Non-chelation control in allylations of α-oxy ketones using group-14 allylatranes
Yuya Tsutsui, Kokoro Shiga, Akihito Konishi, Makoto Yasuda

TL;DR
This paper introduces a new method for anti-selective allylation of α-oxy ketones using group-14 allylatranes, enabling the synthesis of anti-1,2-diols with high yield and selectivity.
Contribution
The study presents the first exploration of anti-selective allylation via a non-chelation pathway using group-14 allylatranes.
Findings
Allylatranes with a group-14 element center enable anti-selective allylations of α-oxy ketones.
The method achieves high yields and excellent diastereoselectivity for anti-1,2-diols.
The non-chelation pathway is supported by experimental and theoretical evidence.
Abstract
Stereoselective nucleophilic additions to α-substituted carbonyl compounds are a crucial area of contemporary research in organic chemistry. Of the various advancements in π-facial selectivity in addition reactions of carbonyl compounds, the (polar) Felkin-Anh model and the chelation model are well recognized for accurately explaining the selectivity of the allylic products. For reactions that involve α-oxy carbonyl groups - known for their broad applications in natural-product synthesis and as effective building blocks in organic synthesis - the stereoselective reaction typically follows the chelation model, favoring syn-selective addition. In contrast to the well-established syn-selective additions of α-oxy carbonyls, anti-selective additions through a non-chelation pathway remain largely unexplored. In this study, we present the anti-selective allylation of α-oxy ketones using…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
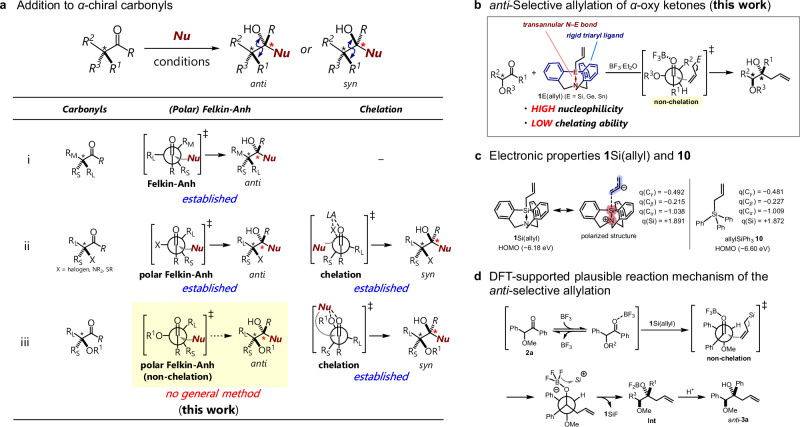 Figure 1
Figure 1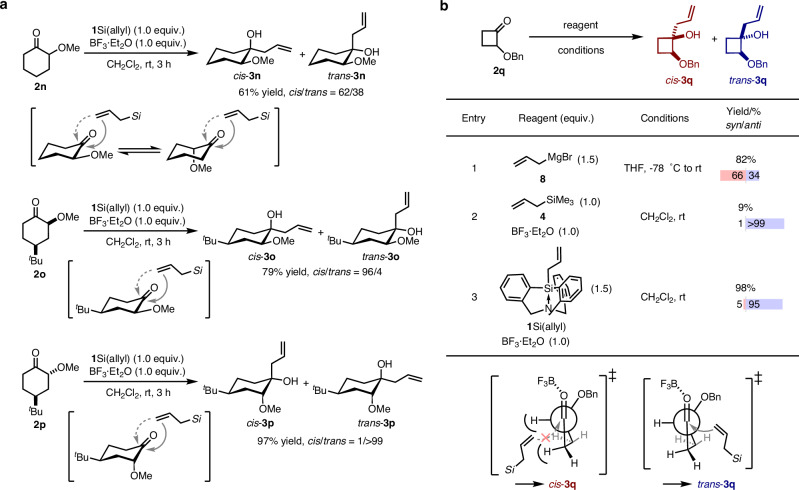 Figure 2
Figure 2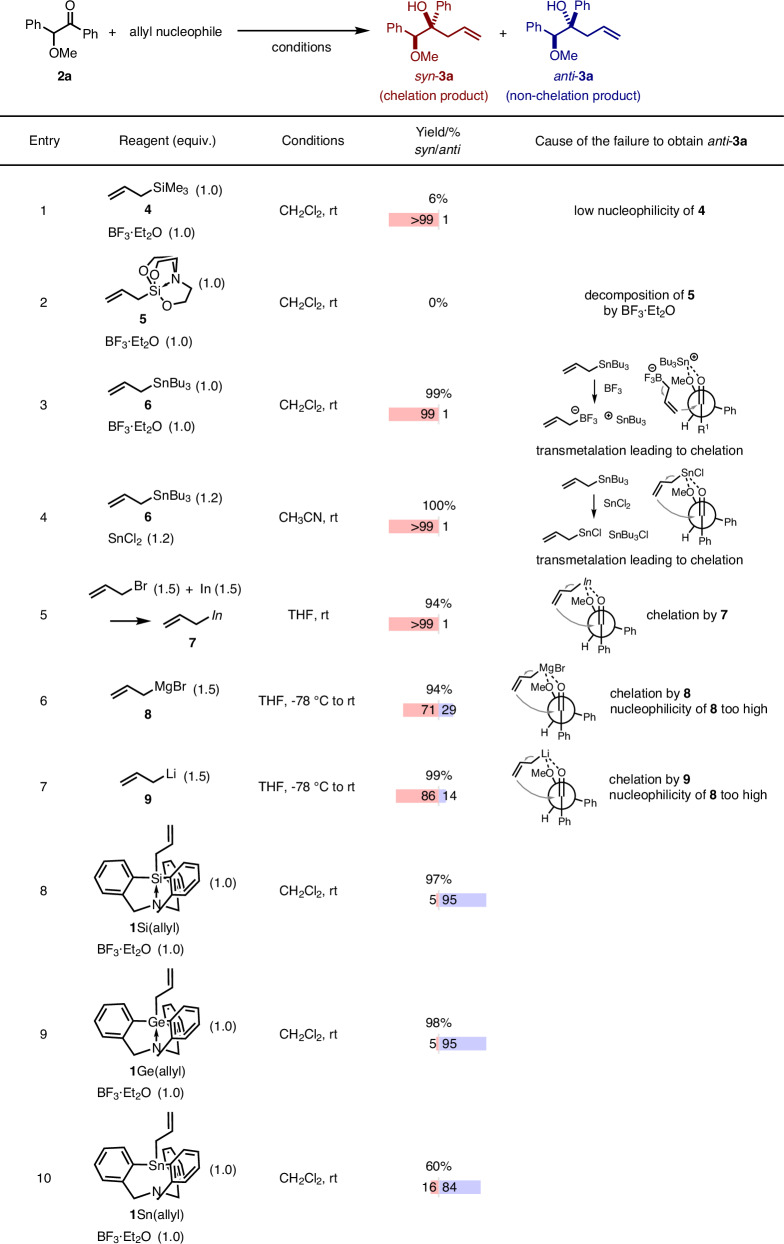 Figure 3
