Women’s health is a team effort: probiogenomics supports the development of a multi-species vaginal probiotic
Chiara Maria Calvanese, Vincenzo Valentino, Annachiara De Prisco, Serena Allesina, Angela Amoruso, Francesca Deidda, Annalisa Visciglia, Danilo Ercolini, Marco Pane, Francesca De Filippis

TL;DR
This study explores how different Lactobacillus species from the vaginal microbiota can work together in probiotics to improve women's health.
Contribution
The study identifies complementary roles of four Lactobacillus species and supports the development of multi-species vaginal probiotics.
Findings
Vaginal Lactobacillus strains are genomically adapted to promote persistence and fight pathogens.
L. gasseri and L. crispatus show complementary genetic traits for vaginal health.
Lm. fermentum may play a role in the gut-vagina axis through oral supplementation.
Abstract
The healthy vaginal microbiota is typically dominated by Lactobacillus species, while the dominance of different taxa often signals dysbiosis. Vaginal probiotics offer a promising therapeutic avenue to restore microbial balance and prevent recurrent infections. Using probiogenomics, this study investigated 19 novel strains belonging to four Lactobacillus species (Lactobacillus crispatus, Lactobacillus gasseri, Lactobacillus paragasseri, Limosilactobacillus fermentum) isolated from the vaginal environment of fertile and menopausal women. Through genomic screening and comparative genomics, we identified complementary roles between these species in activities crucial to women’s health. Our results indicate that vaginal Lactobacillus strains are genomically adapted to this niche, promoting persistence and pathogen-fighting. Furthermore, we demonstrated the potential ability of vaginal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
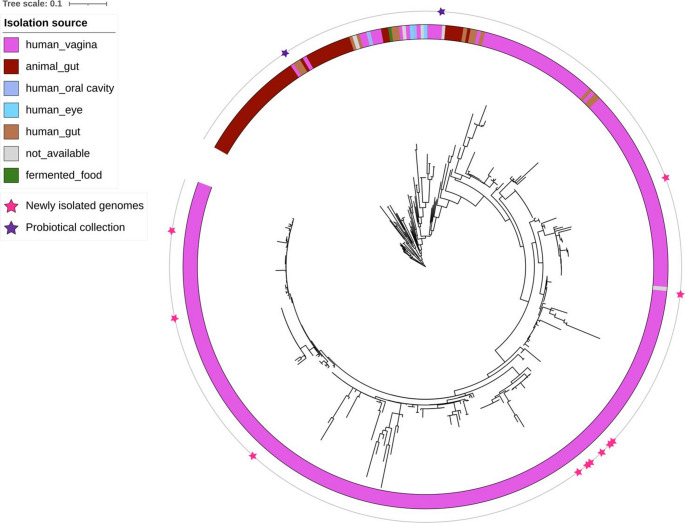 Figure 1
Figure 1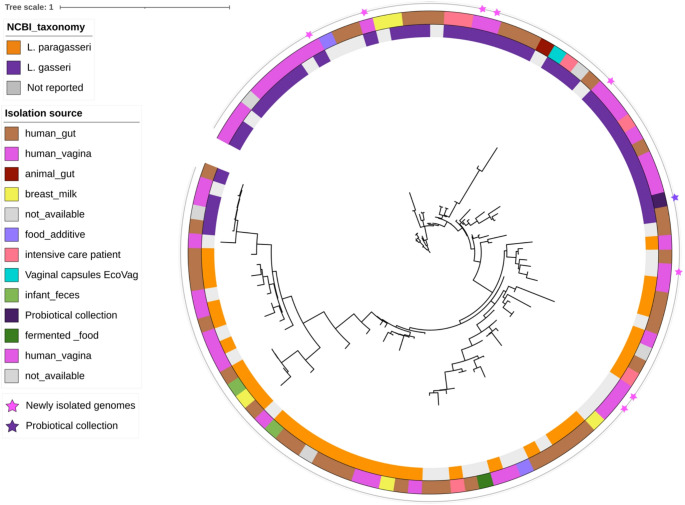 Figure 2
Figure 2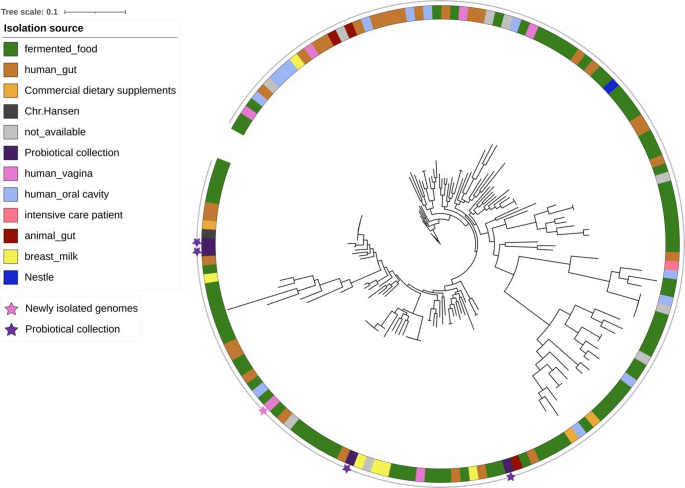 Figure 3
Figure 3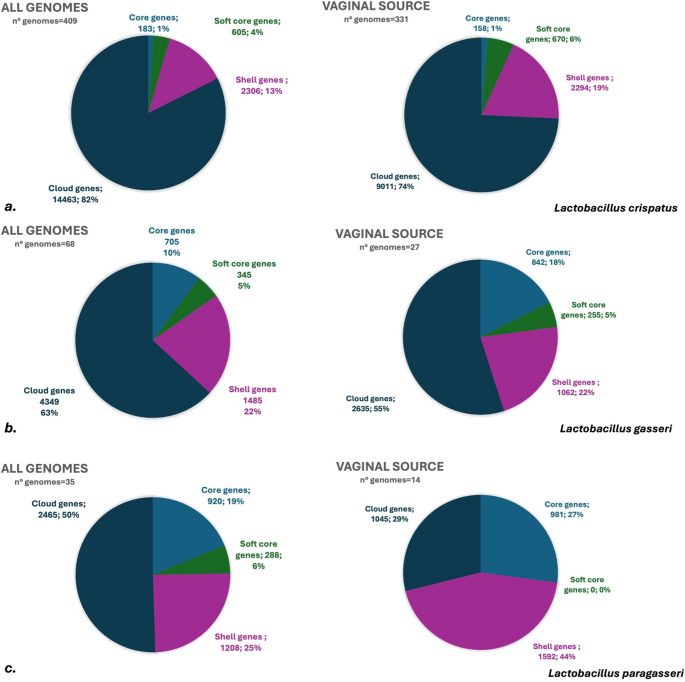 Figure 4
Figure 4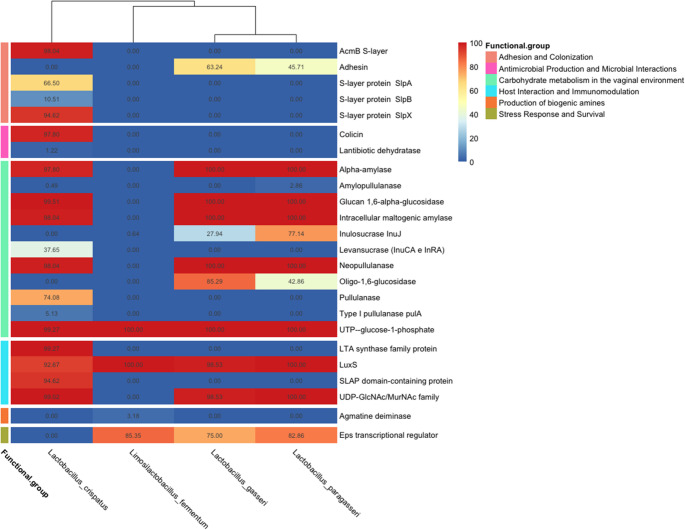 Figure 5
Figure 5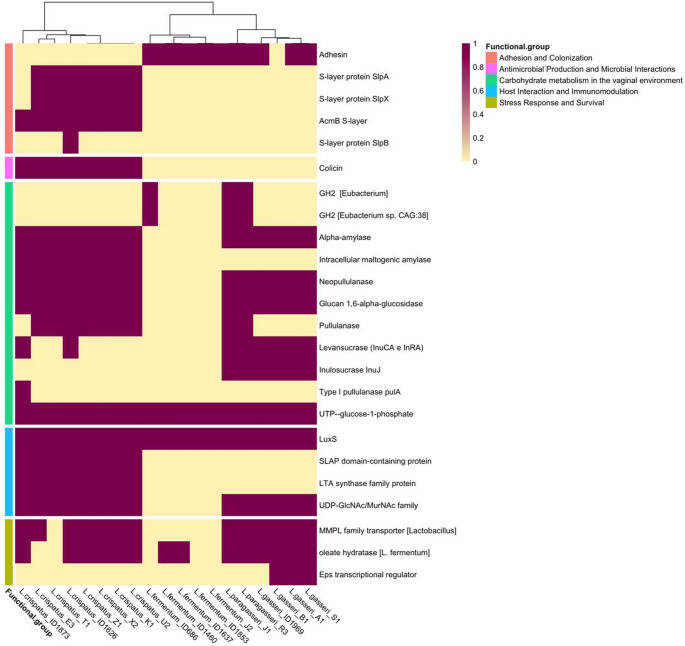 Figure 6
Figure 6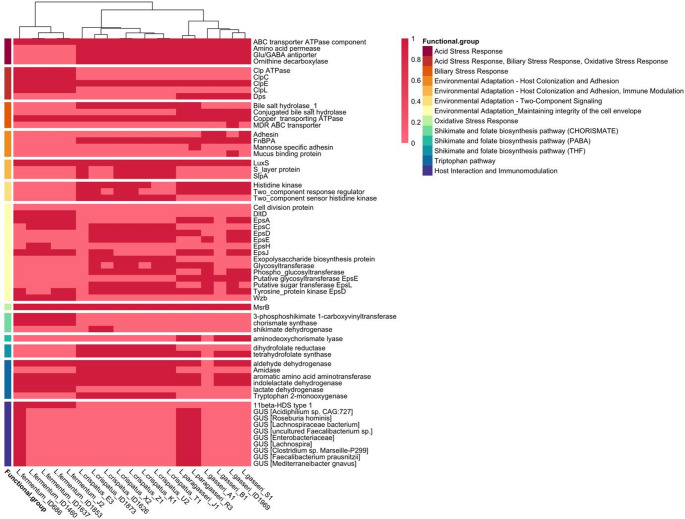 Figure 7
Figure 7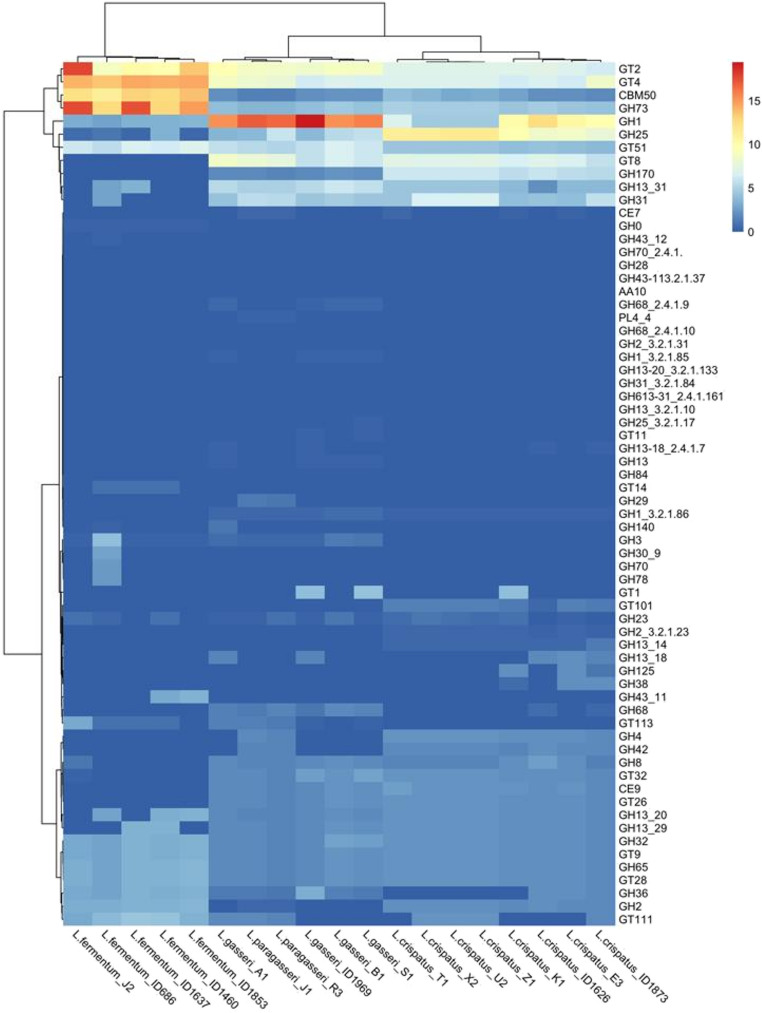 Figure 8
Figure 8- —Università degli Studi di Napoli Federico II
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsReproductive tract infections research · Gut microbiota and health · Probiotics and Fermented Foods
Introduction
