlying glomerular lesion when tissue diagnosis is…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1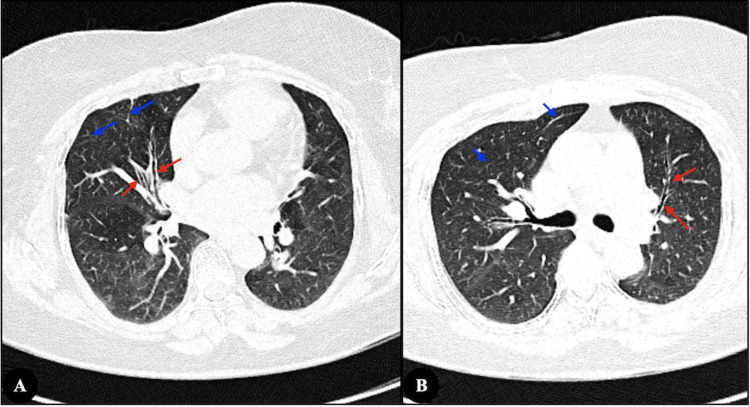 Figure 2
Figure 2| Test | Day 1 | Day 30 | Day 60 | Day 90 | Day 120 | Day 180 | Day 240 | Normal Range |
| Hemoglobin (Hb) | 15.4 | 13.5 | 12.5 | 12.6 | 12.2 | 13.0 | 12.3 | 12.0 to 15.5 g/dL |
| Total Leucocyte Count (WBC) | 10600 | 10000 | 7000 | 7500 | 8500 | 11000 | 7800 | 4000-11000 Cells/cumm |
| Platelet Count | 4.2 | 3.0 | 3.0 | 2.8 | 2.8 | 3.2 | 2.8 | 1.5 - 4.5 Lakh /cumm |
| Blood Urea | 20 | 22 | 21 | 14 | 20 | 22 | 11 | 6 to 21 mg/dL |
| Creatinine | 0.7 | 0.5 | 0.7 | 0.7 | 0.6 | 0.6 | 0.6 | 0.6 to 1.1 mg/dL |
| Serum Bilirubin (Total) | 1.1 | 1.0 | 0.6 | 0.9 | 0.5 | 1.0 | 1.1 | 0.2 - 1.3 mg/dl |
| Prothrombin Time | 12.5 | 12.5 | 12.2 | 11.7 | 11.9 | 12.0 | 11.3 | 9.8-12.1 |
| INR | 1.1 | 1.1 | 1.07 | 1.0 | 0.91 | 1.02 | 1.02 | 0.6-1.5 |
| Serum Calcium | 8.5 | 8.6 | 8.4 | 8.2 | 7.8 | 8.4 | 8.9 | 8.6-10.2 mg/dl |
| Serum Phosphorus | 3.5 | 3.6 | 3.4 | 3.2 | 3.6 | 3.7 | 3.8 | 2.5-4.5 mg/dl |
| Total Protein | 6.1 | 5.9 | 6.4 | 7.0 | 8.2 | 7.0 | 6.1 | 6.4-8.3 g/dl |
| Serum LDH | 245 | - | - | - | - | - | 236 | 120-246 U/L |
| Serum Albumin | 3.4 | 3.2 | 3.6 | 3.7 | 4.7 | 3.8 | 3.6 | 3.5-5.2 g/dl |
| Serum Triglycerides | 138 | 184 | - | - | - | - | 144 | < 161.0 mg/dl |
| Serum Cholesterol | 173 | 206 | - | - | - | - | 201 | 200-239 mg/dl |
| Serum HDL Cholesterol | 65 | 70 | - | - | - | - | 54 | 42-88 mg/dl |
| Serum VLDL Cholesterol | 28 | 37 | - | - | - | - | 20 | 2-30 mg/dl |
| Direct LDL Cholesterol | 98 | 120 | - | - | - | - | 101 | <100 mg/dl |
| C- Reactive Protein | <5 | - | - | - | 7.7 | 8.8 | <5 | <10 mg/dl |
| Urine Appearance | Turbid White | Turbid White | Dark Yellow | Pale Yellow | Turbid White | - | Turbid white | None |
| Urine Protein | +++ | +++ | +++ | ++ | +++ | - | +++ | None |
| Urine Epithelial Cells | Occasional | Occasional | 2-4/hpf | 2-4/hpf | Occasional | - | Occasional | None |
| Urine Pus Cells | 24-24/hpf | Occasional | 2-4/hpf | 8-10/hpf | Occasional | - | Occasional | None |
| Urine RBC | Plenty/hpf | Nil | Nil | Nil | Plenty/hpf | - | Nil | None |
| Urine Sugars | Nil | Nil | Nil | Nil | Nil | - | Nil | None |
| Urine Cast/Crystals | Nil | Nil | Nil | Nil | Nil | - | Nil | None |
| Urine for Dysmorphic RBC | - | - | - | Not Detected | - | - | Not Detected | Not Detected |
| Urine Microalbumin | - | 6.77 | 705 | - | - | - | 0-20 mg/L | |
| Urine Protein | - | 142 | - | - | - | 0-14 mg/dL | ||
| Urine Creatinine | - | 74.30 | 92 | - | - | - | 28-217 mg/dl | |
| Urine Albumin-Creatinine Ratio | - | 9.11 | 766 | - | - | - | 0-0.20 mg/g creatinine | |
| Urine Protein/Creatinine Ratio | - | - | 1.33 | - | - | - | 0.00-0.20 | |
| Urine Triglycerides | 55 | - | - | < 10 mg/dl | ||||
| Urine Chyle | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| 24-Hour Urinary Protein | 21000 | 20304 | - | - | - | - | 18000 | 42 - 225 mg/day |
| Urine Culture | Sterile | Sterile | Sterile | Sterile | Sterile | Sterile | Sterile | Sterile |
| Sputum AFB (2 Days) | Negative | - | Negative | - | - | Negative | - | Negative |