Figure 3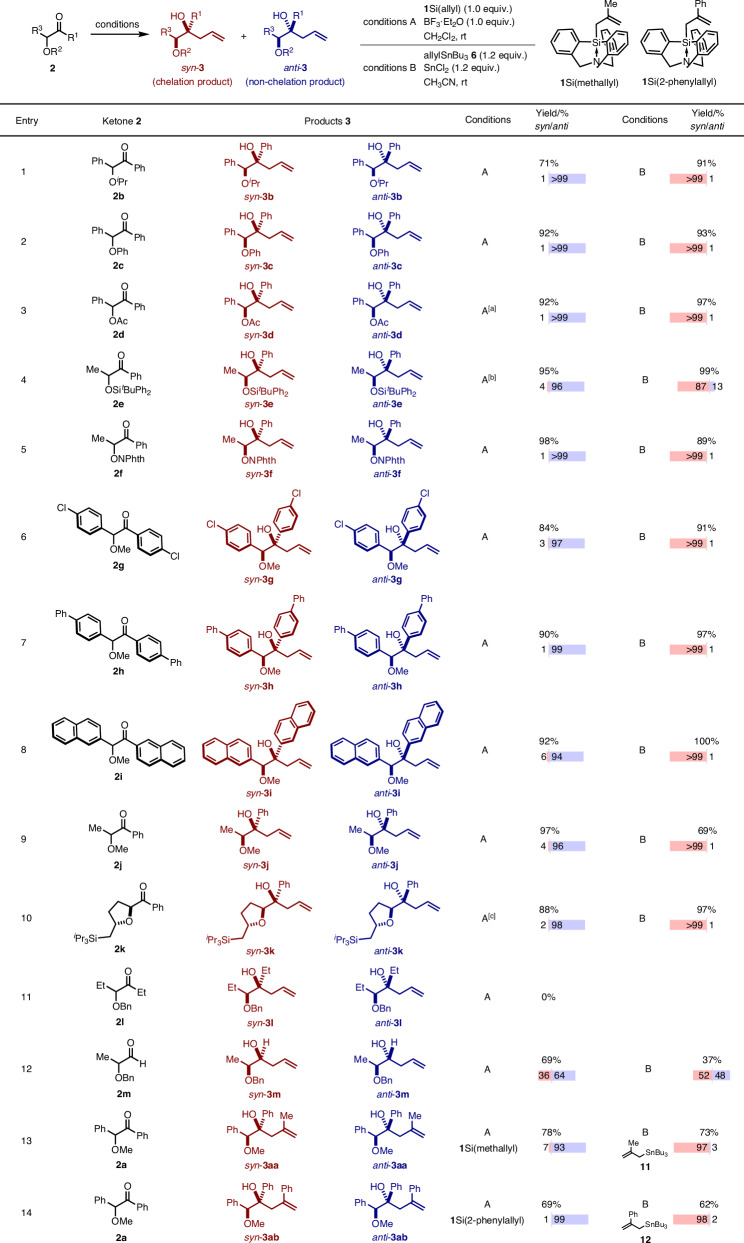 Figure 4
Figure 4- —https://doi.org/10.13039/501100001700Ministry of Education, Culture, Sports, Science and Technology (MEXT)
- —https://doi.org/10.13039/501100003382MEXT | JST | Core Research for Evolutional Science and Technology (CREST)
- —https://doi.org/10.13039/501100001691MEXT | Japan Society for the Promotion of Science (JSPS)
- —https://doi.org/10.13039/501100002241MEXT | Japan Science and Technology Agency (JST)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic C–H Functionalization Methods · Catalytic Cross-Coupling Reactions · N-Heterocyclic Carbenes in Organic and Inorganic Chemistry
Introduction
Controlling the π-facial selectivity in addition reactions to carbonyl compounds is of crucial importance in stereoselective organic synthesis^1–4^. A common case is that addition reactions to carbonyl compounds with a substituent at the α-position results in diastereomers. This stereochemical outcome is accurately explained by the Felkin-Anh model^5,6^, which illustrates the relationship between the conformation around the α-carbon and the attacking nucleophile. The nucleophile reacts from the direction opposite to the largest α-carbon substituent (R_L_), and when the nucleophile approaches the carbonyl carbon at the Bürgi-Dunitz angle^7^, R_L_ avoids steric repulsion orienting away from it, thus avoiding steric repulsion with the medium-sized substituent (R_M_), favoring a transition state with the nucleophile closer to the smallest substituent (R_s_) (Fig. 1ai). If there is an electronegative atom (X) at the α-position, the conformation of the transition state is changed. The C–X bond (X = halogen, N, or S) adopts an orthogonal conformation to the carbonyl group due to the stabilizing effect of the hyperconjugation between the σC–X and πC=O orbitals (polar Felkin−Anh Model)^8–11^. Thus, the nucleophile