While the gut and oral microbiome have been extensively studied, the role of vaginal microbiome in women’s health is gaining significant attention only recently. Indeed, recent research efforts highlighted that the vaginal microbiome directly influences the risk of infections, reproductive issues, and other health conditions [1–3]. Human vagina is a complex environment, with a unique microecosystem that changes throughout a woman’s life due to hormonal fluctuations, immune responses, age, and external factors like infections and antibiotics [4, 5]. The healthy adult vaginal microbiota is typically dominated by Lactobacillusspecies, while a more diverse microbial community often indicates compromised health [6]. Large cohort studies allowed to categorize the vaginal microbiome into five Community State Types (CSTs), each characterized by different dominant species: CTS-I is dominated by Lactobacillus crispatus, CTS-II by Lactobacillus gasseri, CTS-III is associated with transitions and dominated by Lactobacillus iners and CTS-V by Lactobacillus jensenii. CTS-IV has a more heterogeneous community of strict and facultative anaerobes (Gardnerella, Atopobium, Mobiluncus, Prevotella, etc.) and it is often associated with bacterial vaginosis [1, 7–9]. Indeed, an imbalance in the vaginal microbiome, known as bacterial vaginosis (BV), is a common issue, particularly in women during reproductive age, and it is often linked with the dominance of anaerobic bacteria and a reduction in*Lactobacillus *[6]. Other conditions, like vulvovaginal candidiasis (VVC) and streptococcal infections, are also connected with disruptions in the vaginal microbial communities [10]. A dominance of non-Lactobacillusspecies was also found in post-menopausal women, and it was associated with vaginal dryness [11]. Antibiotic treatments for vaginal infections can also have negative side effects, including disrupting the beneficial vaginal microbiota and increasing the risk of recurrent infections and antibiotic resistance. For this reason, scientific and commercial interest towards the development of alternative therapies increased. Vaginal probiotics, administered either topically or orally, are currently promising to restore the balance in the vaginal microbial community and preventing recurrent infections by inhibiting pathogen growth [12]. Since Lactobacillus species are crucial for a healthy vaginal environment, there is a growing interest in their role and potential benefits [5]. Indeed, potential probiotic candidates for topical use should show some key attributes, such as promoting vaginal epithelial cell membrane integrity, adhering to the