Proteolytic dysregulation in the skin: insight from rare monogenic skin diseases
Zhongtao Li, Sheng Wang, Diana C. Blaydon, David P. Kelsell

TL;DR
This paper reviews how protease dysregulation contributes to rare skin diseases and explores potential treatments.
Contribution
The paper provides insights into protease-related mechanisms in monogenic skin diseases and highlights emerging therapeutic approaches.
Findings
Protease and protease inhibitor dysregulation is linked to monogenic skin diseases.
Disease gene discoveries have enhanced understanding of skin homeostasis and barrier function.
Emerging therapies target protease-related mechanisms in these diseases.
Abstract
Proteases are essential enzymes that, through the breakdown of proteins, regulate many aspects of tissue homeostasis including barrier function, cellular signaling, and tissue repair mechanisms in organisms. Disease gene discovery in a number of monogenic skin diseases has deepened the knowledge of how proteases and protease inhibitors can regulate skin homeostasis, keratinocyte desmosome-mediated cell adhesion, and epidermal barrier function. This short review details the association of protease dysregulation with monogenic skin diseases, postulated disease mechanisms, and emerging therapeutic strategies.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
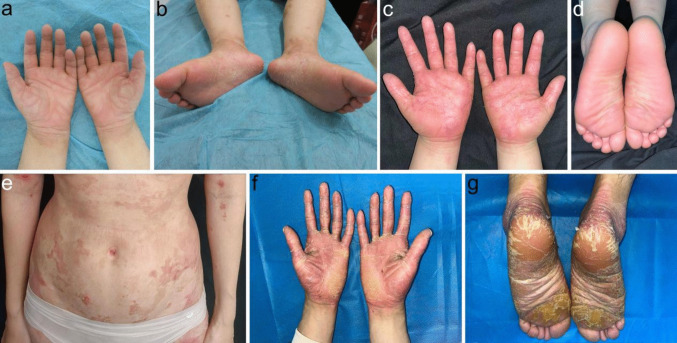 Figure 1
Figure 1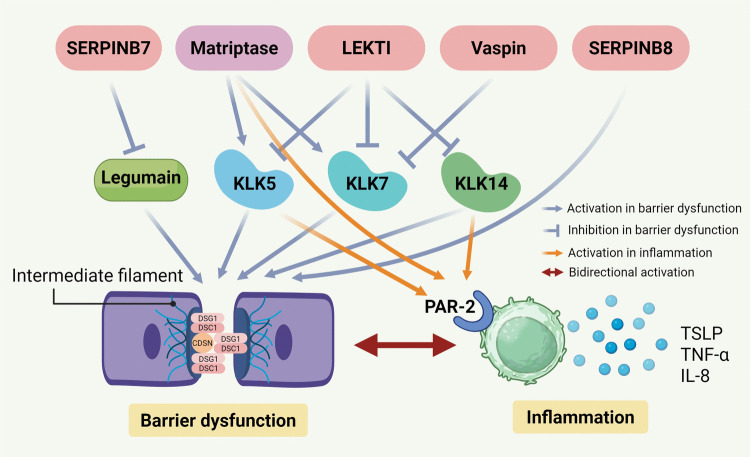 Figure 2
Figure 2- —Qimingxing Research Fund for Young Talents
- —https://doi.org/10.13039/501100000265Medical Research Council
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSkin and Cellular Biology Research · Coagulation, Bradykinin, Polyphosphates, and Angioedema · Dermatological and Skeletal Disorders
Introduction
The skin is the largest organ of the human body and is constantly exposed to the external environment. The outer epidermal layer plays a vital role in forming the protective skin barrier and is, in part, reliant on the integrated structure and function of intercellular adhesion and cell communication hubs; these are the desmosomes, gap, adherens, and tight junctions (Cohen-Barak et al. 2020; Jensen and Proksch 2009). However, the epidermis constantly undergoes a process of renewal whereby adhesions between the outermost, dead keratinocytes (corneocytes) need to be cleaved in a controlled manner to allow corneocyte shedding from the stratum corneum (termed desquamation). This desquamation process is a highly regulated process that requires a careful balance of proteases and their respective inhibitors (Stamatas 2024; Cohen-Barak et al. 2022).