approaches from the direction opposite to the X substituent (Fig. 1aii left). On the other hand, when a chelating Lewis acid (LA)^12^ or hydrogen bond^13,14^ are present with either α-halogenated carbonyls or α-amino carbonyls, the reaction proceeds via the chelation model^15–19^, where the stereoselectivity is the opposite to that of the Felkin-Anh Model. Here, the conformation where the LA chelates with the coordinating electronegative group (X) and the carbonyl oxygen is preferred, and the nucleophile approaches in a manner that avoids steric repulsion with R_L_ (Fig. 1aii right). For reactions involving α-oxy carbonyl groups, which have broad applications in natural-product synthesis and are effective building blocks in organic synthesis, the stereoselective reaction following the chelation model is strongly effective (Fig. 1a-iii right). One of the diastereoselectivities can be easily controlled because the oxygen groups, which are highly coordinating, readily chelate to nucleophiles. However, obtaining stereoselective outcomes according to the Felkin-Anh model with α-oxy carbonyl compounds has been virtually impossible, except in a few limited cases^2,10^, and has remained a long-standing challenge (Fig. 1b). Non-chelation control in the nucleophilic addition to α-oxy carbonyls is the final challenge to be solved in the Felkin-Anh model for the addition of nucleophiles to carbonyl compounds.Fig. 1. Overview for the diastereoselective additions to α-chiral carbonyls and allylations using atrane-type reagent.a Summary for the diastereoselective addition to α-chiral carbonyls. b anti-Selective allylation of α-oxy ketones by 1E(allyl) (E = Si, Ge, or Sn; this work). c Calculated electronic properties of 1Si(allyl) (left) and allylSiPh_3_ 10 (right). d DFT-supported plausible reaction mechanism for the anti-selective allylation using 1Si(allyl).