vaginal epithelium, and producing beneficial metabolites [13]. On the other side, while biofilm formation promotes the colonization and persistence of microbial strains in the vaginal environment, if it is not well balanced, it may interfere with sperm motility and fertility [1, 14, 15]. Another important trait is effective glycogen metabolization. Glycogen, abundantly found in vaginal fluid, is metabolized by vaginal lactobacilli, lowering pH and hindering pathogens [16]. On the contrary, an undesired trait is the production of biogenic amines, to prevent malodor and increase in pH, leading to dysbiosis [17, 18].
The use of probiogenomics is revolutionizing the identification of novel probiotic strains [19]. Indeed, a wide genetic profiling of multiple strains may support the discovery of novel potential probiotics, screening in time-consuming and expensive laboratory tests only those strains that have been previously selected in silico. Based on this principle, this study aims to identify new, effective probiotics for improving women’s vaginal health. In particular, we explored the hypothesis that a combined use of multiple lactobacilli species may be more effective. Starting from a genomics screening of vaginal isolates and comparative genomics of these strains with other isolates from the same species, we identified a complementary role of different vaginal species in key activities that may be connected with women’s health, highlighting the promising avenue for the development of a multi-species probiotic product.
Materials and methods
Sampling and strains isolation
Vaginal swabs (Clinicswab^LTS^, APTACA S.p.A.) were self-collected by fifteen fertile and ten menopausal women (18–55 years old). The eligibility was defined according to the following inclusion criteria: (1) no symptoms of vaginal or urinary tract infection; (2) no menstrual period; (3) no use of local antibiotics and antifungals in the last six months. Women were instructed about the aim of the study and signed a written consent to participate. Women who did not meet the inclusion criteria were excluded from the study.
Swabs were striked on De Man, Rogosa and Sharpe agar (MRS, Oxoid) supplemented with 0.05% L-cysteine for lactobacilli isolation [20]. Plates were incubated anaerobically for 48 h at 37 °C. Morphologically different colonies were isolated. The isolates that were Gram-positive, catalase negative and rod-shaped were considered. Isolates were preliminary identified using MALDI-TOF Biotyper Sirius (Bruker), according to the manufacturer’s standard protocol [21].