| Sputum C/S | Growth of a non-pathogenic organism | - | Growth of a non-pathogenic organism | - | - | - | - | Growth of a non-pathogenic organism |
| Sputum Gram Stain | Pus cells >25, epithelial cells 10-25, numerous GPCs in clusters, chains, pairs, moderate GNB seen | - | No pus cells or epithelial cells, numerous GPCs in clusters, chains, pairs | - | - | - | - | Sterile |
| Urine AFB | Negative | - | Negative | - | - | - | - | Negative |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLymphatic Disorders and Treatments · Amyloidosis: Diagnosis, Treatment, Outcomes · Renal Diseases and Glomerulopathies
Introduction
Chylous urine most often reflects a lymphatico-urinary fistula from filarial disease or non-parasitic lymphatic obstruction and can mimic nephrotic syndrome when accompanied by edema and heavy proteinuria [1,2]. Turbid white urine may also arise from lipid-rich nephrotic urine, infection, or crystals (“pseudochyluria”) [2,3]. Chronic bronchiectasis is a recognised cause of secondary (AA) renal amyloidosis presenting with proteinuria and progressive renal dysfunction [4]. In settings where filariasis, tuberculosis, and chronic infections are endemic and access to repeat renal biopsy or mass spectrometry is limited, distinguishing chyluria, pseudochyluria, and amyloidosis can be difficult. We report a patient with intermittent white urine, massive proteinuria, preserved renal function, and bronchiectasis in whom extensive evaluation for chyluria and amyloidosis remained negative.
Case presentation
A 47-year-old woman presented with 6 months of intermittent, strikingly white, milky urine and progressive bilateral pitting pedal edema. There was no dysuria, colicky pain, fever, weight loss, or known lymphatic disease. She had a several-year history of chronic cough with intermittent purulent sputum; earlier cultures had grown mixed bacteria and Candida species. Screening for secondary immunodeficiency (HIV, glycemic status, and complete blood counts) was normal, and there was no history of recurrent severe infections or immunosuppressive therapy.
Blood pressure and systemic examination were unremarkable, apart from edema. Initial investigations showed a normal complete blood count and liver and renal function tests. A 24-h urine sample demonstrated nephrotic-range proteinuria of 20.3 g/day. Spot urinalysis repeatedly revealed turbid urine, 2+-3+ protein, microscopic hematuria, and pyuria without casts (Table 1, Figure 1).
Physical appearance of urine(A) Initial sample showing dense, white-yellow, turbid urine at presentation; (B) Follow-up sample after six months showing persistent white-yellow turbidity
Ultrasonography showed normal-sized kidneys with preserved corticomedullary differentiation and mobile linear echogenic strands in the bladder, raising the possibility of chyluria. CT abdomen was otherwise normal. High-resolution CT chest demonstrated bilateral lower-lobe air trapping, fibrotic bands with traction bronchiectasis in the right middle lobe and lingula, and calcified mediastinal nodes, consistent with long-standing post-infective bronchiectasis (Figure 2).