The allylation of α-oxy carbonyls frequently follows the Chelation model, yielding syn products where the vicinal hydroxyl groups are positioned on the same side relative to the carbon-chain backbone^20^. When using Sn-^21,22^, In-^23,24^, Ga-^25^, Si-^26,27^, and B^28^-based allyl nucleophiles, the rigid conformation of the transition state provides syn products. Another approach involves the use of chelating Lewis acids, such as TiCl_4_^29–31^, SnCl_4_^30,31^, or a combined InCl_3_–chlorosilane^32^. However, anti-selective allylation via a non-chelation pathway is still relatively unexplored. In a pioneering study, Reetz et al. have reported the anti-selective allylation of α-oxy aldehydes through non-chelation control using allylSiMe_3_ and a monodentate Lewis acid (BF_3_·Et_2_O)^33^. Still, examples of anti-selective allylations are limited and there is room for significant further improvement. Establishing anti-selective stereocontrol in allylations of α-oxy ketones is particularly challenging, considering that ketones are less electrophilic than aldehydes and therefore require a stronger nucleophile for addition. However, enhanced nucleophilicity increases the Lewis acidity of the allyl metal, thus promoting chelation. Achieving anti-selective allylations requires balancing low chelation ability with high nucleophilicity, but the trade-off between these properties hampers the realization of this goal. Using highly coordinated nucleophiles^34^ is one of the most promising solutions to realize the allylation of α-oxy ketones under non-chelation control. Our group has previously investigated group-14-element atrane-type cationic species 1E^+^ (E = Si, Ge, or Sn)^35^. The atrane framework contains a highly coordinated element center due to the intramolecular transannular N–E bond, and thus exhibits significantly enhanced nucleophilicity. The rigid triaryl atrane structure also reduces undesired chelation effects. Thus, we designed allylatranes 1E(allyl) (E = Si, Ge, or Sn) for the anti-selective allylation of α-oxy ketones (Fig. 1b). The synthesis of 1E(allyl) is summarized in Fig. S1.
Results
As a model reaction, we performed the allylation of benzoin methyl ether 2a (Table 1). When allylSiMe_3_ 4 was used in the presence of BF_3_·Et_2_O, syn-product 3a was obtained. The syn-selectivity may be due to either the generation of a high-coordinated or a cationic Si species^36^ the applied reaction conditions. The low yield (6%) of this reaction is caused by the low nucleophilicity of 4 (Table 1, entry 1). Oxygen-ligated allylsilatrane allylSi(OC_2_H_4_)3_N 5, which exhibits heightened nucleophilicity^37,38^, was an ineffective allylation reagent (Table 1, entry 2). Nuclear-magnetic-resonance (NMR) measurements revealed that 5 decomposes rapidly in the presence of BF_3·Et_2_O (Fig. S10). Allyltributylstannane allylSnBu_3_ 6 furnished the chelation-controlled product syn-3a in 99% yield with high diastereoselectivity (syn/anti = 99/1; Table 1, entry 3). Here, stannyl cation species generated by transmetalation with BF_3_·Et_2_O likely activate the substrate in a chelation-controlled manner^39,40^. Treating 2a with allylSnBu_3_/SnCl_2_^41^ or allylindium^23,24,42^ provided product syn-3a in high yield with excellent syn-selectivity (syn/anti = >99/1; Table 1, entries 4 and 5) via the chelation pathway. The reactions with allylMgBr 8 or allylLi 9 proceeded without diastereoselectivity due to strong chelation combined with too high nucleophilicity (Table 1, entries 6 and 7)^43,44^. However, we were pleased to find that the desired homoallylic alcohol (3a) was produced using newly synthesized 1Si(allyl) in 97% yield with high anti-selectivity (syn/anti = 5/95; Table 1, entry 8). The stereochemistry of the allylated product anti-3a was confirmed using single-crystal X-ray diffraction analysis after silylation of the hydroxy group. Allylgermatrane 1Ge(allyl) also furnished anti-3a with nearly perfect selectivity (syn/anti = 5/95; Table 1, entry 9). A decrease in diastereoselectivity and yield was observed with 1Sn(allyl) (syn/anti = 14/86, 60%; Table 1, entry 10), because the nucleophilicity of 1Sn(allyl) was too high. The high stability of 1E(allyl) with BF_3_·Et_2_O is likely due to the relatively strong E–C_Ar_ bonds and because the chelate effect of the cage-shaped structure suppresses decomposition. Other allyl nucleophiles were not effective in this anti-selective allylation (Table S2).Table 1. Allylation of benzoin methyl ether 2aYields and diastereomeric ratios of the products were determined by ^1^H NMR measurements using 1,1,2,2-tetrachloroethane as an internal standard. Bold numbers represent compound numbers