Isolate deduplication, genome sequencing and analysis
Isolates from the same species were deduplicated using (GTG)5-rep-PCR fingerprinting [22]. DNA from the colonies was extracted using the Wizard^®^ Genomic DNA Purification Kit (Promega). After this step, 20 strains were kept for the following analyses (Table 1). Whole Genome Sequencing (WGS) was carried out on Illumina NovaSeq platform, leading to 2 × 150 bp, paired-end reads. Reads were assembled through the SPAdes pipeline (version 4.1.0) [23] and contigs > 1000 bp were filtered using Seqtk [24]. Taxonomic identification was obtained using PhyloPhlan 3.0 [25].Table 1. Taxonomic identification of vaginal strains isolated in this studyStrain originStrainProbiotical Internal Collection CodeMALDI-TOF identificationWGS identificationThis studyE3770/2Lactobacillus crispatus**Lactobacillus crispatusThis studyT1770/4Lactobacillus crispatus**Lactobacillus crispatusThis studyU2ID 2026Lactobacillus crispatus**Lactobacillus crispatusThis studyZ1770/6Lactobacillus crispatus**Lactobacillus crispatusThis studyK1770/3Lactobacillus crispatus**Lactobacillus crispatusThis studyX2770/5Lactobacillus crispatus**Lactobacillus crispatusProbiotical collectionID_1873DSM 24619Lactobacillus crispatus**Lactobacillus crispatusProbiotical collectionID_1626DSM 24439Lactobacillus crispatus**Lactobacillus crispatusThis studyS1ID 2028Lactobacillus gasseri**Lactobacillus gasseriThis studyB1772/3Lactobacillus gasseri**Lactobacillus gasseriThis studyA1770/7Lactobacillus gasseri**Lactobacillus gasseriProbiotical collectionID_1969DSM 32405Lactobacillus gasseri**Lactobacillus gasseriThis studyR3770/9Lactobacillus paragasseri**Lactobacillus paragasseriThis studyJ1ID 2029Lactobacillus paragasseri**Lactobacillus paragasseriThis studyJ2770/11Limosilactobacillus fermentum**Limosilactobacillus fermentumProbiotical collectionID_686DSM 32277Limosilactobacillus fermentum**Limosilactobacillus fermentumProbiotical collectionID_1460DSM 18297Limosilactobacillus fermentum**Limosilactobacillus fermentumProbiotical collectionID_1853DSM 26956Limosilactobacillus fermentum**Limosilactobacillus fermentumProbiotical collectionID_1637DSM 19187Limosilactobacillus fermentum**Limosilactobacillus fermentum
Phylogenetic analysis
We included in the comparative analysis publicly available genomes of the four isolated species, namely Lactobacillus crispatus (n = 396), L. gasseri (n = 62), L. paragasseri (n = 32) and Limosilactobacillus fermentum (n= 156). Genomes of these species were downloaded from the NCBI database [26]. We manually retrieved the “isolation source” from NCBI or from the original study (Online Resource1).
Phylogenetic tree for each species (including new isolates and NCBI genomes) was developed using the PhyloPhlAn 3.0 pipeline [25] and visualized using iTOL v6 [27]. The Average Nucleotide Identity (ANI) values between each pair of genomes was calculated using fastANI (version 1.0) [28]. Classical Multidimensional Scaling (MDS, cmdscale R function) was carried out on the ANI distance matrix and plots were visualized using the ggplot2 R package.
Pangenome and gene onthology (GO) analysis
Coding sequences (CDS) of each genome were predicted using Prokka (version v1.11) [29]. Pangenomes of each species was computed using the Roary pipeline [30]. Then, we used the Gene Ontology database [31], to search for the key GOs in core/soft-core and shell/cloud genes of the different species using UNIPROT 2023_04 [32], including in the analysis only genomes from vaginal source. GOs terms of interest for vaginal lactobacilli were considered as described in the supplementary material of the work by Bhattacharya et al. [13].
Functional analysis through the alignment of predicted genes against protein reference sequences
Predicted genes in the analyzed genomes were aligned against key enzymes of vaginal interest using DIAMOND blastx (version 2.1.11) [33]. A hit was considered when displaying at least 80% of identity on at least 50% of the query length. Protein sequences of key genes were downloaded from NBCI. Genes searched included those related to the adaptation in the vaginal niche, to the production of beneficial metabolites and to the resistance to gastrointestinal passage, selected through screening of the scientific literature using different combinations of keywords in SCOPUS search: ‘vaginal adaptation microbial enzymes’, “Lactobacillus role in women’s health”, “vaginal fitness”, “glycogen metabolism”, “vaginal health”, “pathogen inhibition”, “genomic analysis”, “vaginal dysbiosis”, “sialidase activity”, “biofilm formation”, “immune modulation”, “probiotic for vaginal health”, “probiotic genetic traits”, “Lactobacillus crispatus enzymes”, “adhesion disruption enzymes vaginal bacteria”, “microbial degradation of vaginal mucus”, “adhesion to vaginal epithelium”, “bacterial vaginosis”, “bacteriocin production”, “folate metabolism”, “tryptophan metabolism”. Genes included in the custom database and NCBI accessions are reported in Online Resource 2.
The ability of genomes to degrade different carbohydrates sources was estimated by aligning predicted genes against the Carbohydrate-Active Enzyme database [34]. The presence of known antibiotic resistance (AR) genes involved in resistance to antibiotics was evaluated using TORMES, that search against CARD, AMRfinder and ARGANNOT databases considered [35]. The presence of plasmids in the newly sequenced genomes was verified using Plasmid Finder Tool [36].
Heatmaps were obtained from presence-absence gene matrices using the R package ‘pheatmap’.
Results
Comparative genomics reveals genomic adaptation to specific environmental niches
Isolate identification obtained by MALDI-TOF [21] was confirmed using WGS, that is the final one considered for further analyses (Table1).
Through a phylogenetic analysis, although most of L. crispatus genomes available are of vaginal origin, we can see that the host (human or animal) drives the clustering of the strains (Fig. 1). In addition, we observed 3 different genome clusters within L. crispatusspecies, suggesting the presence of putative different sub-species, as previously reported [37]. Two clusters exclusively included strains of vaginal source, while the other comprised strains from mixed isolation sources. Interestingly, strains newly isolated in this study fell in both the sub-species.Fig. 1. Phylogenetic tree of Lactobacillus crispatus genomes. The outer ring is coloured according to the isolation. The genomes newly isolated in this study and those provided by Probiotical are indicated by stars
We found that several L. gasseri/L. paragasserigenomes present in NCBI were mislabeled and positioned inconsistently into the phylogenetic tree. Therefore, for the final identification of NCBI strains of these species, we referred to that reported by Ene et al. [38].