High-resolution computed tomography of the chest(A) Traction bronchiectasis in the medial segment of the right middle lobe (red arrows) with adjacent fibrotic streaks (blue arrows); (B) Traction bronchiectasis in the superior lingular segment of the left upper lobe (red arrows) with associated fibrotic bands (blue arrows)
Urine for chyle was negative on repeated testing, including an early-morning fasting sample. In the same fasting specimen, urinary triglycerides were markedly elevated (55.9 mg/dL; reference <10 mg/dL). Filarial antigen, urine, and sputum AFB smears, urine filarial antigen testing, urine and sputum AFB smears, urine GeneXpert MTB/RIF assay (cartridge-based nucleic acid amplification test), and Mantoux test were all negative; an interferon-γ release assay (QuantiFERON-TB Gold) could not be performed because of financial constraints. A native kidney biopsy was non-representative, with only scant renal cortex (three preserved glomeruli) and no immune deposits on immunofluorescence (IgG, IgM, IgA, C3, C1q, κ, λ all negative). Serum free light chains were within the normal ratio (κ 20.9 mg/L, λ 14.9 mg/L, κ/λ 1.40), and serum anti-PLA2R antibodies were negative (<2 RU/mL). Abdominal fat-pad biopsy was negative for amyloid on Congo red staining, and serum amyloid A was just above the normal range. After counselling about further options, the patient declined a repeat renal biopsy or additional invasive procedures.
She was treated conservatively with telmisartan 40 mg once daily, dapagliflozin 5 mg once daily, torsemide 10 mg on alternate days, and dietary sodium restriction. Pedal edema improved within a few weeks and remained controllable with intermittent low-dose diuretics. During approximately nine months of outpatient follow-up, serum creatinine stayed stable at 0.6-0.8 mg/dL and serum albumin rose from 3.2 g/dL to 4.4-4.7 g/dL, while 24-hour urinary protein showed only a modest decline (from about 21 g to 18 g/day).
Urine protein electrophoresis was not performed because the test is not routinely available at our center, and in view of normal serum free light chains and κ/λ ratio and the absence of clinical or laboratory features of plasma cell dyscrasia, the pre-test probability of monoclonal proteinuria was considered low.
Despite this biochemical stabilization, she continued to experience intermittent yellow-white turbid urine with persistent 3+ dipstick proteinuria. Based on the overall clinical picture, the final working diagnosis was intermittent pseudochyluria with nephrotic-range proteinuria and preserved renal function in the setting of chronic bronchiectasis, managed with ongoing conservative therapy and regular clinical-biochemical follow-up.
Discussion
This case highlights that milky urine with massive proteinuria does not automatically equate to primary glomerular disease or classic chyluria [1-3]. Negative chyle tests in repeated fasting samples, absence of filarial exposure or obstructive lymphatic pathology, and preserved serum albumin despite very high protein losses argue against overt chyluria [1,2]. Elevated urinary triglycerides in a fasting specimen confirm lipid-rich urine but are not pathognomonic for chyle and may also reflect lipiduria from nephrotic-range proteinuria [3]. The combination of intermittent white urine, raised urinary triglycerides, and negative chyle tests favours a mixed mechanism of lipid-rich nephrotic urine with possible low-grade lymphatic leakage that remains below the sensitivity of routine chyle assays [1-3].
In our patient, several features ultimately favoured pseudochyluria over frank chyluria despite the markedly elevated urinary triglycerides. All qualitative “chyle” assays, including early-morning fasting samples, repeatedly reported chyle as absent, and lymphocyturia was not documented. There was no demonstrable filarial infection, lymphatic obstruction, or retroperitoneal mass on imaging that could explain a lymphatico-urinary fistula. Clinically, the picture was dominated by massive glomerular proteinuria with subsequent normalization of serum albumin and stable renal function, which is compatible with lipid-rich nephrotic urine rather than sustained, high-volume chyle loss. The patient also noted that urine turbidity increased after oral protein supplements, suggesting that the “milky” appearance was at least partly driven by protein- and lipid-laden nephrotic urine. For these reasons, we used the pragmatic label of intermittent pseudochyluria on a background of nephrotic-range proteinuria, while acknowledging that a low-grade lymphatic leak cannot be completely excluded [1-3].