To investigate the electronic properties of 1E(allyl), we calculated its molecular orbitals (Fig. 1c). The calculated energy of the HOMO of 1Si(allyl) (−6.18 eV) suggests that its nucleophilicity is higher than that of allylSiPh_3_ 10 (−6.60 eV). The high nucleophilicity of 1Si(allyl) is also supported by ^13^C NMR and natural-bond-orbital (NBO) analyses (Table S6). The difference in NBO charge between C_β_ and C_γ_ was higher for 1Si(allyl) ( | q(C_β)–q(Cγ)| = 0.277) than for 10 (|q(Cβ)–q(Cγ)| = 0.254). Meanwhile, a second-order-perturbation analysis indicated that the hyperconjugation from the σ(Si–Cα) bond to the π*(Cβ_ = C_γ_) bond is less effective in 1Si(allyl) (ΔE = 3.84 kcal/mol) than in 10 (ΔE = 5.61 kcal/mol), commensurate with weak electronic communication between the allylic moiety and the group-14-element center of 1Si(allyl). Thus, the strong nucleophilic character of 1Si(allyl) should reflect the charge localization rather than the stereoelectronic effects. Notably, the results of the NBO analyses indicate that the nucleophilicity of 1Si(allyl) is similar to that of allylsilatrane 5, which rapidly decomposes under the applied reaction conditions. This highlights the balance of reactivity and stability in 1Si(allyl), facilitated by its rigid triaryl-based structure. The stabilization of the silyl center by transannular interactions would also enhance the allyl-anion character. A crystallographic analysis confirmed that the molecular geometry of 1Si(allyl) exhibits a nearly trigonal-bipyramidal structure with a five-coordinated silicon center. Most allylic reagents with high nucleophilicity feature a strong Lewis-acidic metal center, which facilitates the allylation of α-oxy ketones through a chelation pathway, resulting in syn-products^9,31,33^. In contrast to the traditional methods for achieving syn-selective allylations, 1E(allyl) enhances the nucleophilicity of the allylic group while simultaneously reducing the Lewis acidity of the metal center. This careful balance between high nucleophilicity and weak Lewis acidity in 1E(allyl), achieved through an atrane-type framework, allows for the distinctive anti-selective allylations of α-oxy ketones.
DFT calculations at the B3LYP-D3/6-31 G*/SMD (dichloromethane)//B3LYP-D3/6-31 G* level support the observed anti-selectivity via a non-chelation pathway (Fig. S16). A plausible mechanism for the allylation of 2a with allylsilatrane 1Si(allyl) is shown in Fig. 1d. First, ketone 2a coordinates to BF_3_. The activated ketone then interacts with 1Si(allyl), forming a carbon–carbon bond. The estimated transition state leading to the anti-product is by 6.4 kcal/mol more stable than that leading to the corresponding syn-product. This enhanced stability results from steric repulsion between the atrane moiety of 1Si(allyl) and the phenyl group of 2a. Interestingly, the conformation in the transition state resembles the Cram-type mode, in which the α-methoxy group is located antiperiplanar relative to the carbonyl group of 2a activated by BF_3_, rather than following the polar Felkin–Ahn model. The allylation of 2a with the bulky 1Si(allyl) and the small Lewis acid BF_3_ is therefore likely to favor the Cram-type conformation over the initially anticipated polar Felkin–Ahn conformation. Subsequently, one of the fluorine atoms on the boron atom is captured by silyl cation 1Si^+^, which results in the formation of the allyl product Int and 1SiF. NMR monitoring confirmed the generation of Int and 1SiF (Fig. S12). Finally, protonation by quenching yields the homoallylic alcohol anti-3a. There is a possibility that an in-situ exchange of the allyl moiety between 1Si(allyl) and BF_3_·Et_2_O occurs to form allylBF_2_·Et_2_O, albeit that a signal for allylBF_2_·Et_2_O was not observed in the NMR experiments. Therefore, it is considered that this anti-selective allylation proceeds via a non-chelation pathway. Further theoretical calculations to investigate the pathway in detail are currently in progress in our group. The high stability of 1Si(allyl) derived from the atrane structure suppresses the undesired in-situ exchange process.