Indeed, in Fig. 2 we can clearly see the separation of the genomes of L. gasseri and L. paragasseri on the tree. However, we did not find a clear genome clustering according to the isolation source within these species (Fig. 2).Fig. 2. Phylogenetic tree of Lactobacillus gasseri and Lactobacillus paragasserigenomes. The inner ring is coloured according to the species-level identification (as reported by Ene et al. [38]). The outer ring is coloured according to the isolation source The genomes newly isolated in this study and those provided by Probiotical are indicated by stars
Finally. we did not observe a genome clustering driven by the strain origin for Limosilactobacillus fermentum, (Fig. 3). However, it has to be pointed out that the dataset included only few genomes of vaginal source, (n = 5 out of 156; 1 newly isolated in this study)Fig. 3. Phylogenetic tree of Limosilactobacillus fermentum. The ring is coloured according to the isolation source The genomes newly isolated in this study and those provided by Probiotical are indicated by stars
Pangenomic insights demonstrate different genetic evolutionary dynamics in the main vaginal lactobacilli species
The pangenome refers to the complete set of genes present in all individual strains or isolates of a particular species. This gene pool is divided into two main categories: core genes, which are shared by all the members of the species, and accessory genes, which are present in only some individuals.
A single genome sequence captures a specific moment in the history of a lineage. While some genes and mutations have a long-shared history and are predisposed to persist within a single species over generations, others are temporary, and the gene loss/acquisition events may be boosted by environmental factors [39]. In the pangenome analysis, genomes from L. crispatus (n = 396; 13 newly isolated in this study), L. gasseri (n = 62; 6 newly isolated in this study), L. paragasseri (n = 32; 3 newly isolated in this study) were included. In order to exclude biases due to the different isolation sources of the strains considered, we further calculated the pangenome considering exclusively genomes of vaginal origin (Fig. 4). For this reason, we excluded the genomes of Lm. fermentum, since only 5 genomes of vaginal origin were present in NCBI.Fig. 4. Comparison and distribution of the percentage of core, soft-core, shell and cloud genes in the species L. crispatus (a) L. gasseri (b) and L. paragasseri (c) from vaginal origin
We observed that L. paragasseri retained the highest percentage (25%) of core/soft-core genes (defined as present in = > 99–100% and 95%−99% of the genomes, respectively), while L. crispatus retained the lowest number of core genes (core + soft-core; 5%) (Fig. 4). Consistently, L. crispatus had the highest percentage (82%) of cloud genes, that is genes present in < 15% of the genomes considered, although this may be affected by the higher number of genomes available for this species. Considering only the genomes of vaginal origin, an increase in core genes was observed for L. gasseri and L. paragasseri, suggesting the presence of niche-specific functions, related to the adaptation to the vaginal environment. As expected, the situation remained unchanged for L. crispatus, since 81% of the genomes included in this study were of vaginal origin (Fig. 4).
The distributions of key GOs within the pangenomes are summarized in Online Resource 3, while a full list of the key GOs found in the pangenomes is reported in Online Resource 4. From the intersection of the core genes of the three species (Online Resource 3), we did not find any GOs relevant for the vaginal environment (among those highlighted by Bhattacharya et al. [13], but rather the three species only share primary biological functions (e.g., ATP hydrolysis activity, proteolysis, glycolytic process, etc.). In the core of L. paragasseri strains, we found the biosynthesis of fatty acids, associated with the integrity of the vaginal cell membranes [40]. In L. gasseri core, the biosynthesis of organic acids and hydrogen peroxide were identified, that are relevant for the antimicrobial and anti-pathogenic action. Finally, the core genome of L. crispatus is characterized by the GO related with bacteriocin biosynthesis.
Differently, in the cloud of L. gasseri and L. paragasseri (Online Resource 3) we found key GOs that are related with adhesion, aggregation and synthesis of exopolysaccharides, while the three species share in their cloud genome hydrolase and transferase activities involved in the synthesis of biosurfactants, that may help preventing microbial infections.
Functional screening highlights genomic traits linked with fitness in the vaginal niche and species-specific mechanisms
We firstly screened genomes of vaginal origin for the presence of genes related to the adaptation and persistence in the vaginal environment, grouped in the following functional classes: (i) Adhesion and Colonization; (ii) Antimicrobial Production and Microbial Interaction; (iii) Carbohydrate metabolism in the vaginal environment; (iv) Host interaction and Immunomodulation; (v) Stress Response and Survival (Fig. 5). Twenty-four (out of 59) of the genes included in the manually curated database (Online Resource 2) were found. L. crispatus genomes have the highest prevalence of these genes (12 out of 24 genes were present in > 93% of the genome), as well as the largest number of genes involved in immunomodulation, adhesion to cervicovaginal epithelial cells and antagonism against pathogens of the urogenital tract. This support the observation that L. crispatus dominated CS are the most frequent in women and are associated with a general vaginal health. We observed a very similar profile in L. gasseri and L. paragasseri in the context of vaginal adaptation. As shown in Fig. 5, the two species have a high genomic overlap, confirming a close evolutionary relationship [41]. It is therefore interesting to investigate the differences at the strain level. Both species have a lower gene repertoire thanL. crispatus for activities potentially linked with host interaction and immunomodulation. Similarly, these two species have fewer genes, among those examined, involved in the adhesion and colonization processes. In contrast, the gene encoding the transcriptional regulator Eps has been identified in the genomes of L. gasseri and L. paragasseri, while it is absent in L. crispatus. On the contrary, the lowest prevalence of genes was observed in Lm. fermentum genomes, where all the genes found were those involved in biofilm formation and co-aggregation. In addition, genes related to the production of biogenic amines, such as cadaverine, putrescine and trimethylamine, that are a negative trait for a vaginal strain, since are responsible for the increase of vaginal pH, promoting dysbiosis and unpleasant odors, were not found in the genomes screened.Fig. 5. Prevalence (%) of genes associated with vaginal adaptation in the genomes of lactobacilli
Among the enzymes involved in bacterial carbohydrate metabolism in the human vagina, the absence of sialidase stands out in all the species analyzed. Bacterial sialidase degrades glycans in vaginal mucus by removing sialic acid. The presence of this enzyme may offer limited nutritional benefits for vaginal lactobacilli but carries the risk of compromising the integrity of the protective mucus; its absence promotes mucus stability but limits access to specific nutritional sources.