Chronic bronchiectasis prompted concern for AA amyloidosis, which can present with heavy proteinuria and progressive kidney dysfunction [4]. The underlying cause of her non-cystic fibrosis bronchiectasis could not be definitively established. The HRCT pattern of traction bronchiectasis with fibrotic bands and calcified mediastinal lymph nodes is most compatible with old post-infective disease (tuberculous or severe bacterial), and HIV, diabetes, and overt secondary immunodeficiency were excluded clinically and biochemically. However, a full etiologic workup for primary immunodeficiency, cystic fibrosis, primary ciliary dyskinesia, or allergic bronchopulmonary aspergillosis was not feasible in this resource-limited setting, and we acknowledge this uncertainty as a limitation. The combination of stable creatinine, normalized serum albumin, near-normal inflammatory markers, only minimally raised serum amyloid A, a non-diagnostic renal biopsy, and a negative fat-pad Congo red stain makes extensive, clinically overt systemic AA amyloidosis unlikely at present, while recognising that very early or focal AA disease cannot be fully excluded [4,5]. A normal serum amyloid A level reflects the absence of a strong ongoing inflammatory drive at the time of testing, but does not on its own rule out pre-existing AA deposits [4,5]. Likewise, negative serum free light chains with a normal κ/λ ratio reduce the likelihood of AL amyloidosis [6]. The absence of anti-PLA2R antibodies argues against classic primary membranous nephropathy, although other podocytopathies (including PLA2R-negative membranous nephropathy, minimal-change disease, or FSGS) cannot be conclusively distinguished without adequate renal tissue [5].
In resource-limited settings, where repeat biopsies, laser microdissection, and mass spectrometry are not readily available, a pragmatic approach combining urine chemistry (including urinary triglycerides), imaging, targeted serology (anti-PLA2R, serum free light chains), and low-risk tissue (fat-pad) biopsy can help narrow the differential. A key limitation of this report is the absence of a definitive histologic diagnosis, as further biopsies (repeat kidney or bladder) were not performed because of patient preference, perceived risk-benefit, and financial constraints. In addition, we could not obtain urine protein electrophoresis, which would have better characterised the protein profile, but was not feasible because of cost and limited local availability; however, the normal serum free light chain assay and clinical context made significant monoclonal proteinuria unlikely [6]. Our patient, therefore, exemplifies a real-world diagnostic dilemma at the interface of chyluria, pseudochyluria, and glomerular proteinuria, managed conservatively while avoiding unnecessary immunosuppression.
Conclusions
This case highlights how intermittent “white urine” with nephrotic-range proteinuria and preserved renal function can pose a major diagnostic dilemma. Systematic evaluation excluded filarial chyluria, urinary tract obstruction, AA amyloidosis, AL amyloidosis, and primary membranous nephropathy, despite the patient’s background of chronic bronchiectasis and markedly elevated urinary triglycerides. We finally favoured a mixed mechanism of intermittent lipid-rich (pseudochylous) urine with an as-yet unclassified glomerular proteinuric disorder, managed conservatively with maximal antiproteinuric therapy and treatment of underlying bronchiectasis. The case underlines the need to approach “milky urine” analytically, to avoid prematurely labelling it as filarial chyluria or amyloidosis, and to integrate urine chemistry, imaging, limited histology, and serologic markers in resource-limited nephrology practice.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chyluria: a mimicker of nephrotic syndrome Ann Saudi Med Kaul A Bhadhuria D Bhat S Sharma RK Karoli R Gupta A Prasad N 5935953220122339602210.5144/0256-4947.2012.593PMC 6081118 · doi ↗ · pubmed ↗
- 2Chyluria with massive proteinuria. Do not reach for the biopsy gun!Saudi J Kidney Dis Transpl Sudrania MK Valson AT Dangi AD Kekre NS 140714103120203356545510.4103/1319-2442.308357 · doi ↗ · pubmed ↗
- 3Chyluria presenting as milky urine and nephrotic-range proteinuria Kidney Int Cheng JT Mohan S Nasr SH D'Agati VD 151815227020061687124610.1038/sj.ki.5001703 · doi ↗ · pubmed ↗
- 4Secondary renal amyloidosis in a 13-year-old girl with bronchiectasis Korean J Pediatr Yang EA Lee DW Hyun MC Cho MH 7707735320102118995410.3345/kjp.2010.53.7.770PMC 3004490 · doi ↗ · pubmed ↗
- 5The anti-PLA 2R antibody in membranous nephropathy: what we know and what remains a decade after its discovery Kidney Int van de Logt AE Fresquet M Wetzels JF Brenchley P 129213029620193161106810.1016/j.kint.2019.07.014 · doi ↗ · pubmed ↗
- 6Evaluation of the serum-free light chain test in untreated patients with AL amyloidosis Haematologica Bochtler T Hegenbart U Heiss C 4594629320081828713710.3324/haematol.11687 · doi ↗ · pubmed ↗