The anti-product was obtained with high selectivity in high yield from a variety of α-oxy ketones using 1Si(allyl) (conditions A, Table 2). The stereochemistry of anti-3 was determined by comparison with the NMR spectrum of syn-3, which was prepared using our reported method employing Sn(II) chloride for chelation-control (conditions B, Table 2)^41^. The reactions conducted under conditions A with α-isopropoxy (2b), α-phenoxy (2c), or α-acetoxy (2****d) ketones afforded products 3b–d with high anti-selectivity (syn/anti = 1/ > 99) (conditions A, Table 2, entries 1 – 3). Under chelation control, the syn-products were obtained with perfect selectivity (conditions B, Table 2, entries 1 – 3). A bulky α-silyloxy-substituted ketone (2e) gave the desired product (3e) with high diastereoselectivity (syn/anti = 4/96) in 95% yield (conditions A, Table 2, entry 4). The selectivity using 1Si(allyl) is better than that obtained using allylMgBr (syn/anti = 15/85) as reported by Woerpel^45^. In the case of the Sn(II) system, the selectivity was slightly lower, but the syn-product was still obtained with high selectivity (syn/anti = 87/13, conditions B, Table 2, entry 4). The α-aminoxy-substituted ketone (2****f) gave the anti-product in 98% yield with great anti-selectivity (syn/anti = 1/ > 99, conditions A, Table 2, entry 5). The reaction also gave good yields and diastereoselectivities with p-chloro- or p-phenyl-substituted benzoin methyl ethers (2g, 2h) and a naphthyl ketone derivative (2i) (conditions A, Table 2, entries 6–8). Furthermore, the allylations of α-methyl-α-methoxy ketones 2j and 2k with a 2,5-disubstituted trans-tetrahydrofuran motif afforded 3j and 3k with nearly perfect anti-selectivity (syn/anti = 4/96 and 2/98) (conditions A, Table 2, entries 9–10). The selectivity of 3k was dramatically improved relative to a previous study with allylMgCl (syn/anti = 29/71)^46^. Unfortunately, the desired product was not obtained from the allylation of α-oxy aliphatic ketone 2l (conditions A, Table 2, entry 11), as the allylation of aliphatic ketones requires an allyl nucleophile with higher nucleophilicity; using 1Sn(allyl) instead of 1Si(allyl) had no effect. The allylation of α-oxy aldehyde 2m proceeded with low diastereoselectivity (syn/anti = 36/64, conditions A, Table 2, entry 12). The nucleophilicity of 1Si(allyl) toward α-oxy aldehydes is assumed to be too high. The allylsilatrane derivatives 1Si(methallyl) and 1Si(2-phenylallyl) gave the desired adducts with nearly perfect diastereoselectivity. Methallylsilatrane 1Si(methallyl) selectively afforded anti-3aa (syn/anti = 7/93) in 78% yield (conditions A, Table 2, entry 13). The allylation with 1Si(2-phenylallyl) also furnished anti-3ab with high selectivity (syn/anti = 1/99) in 69% yield (conditions A, Table 2, entry 14). It was also confirmed that the syn-products could be obtained from the aforementioned examined substrates using the corresponding allylstannane derivatives and Sn(II) chloride. Thus, the use of 1Si(allyl) (via a non-chelation path) and the Sn(II) system (via a chelation path) affords a high degree of diastereocontrol in the allylations of α-oxy ketones.Table 2. Substrate scope of α-oxy carbonyls in diastereoselective allylationsConditions A: 1Si(allyl) (1.0 eq.), BF_3_·Et_2_O (1.0 eq.), and CH_2_Cl_2_ at room temperature. Conditions B: allylSnBu_3_ 6 (1.2 eq.), SnCl_2_ (1.2 eq.), and CH_3_CN at room temperature. Yields and diastereomeric ratios of the products were determined using ^1^H NMR measurements using 1,1,2,2-tetrachloroethane as an internal standard. [a] Reaction was conducted at 0 °C. [b] 1Si(allyl) (1.5 equiv.). [c] Reaction was conducted at −20 °C. Bold numbers represent compound numbers.