We screened the newly isolated genomes for the presence of the 59 genes involved in vaginal adaption (Online Resource 2) (Fig. 6). We confirmed that our L. crispatus strains exhibited the highest prevalence of the genes searched. L. gasseri and L. paragasseri displayed a highly similar gene profile. Consistently with previous observations, Lm. fermentum presented the lowest prevalence of the analyzed genes, particularly those associated with biofilm formation and co-aggregation. Variability was observed in the presence of genes involved in Carbohydrate Metabolism in the Vaginal Environment and Stress Response and Survival (Fig. 6). This suggests distinct mechanisms of adaptation to the potentially dynamic conditions of the vaginal milieu and indicates that each species may apply slightly different metabolic strategies for utilizing available resources. The absence of sialidase-related genes across all strains was noted, suggesting that direct degradation of vaginal mucus via this enzyme may not be a common strategy among these lactobacilli. In addition, genes related with the fatty acid response mechanism, important when vaginal bacterial vaginosis may be treated with oleic acid as alternative to antibiotics [42], were found in all the strains.Fig. 6. Prevalence of genes associated with vaginal adaptation in the genomes of the newly isolated strains
Lactobacillus crispatus and Lactobacillus gasseri are good candidates as oral probiotics targeting women’s health
We further screened the genomes of newly isolated strains for the presence of genes related to key probiotic factors, assuming a potential oral use of our strains. The genes screened included those for the resistance to stress and gastrointestinal (GIT) passage and the production of beneficial metabolites in the gut (e.g., folate and tryptophan metabolism) (Fig. 7). To reach the colon, oral probiotics must be able to survive gastric acidity, bile salts in the small intestine, and oxidative stress, through a variety of resistance mechanisms or active stress responses. Many of the analyzed strains harbour genes involved in resistance to acid stress (broadly present in all except Lm. fermentum genomes), biliary stress (present in L. crispatus and L. paragasseri, but absent in L. gasseri and Lm. fermentum), and oxidative stress (methionine sulfoxide reductase MsrB reverses the loss of the biological activity of proteins caused by the oxidation of methionine to methionine sulfoxide and, therefore, contributes to the protection of bacteria against oxidative damages, and it was present in all the genomes). Although all the strains potentially have genes involved in different resistance mechanisms, Lm. fermentum strains were those showing the lowest occurrence, thus potentially performing worse during GIT passage.Fig. 7. Occurrence of genes associated with probiotic factors in the genomes of the strains newly isolated in this work
We also explored the potential of the strains to produce folates, particular important for women during pregnancy. Chorismate is an important biochemical intermediate, as it acts as a precursor in the biosynthesis of aromatic amino acids, but also of indole, indole derivatives, tryptophan and folic acid [43]. The genes coding for 3-phosphoshikimate-1-carboxyvinyltransferase and chorismite synthase, final enzymes of the shikimate pathway that leads to chorismate production, were observed exclusively in Lm. fermentum genomes (Fig. 7). On the contrary, all the strains, except Lm. fermentum, had the gene coding for tetrahydrofolate synthetase, involved in the production of tedrahydrofolate (THF), while dihydrofolate reductase and shikimate dehydrogenase were exclusively found in L. crispatus. The first one is involved in the final step of tetrahydrofolate biosynthesis, which is the active form of folic acid for humans, while the second one catalyzes the fourth step of the shikimate pathway that leads to the production of precursors necessary for folate biosynthesis in bacteria and is absent in mammals. L. gasseri and L. paragasseristrains share the aminodeoxychorismate lyase gene, which leads to the production of para-aminobenzoic acid (PABA), an important intermediate in the bacterial synthesis of folate [43].
Another important feature is the synthesis of the indole metabolites of tryptophan (Fig. 7), which are important both at intestinal (involved in immune modulation, barrier integrity, bacterial communication, protection from stress) and at systemic (benefits on mental health, since they are involved in serotonin and melatonin biosynthesis) [44]. L. gasseri A1, L. crispatus C3 and L. crispatus Q1 did not show any of the mapped genes involved in tryptophan metabolism. All other genes were present in all the genomes except the lactate dehydrogenase, which was present only in Lm. fermentum genomes, indicating that only this strain harbored the complete pathway of indolic 3-lactic acid (ILA) production (Fig. 7). Tryptophan 2-monooxygenase, leading to the production of indole-3-acetamide (IAM), was present only in L. crispatus genomes and, coupled with the presence of amidase and aldehyde dehydrogenase, complete the metabolic pathway for indolic 3-acetic acid (IAA) production in these strains.
We further screened the genomes for the presence of genes related with beneficial or detrimental activities for women health, possibly explicated in the human gut, thus supporting their oral administration. GUS (β-glucuronidase) genes code for an enzyme that deconjugate estrogens and are involved in female estrobolome and hormonal disorders [45], were found in all the newly isolated L. paragasseri strains and in Lm. fermentumID 686, while were absent in other strains. Moreover, hydroxysteroid dehydrogenase (HSD) genes [46], involved in steroid degradation, were observed only in Lm. fermentum strains.
No known gene potentially involved in antibiotic resistance were found in lactobacilli strains isolated in this study. In addition, we screened the genomes for the presence of plasmids. Plasmids were detected in 12 out of 19 genomes (Online Resource 5). However, mapping genes present on plasmids against our database of key genes involved in the vaginal fitness, we did not find any matches, suggesting that these activities are well conserved among vaginal-origin lactobacilli.