Proteases are enzymes that catalyze the hydrolysis of peptide bonds in proteins and polypeptides. More than 500 proteases have been identified in the human genome (Donzelli et al. 2023) and are categorized according to their catalytic mechanisms, into serine, cysteine, aspartic, threonine proteases, and metalloproteases, though some categories contain families with two or more catalytic types (Fan et al. 2017). Among them, over 100 proteases are expressed in human skin with emerging different biological functions including epidermal barrier homeostasis, maintaining keratinocyte adhesion and regulation of cell signaling including those related to inflammation (Stewart-McGuinness et al. 2022). Many of these biological functions within the skin have been revealed from genetic and cell biology studies of rare monogenic disorders of epidermal keratinization (Table 1) (Has 2018). These include palmoplantar keratoderma (PPK; hyperkeratosis/callus formation on the palm and sole epidermis) and peeling skin syndrome (PSS; epidermal peeling of the palm and sole epidermis) (Fig. 1). These epidermal differentiation disorders (EDD) represent inherited disorders of keratinization characterized by abnormal epidermal differentiation, encompassing those that largely affect the palmoplantar (palm and sole skin, pEDD) and those that associate with nonsyndromic (nEDD) or syndromic skin conditions (sEDD) (Sprecher et al. 2025; Akiyama et al. 2025; Paller et al. 2025). This new nomenclature includes the causative gene associated with each specific EDD condition.
Table 1. Protease and protease inhibitors in hereditary skin diseasesDiseaseGene-based nameClinical manifestationMIMInheritanceProteaseProtease inhibitorInactive proteaseReferenceNagashima-type PPKSERPINB7-pEDDMild diffuse hyperkeratosis with erythema, skin peeling, aquagenic whitening, palmoplantar hyperhidrosis on the palms and soles accompanied with odor615598ARLegumainSERPINB7/ Kubo et al. 2013SERPINA12-related PPKSERPINA12-pEDDDiffuse hyperkeratosis with erythema, skin peeling, aquagenic whitening, palmoplantar hyperhidrosis on the palms and soles accompanied with odor/ARKLK7Vaspin/ Mohamad et al. 2020Exfoliative ichthyosis/PSS5SERPINB8-pEDDSkin peeling, hyperkeratotic plaques, and erythema on the palms and soles617115ARFurinSERPINB8/ Pigors et al. 2016Netherton syndromeSPINK5-sEDDSkin desquamation, atopic manifestation, hair abnormality256500ARKLK5KLK7KLK14LEKTI/ Chavanas et al. 2000Ichthyosis-hypotrichosis syndromeST14-sEDDDiffuse scaling and hypotrichosis on the body, photophobia602400ARMatriptase// Alef et al. 2009Papillon-Lefèvre syndromeCTSC-pEDDSymmerical diffuse PPK, early loss of teeth, destructive periodontitis, and recurrent pyogenic infections245000ARCathepsin C// Toomes et al. 1999; Ghanei et al. 2021Haim-Munk syndromeCTSC-pEDD-arachnodactylyRed, scaly, thickened skin on the palms and soles, alongside pus-producing skin infections, periodontitis, pes planus, onychogryphosis, arachnodactyly and acroosteolysis245010// Hart et al. 2000; Chitsamankhun et al. 2024Keratolytic winter erythemaCTSB-pEDDRecurrent palmoplantar erythema and epidermal peeling, accompanied with itching, hyperhidrosis and odor, often worsen in winter148370ADCathepsin B// Hull et al. 2013; Ngcungcu et al. 2017Acral peeling skin syndrome with exfoliative ichthyosis/PSS4CSTA-nEDDDry, scaly skin over most of the body with coarse peeling of nonerythematous skin on the palms and soles, which is exacerbated by excessive moisture and minor trauma607936ARCathepsin L Cathepsin VCystatin A/ Blaydon et al. 2011aEctodermal dysplasia 15, hypohidrotic/hair typeCST6-sEDDHypotrichosis, dry skin, eczema, blepharitis, photophobia and impaired sweating618535ARCathepsin LCathepsin VLegumainCystatin M/E/van den Bogaard et al. 2019Keratosis