Then, the reactivity of allylsilatrane 1Si(allyl) with cyclic α-oxy ketones was explored (Fig. 2a). In the case of 2-methoxycyclohexanone 2n, low diastereoselectivity was observed (cis/trans = 62/38), probably due to the flexible conformational orientation of the methoxy group. In contrast, the allylations of conformationally fixed cyclohexanones 2o and 2p with a ^t^Bu group at the 4-position, selectively yielded diastereomers via an equatorial attack. For the allylation of 2p, the product 3p showed the trans-configuration, which is identical to that generated under the reported chelation conditions involving a twist-boat conformation in the transition state^47^. These results indicate that 1Si(allyl) acts as a bulky allylic nucleophile, favoring nucleophilic attack from the less hindered equatorial position under our non-chelation conditions. The products obtained using 1Si(allyl), i.e., cis-3o and trans-3p, were identical to those reported by Paquette using allylindium^48^. Paquette has proposed a transition state with a twist-boat conformation to maximize the O–In interactions, whereas our method involves a simple equatorial attack. Furthermore, the steric effect of 1Si(allyl) was evident in the allylation of α-oxy cyclobutanone 2q (Fig. 2b). The resulting allylated product (3q) is an important synthetic intermediate for the substructure of a β-secretase modulator candidate. When 2q was treated with allylMgBr 8, the diastereoselectivity was low (cis/trans = 66/34; Fig. 2b, entry 1)^49^. Treatment with allylSiMe_3_ 4 in the presence of BF_3_·Et_2_O gave the desired product (trans-3q) with high diastereoselectivity (cis/trans = 1/ > 99; Fig. 2, entry 2), albeit that the yield was disappointing. Silatrane 1Si(allyl) afforded trans-3q in high yield and high diastereoselectivity (cis/trans = 5/95). The stereochemistry of 3q was determined by acetalization of the diols obtained from 3q (Fig. S9). Cyclobutanone 2q has a rigid conformation due to its four-membered ring and a transition state that minimizes steric repulsion between the hydrogens on the cyclobutane ring and 1Si(allyl) would be favored. Allylsilatrane 1Si(allyl) is expected to demonstrate efficacy in natural-product synthesis and pharmaceutical synthesis.Fig. 2. Diastereoselective additions to α-oxy cyclic carbonyls.a Allylation of α-oxy cyclohexanones 2n–2p. b Allylation of α-oxy cyclobutanone 2q.
Methods
General procedure for anti-selective allylations using 1Si(allyl)
In a nitrogen-filled glovebox, a mixture of BF_3_ ⋅ Et_2_O (0.2 mmol) and ketone 2 (0.2 mmol) in dichloromethane (2 mL) was treated with 1Si(allyl) (0.2 mmol). After the reaction mixture was stirred for 3 h at room temperature, methanol (2 mL) was added to the mixture. All solvents were removed under reduced pressure to give the crude product.
Supplementary information
Transparent Peer Review file Supplementary Information
Source data
Source Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Corey, E. J. The Logic of Chemical Synthesis. (Wiley, 1991).
- 2Otera, J. Modern Carbonyl Chemistry. (Wiley, 2008).
- 3Dickens, T. K. & Warren, S. Chemistry of the Carbonyl Group: A Step-by-Step Approach to Understanding Organic Reaction Mechanisms. (Wiley, 2018).
- 4Anh, N. T. Regio- and stereo-selectivities in some nucleophilic reactions. in Organic Chemistry Syntheses and Reactivity 145–162 (Springer, 1980). 10.1007/B Fb 0048506.
- 5Reetz, M. T. & Kesseler, K. Non-chelation-control in nucleophilic additions to chiral α- and β-alkoxy aldehydes. J. Chem. Soc. Chem. Commun. 1079–1080 (1984) 10.1039/C 39840001079.
- 6Yasuda, M., Haga, M. & Baba, A. Isolation and crystallographic characterization of allylindium species generated from allyl halide and indium(0). Eur. J. Org. Chem. 5513–5517 (2009) 10.1002/ejoc.200900955.