Lactobacilli are equipped with enzyme families that are important for the adaptation and survival in the vaginal niche
We screened the strains for the ability to degrade carbohydrates, searching for Carbohydrates-Active genes (CAZy). Sixty-six CAZy families were identified, 11 of which showed a higher number of genes present: CBM50, GH1, GH13_31, GH25, GH31, GH73, GH170, GT2, GT4, GT8, GT51 (Fig. 8). In the analyzed genomes, the identified CAZy families are predominantly associated with the processing of complex carbohydrates, including structural and storage polysaccharides such as cellulose, chitin, starch, glycogen and peptidoglycan, as well as glycoconjugates. In the context of the vaginal microbiome, GH1 and GH31 families are important for the degradation of glycogen, an energy source for lactobacilli. GH73 and GH25 families act on bacterial peptidoglycan, potentially influencing interactions among different strains sharing the same environmental niche and GH170 includes enzymes which act on particular bonds present in mucins, thus may degrade components of the vaginal mucus. Finally, GT2 and GT4 (including glycosyltransferases that catalyze the polymerization of bacterial exopolysaccharides) are involved in the synthesis of polysaccharides that may influence bacterial adhesion and biofilm formation in the vagina. These enzyme families are therefore relevant for the adaptation and survival of microorganisms in the vaginal niche.Fig. 8CAZymes prevalence in the genomes analysed in this study The color scale indicates the % of genes from each CAZy family found in each genome
We observed a species-specific pattern in the distribution of CAZymes: for example, genes that fall into the GT2, GT4, and GH73 families were prevalent in the genomes of Lm. fermentum. On the contrary, the CAZy GH1 family was particularly abundant in L. gasseri and L. paragasseri, while in the genomes of L. crispatus we found genes that fall into the GH25 family (Fig. 8).
Discussion
The vaginal microbiome has a direct impact on infections, reproductive health and other women’s health issues. A vaginal microbiota dominated by Lactobacillus is generally indicative of a healthy environment, while higher microbial diversity often indicates dysbiosis, which has led to a growing interest in the role and benefits of Lactobacillusin this context. Modulation of vaginal microbiome through the administration of probiotics shows promise in restoring balance and preventing recurrent infections. Genomic screening of putative probiotic strains supports a faster development of well-targeted and personalized probiotic formulations. However, some important features related to probiotic activities may be highly niche-specific, thus suggesting that strains of vaginal origin may perform better in that specific environment than probiotics of different origin. Indeed, although several probiotics targeted to women’s health are available on EU market [47], several of them contain strains from different isolation sources.
In this study, we isolated strains from four different lactobacilli species, namely L. crispatus, L. gasseri, L. paragasseri, and Lm. fermentum. Notably, two other vaginal species, L. iners and L. jensenii, were not found. This is likely due to the limited sample size and the lower prevalence of these species in the vaginal environment, together with a possible bias during isolation, due to the choice of the culture medium. Specifically, L. crispatus and L. gasseri/L. paragasseri are known to dominate the vaginal Community State Types (CST-I and CST-II, respectively), often representing > 50% of the microbial population. L. jensenii, while typical of a healthy vaginal microbiota, is present in approximately 20% of the women, suggesting that a larger cohort would have been necessary for its isolation [48, 49]. We also failed in isolating L. iners, usually associated with less stable or transitional vaginal communities and frequently linked with vaginal dysbiosis [47–49]. WhileLm. fermentumis not typically a dominant species in the gastrointestinal tract or in the vagina, its established probiotic characteristics make it a promising candidate [48, 50–52]. In our study we demonstrated that for L. crispatus, and to a lower extent for L. gasseri and L. paragasseri, a clear genomic adaptation to the vaginal environment occurred. This was not the case for Lm. fermentum strains, although this result may be influenced by the limited number of genomes from strains of vaginal origin available. This was further supported by the pangenome analysis, that showed that L. crispatusretained the lowest number of core (core + soft-core) and the highest number of cloud genes, which suggest the evolutionary diversification of these strains, possibly associated with the adaptation to different niches. Its genome plasticity and ability to evolve may explain why this species is the most frequently found in the vaginal environment as dominant [13]. In addition,L. crispatus is the one with the highest number of genes involved in the adhesion to cervicovaginal epithelial cells and antagonism towards urogenital tract pathogens, further highlighting the high genomic specialization of this species to the vaginal niche and supporting the possible topical use of these strains. Consistently with a previous work on L. gasseri and L. crispatusstrains isolated from human urogenital and gastrointestinal tracts [53], our findings suggest that future probiotic formulations addressed to women’s health should consider the isolation source and carefully evaluate the genetic and phenotypic characteristics of the strains for targeted application in different body sites.
However, a bidirectional communication between the female reproductive system (FRT) and various other organs (e.g., vagina-gut, vaginal-oral, vaginal-brain axes) has been highlighted [54]. Evidence supports the translocation of gastrointestinal *Lactobacillus *strains to the vagina (gut-vagina axis), suggesting that oral probiotic intake may be directly linked with improved vaginal microbiota balance and local immune responses [55]. Direct bacterial translocation from the gut to the vagina has been previously observed [56]. Beyond direct translocation, indirect interactions involving metabolites or immunoglobulin production, as well as the ability of gut microbes of metabolizing estrogens may be also implicated in the gut-vagina communication [52, 57–59].
In order to evaluate the possibility of using our strains in a probiotic blend with oral delivery, we assessed the presence of genomic traits related to GI resistance. All the L. crispatus,* L. gasseri* and L. paragasseri strains have several genes related with active stress response mechanisms required to survive during gastrointestinal transit, except Lm. fermentumstrains, which lack some of them. We further screened the genomes for the presence of genes related with beneficial or detrimental activities for women health and possibly explicated in the human gut, thus supporting their role as oral probiotics. For this aim, we searched for the presence of β-glucuronidase, that may deconjugate estrogen metabolites in the gut. This reactivation allows unbound estrogen to be recirculated through the bloodstream, leading to increased reabsorption of free estrogen and increased risk of cancers and hormonal disorders [45]. These genes were only present in all L. paragasseri and in one strain of Lm. fermentum. Although further in vitro and in vivo studies are necessary to explore the role of β-glucuronidase in women estrobolome, these strains may be potentially involved in hormonal disorders [46].