follicularis spinulosa decalvansCST6-sEDD-cicatricialGeneralised follicular hyperkeratosis, dry skin, progressive cicatricial alopecia mainly on the scalp, facial erythema, folliculitis, and eye symptoms612843AD/ Eckl et al. 2021PLACK syndromeCAST-pEDDPeeling skin, leukonychia, acral punctate keratosis, cheilitis, and knuckle pads616295ARCalpainCalpastatin/ Lin et al. 2015Inflammatory skin and bowel syndromeADAM17-sEDDPerioral and perianal erythema with fissuring and a generalized pustular rash that developed into psoriasiform erythroderma, with flares of erythema, scaling, and widespread pustules, associated with bowel disease614328ARADAM17// Blaydon et al. 2011bTylosis with oesophageal cancerRHBDF2-pEDD PPK, oral and oesophageal leukoplakia, and a significantly high lifetime risk of oesophageal squamous cell carcinoma148500ADADAM17/iRHOM2Blaydon et al. 2012Olmsted syndromeMBTPS2-pEDDBilateral mutilating transgredient PPK and periorificial keratotic plaques with considerable clinical heterogeneity300918XLRS2P// Haghighi et al. 2013IFAP syndrome with or without BRESHECK syndromeMBTPS2-sEDD-IFAPIchthyosis follicularis with atrichia and photophobia with or without additional features (e.g., corneal opacifications, developmental delay, skeletal malformations, ectodermal dysplasia, renal anomalies)308205// Oeffner et al. 2009; Naiki et al. 2012Keratosis follicularis spinulosa decalvansMBTPS2-sEDD-cicatricial alopeciaWidespread hyperkeratotic follicular papules, facial erythema, hypotrichosis and scarring alopecia308800// Aten et al. 2010PPK palmoplantar keratoderma, pEDD palmoplantar epidermal differentiation disorder, nEDD nonsyndromic epidermal differentiation disorder, sEDD syndromic epidermal differentiation disorder, PSS peeling skin syndrome, AR autosomal recessive, AD autosomal dominant, XLR X-linked recessiveFig. 1Clinical manifestations of hereditary skin diseases caused by pathogenic variants in genes encoding proteases or protease inhibitors. a, b Mild erythema and hyperkeratotic desquamation on palm and plantar surfaces, extending to the wrists and ankles in a 10-year-old boy carrying a homozygous pathogenic variant for SERPINB7 c.796C > T. c, d Diffuse erythematous hyperkeratosis of the palms and plantar surfaces, extending to the wrists in an 8-year-old girl carrying a homozygous pathogenic variant for SERPINA12 c.635-7A > G. e Extensive erythematous plaques with double-edged marginal scales on the trunk and extremities in a 23-year-old female carrying compound heterozygous pathogenic variants in SPINK5 c.1048C > T and c.−97_−80del. f, g Erythema and hyperkeratosis on the palms and plantar surfaces in a 21-year-old male caused by compound heterozygous pathogenic variants in CTSC c.800T > C and c.1234T > A
Serine proteases and their inhibitors (serpins)
Serine proteases are present in all living cells (Sil et al. 2024). Kallikrein-related peptidases (KLKs) are the largest family of trypsin- or chymotrypsin-like secreted serine proteases in humans (Meyer-Hoffert 2009). Among them, KLK5, KLK7, and KLK14 are reported to be the most important in desquamation (Ulbricht et al. 2018) and are involved in the cleavage of corneodesmosomes, the main inter-cellular adhesion junctions between corneocytes formed by desmoglein 1 (DSG1), desmocollin 1 (DSC1), and corneodesmosin (CDSN). KLK activity can also regulate the protease-activated receptor 2 (PAR-2) pathway, which in turn modulates pro-inflammatory signals, including thymic stromal lymphopoietin (TSLP), IL-8 and TNF-α (Zani et al. 2022). Overactive KLKs consequently impair the skin barrier and increase inflammation (Pontone et al. 2022). Importantly, epidermal barrier dysfunction and skin inflammation form a self-reinforcing pathogenic loop in inflammatory skin diseases (Beck et al. 