The production of beneficial tryptophan metabolites and folate are important traits for probiotics targeting women’s health. Given the prevalence of folate deficiency, particularly during pregnancy, there is growing interest in developing probiotics capable of synthesizing folate directly in the gut [43]. Our results indicate that none of the newly isolated strains contain the complete pathway for folate biosynthesis. However, each strain possesses one or more key enzymes for producing important intermediates like PABA, chorismate, and THF. This suggests a complementary metabolism among the strains, which supports their potential use in a multi-species probiotic blend rather than as individual strains. Concerning the synthesis of indole metabolites of tryptophan and the two known pathways, onlyLm. fermentum genomes show the complete ILA pathway, while L. crispatusmay synthesize these metabolites through the IAA pathway. The indole derivatives are known to support gastrointestinal homeostasis by maintaining the integrity of the intestinal barrier and modulating anti-inflammatory signaling [60]. Although widely studied in the intestine, their role in the vagina is an emerging field. Evidence suggests that vaginal indoles, including ILA and indole-3-aldehyde, activate the aryl hydrocarbon receptor (AhR) to modulate immune responses and provide protection against vulvovaginal candidiasis [61, 62]. These findings collectively provide a strong rationale for further exploring the role of bacterial tryptophan metabolites, including ILA and potentially IPA, in maintaining vaginal immune homeostasis and preventing dysbiosis. However, we have to point out that this is a preliminary study and several limitations exist, including the relatively small sample size and the possible bias in the strain isolation, due to the choice of the culture medium. Moreover, functional characteristics of the strains are inferred based on genomic data analysis. These predicted functions will need to be validated through in vitro or in vivo experiments in subsequent works. Future studies should also focus on an in-depth analysis of the specific synergistic mechanisms between the different strains (e.g., metabolic complementarity, interactions in gene expression regulation). Finally, the “dual nature” of L. paragasseri would deserve further insights to identify which genes may contribute to the adverse phenotypes.
Conclusion
Recognizing the intrinsic diversity and hormonal variability of the female microbiome, traditional probiotics formulated with generic strains are often suboptimal. For example, hormonal shifts during childbearing years and the decline of estrogen in menopause can significantly alter the vaginal microbiome, increasing the risk of conditions like bacterial vaginosis and urinary tract infections. This highlights the need for targeted probiotic interventions using specific lactobacilli strains capable of effectively colonizing the vagina and providing beneficial activities. Our results demonstrated that strains of vaginal origin are clearly genomically adapted to this niche, giving them higher fitness to persist and counteract pathogens. On the other side, when topical application is not feasible, oral supplementation may also be effective, when selecting the suitable strains. Indeed, we showed that vaginal-origin lactobacilli may potentially survive GI passage and that they may still have beneficial activities in the gut, highlighting the importance of the gut-vagina axis.
Vaginal probiotics containing L. crispatus currently represent the most prevalent formulation commercially available. Our findings confirmed the effective performance of this species within the vaginal niche. However, to meet more specific needs for vaginal health, the genetic characteristics of L. gasseri can be considered equivalent and complementary. Furthermore, the role of L. paragasseri in the vaginal environment presents an intriguing dichotomy: while it exhibits genetic similarities with L. gasseri, it is also characterized by potentially unfavorable traits. In summary, L. crispatus maintains its primary position due to its robust adaptability to this environment, but the previously underappreciated role of L. gasseri emerges, suggesting a potential synergistic interaction between these two species.
Indeed, our results suggest that the combination of multiple species and strains in a probiotic blend may be more effective than a single-strain supplement, since several beneficial activities require metabolic cooperation among the strains.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1 (PDF 227 KB)
Supplementary Material 2 (PDF 249 KB)
Supplementary Material 3 (PDF 628 KB)
Supplementary Material 4 (PDF 159 KB)
Supplementary Material 5 (PDF 99.5 KB)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 14. Auriemma RS, Scairati R, del Vecchio G, et al (2021) The Vaginal Microbiome: A Long Urogenital Colonization Throughout Woman Life. Front Cell Infect Microbiol 11:686167. 10.3389/FCIMB.2021.686167/BIBTEX 10.3389/fcimb.2021.686167 PMC 829085834295836 · doi ↗ · pubmed ↗
- 25. Chen X, Lu Y, Chen T, Li R (2021) The Female Vaginal Microbiome in Health and Bacterial Vaginosis. Front Cell Infect Microbiol 1110.3389/fcimb.2021.631972 PMC 805848033898328 · doi ↗ · pubmed ↗
- 37. France MT, Ma B, Gajer P, et al (2020) VALENCIA: a nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 8:. 10.1186/s 40168-020-00934-610.1186/s 40168-020-00934-6PMC 768496433228810 · doi ↗ · pubmed ↗
- 415. Zhang F, Dai J, Chen T (2021) Role of Lactobacillus in Female Infertility Via Modulating Sperm Agglutination and Immobilization. Front Cell Infect Microbiol 1010.3389/fcimb.2020.620529 PMC 786854533569356 · doi ↗ · pubmed ↗
- 516. Nunn KL, Clair GC, Adkins JN, et al (2020) Amylases in the Human Vagina. 10.1128/m Sphere 10.1128/m Sphere.00943-20PMC 772925633298571 · doi ↗ · pubmed ↗
- 619. Ventura M, O’Flaherty S, Claesson MJ, et al (2009) Genome-scale analyses of health-promoting bacteria: Probiogenomics. Nat Rev Microbiol 7:61–7110.1038/nrmicro 204719029955 · doi ↗ · pubmed ↗
- 725. Asnicar F, Thomas AM, Beghini F, et al (2020) Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using Phylo Phl An 3.0. Nat Commun 11:. 10.1038/s 41467-020-16366-710.1038/s 41467-020-16366-7PMC 723744732427907 · doi ↗ · pubmed ↗
- 832. Bateman A, Martin M-J, Orchard S, et al (2025) Uni Prot: the Universal Protein Knowledgebase in 2025. Nucleic Acids Res 53:D 609–D 617. 10.1093/nar/gkae 101010.1093/nar/gkae 1010 PMC 1170163639552041 · doi ↗ · pubmed ↗