2002) (Fig. 2). Serpins (serine protease inhibitors) are the largest family of all the protease inhibitors and use conformational changes to inhibit proteases. Though most serpins target serine proteases, some have been linked to inhibit papain-like cysteine proteases or caspases (Irving et al. 2002; Ray et al. 1992). To date, five monogenic autosomal recessive skin disorders are associated with dysregulated serine protease activity (Table 1).Fig. 2. Schematic representation of serine protease and serpin regulation in skin barrier function and inflammation (created in https://BioRender.com). Serine proteases and serpins contribute to barrier dysfunction by degrading corneodesmosome components (DSG1, DSC1, and CDSN) and intermediate filaments (blue activation or inhibition arrows). They also activate PAR-2 on immune cells, inducing the release of cytokines such as TSLP, TNF-α, and IL-8, which result in cutaneous inflammation (orange activation arrows). Skin barrier dysfunction and inflammation reciprocally amplify each other, forming a self-reinforcing cycle (red activation arrow). LEKTI, lympho-epithelial Kazal-type related inhibitor; KLK, kallikrein-related peptidase; DSG1, desmoglein 1; DSC1, desmocollin 1; CDSN, corneodesmosin; PAR-2, protease-activated receptor 2; TSLP, thymic stromal lymphopoietin
Nagashima-type palmoplantar keratoderma [NPPK (SERPINB7-pEDD), MIM #615598] is associated with biallelic loss of function (LOF) variants of SERPINB7 (Kubo et al. 2013; Kubo 2025), and SERPINA12-related palmoplantar keratoderma [SPPK (SERPINA12-pEDD)] is linked to biallelic LOF variants in SERPINA12 (Mohamad et al. 2020). SERPINB7-pEDD and SERPINA12-pEDD exhibit overlapping clinical features, including hyperkeratosis with transient erythema (skin redness) extending beyond the palmoplantar margins (Fig. 1a–d). Additional features include palmoplantar skin peeling, aquagenic whitening (upon exposure to water), increased sweating (hyperhidrosis), odor, and prone to fungal infection (Liu et al. 2023; Mohamad et al. 2020). Digenic inheritance of LOF variants in SERPINB7 and SERPINA12 have recently been associated with NPPK in China and Japan suggesting shared biology exists between these two serine protease inhibitors (Liu et al. 2024; Jiang et al. 2025; Ebata et al. 2025; Zhang et al. 2025). Further studies are needed to explore the interplay between SERPINB7 and SERPINA12 in the pathogenesis of NPPK phenotypes.
While cases have also been reported in non-Asian populations (Hannula-Jouppi et al. 2020), SERPINB7-pEDD is the most prevalent hereditary PPK among East Asians due to a high prevalence of founder LOF variants in SERPINB7 with an estimated prevalence of 1.2 per 10,000 in Japanese populations and 3.1 per 10,000 in Chinese populations (Huang et al. 2021). The nonsense variant c.796C > T (p.Arg266*) in SERPINB7 is the most common founder LOF variant in Japanese and Chinese SERPINB7-pEDD patients (Kubo 2025). As to why a high carrier frequency of this LOF allele exists is not known, but it may have conferred some selective heterozygote advantage as postulated for other prevalent recessive gene variants (Man et al. 2007; Zlotogora. 2019; Xu et al. 2019).
Loss of SERPINB7 inhibition may lead to the overactivation of its target protease legumain, a cysteine endopeptidase protease, impairing DSG1, DSC1, and keratin intermediate filaments (Li et al. 2025; Cohen-Barak et al. 2022). SERPINA12 is associated with inhibition of KLK7 activity via vaspin, leading to the decreased levels of KLK7 substrates, DSG1 and CDSN (Mohamad et al. 2020). Their common regulation of the desmosomal cadherin, DSG1, may provide a mechanistic link between these two serpins.
LOF pathogenic variants in SERPINB8 are associated with autosomal recessive exfoliative ichthyosis (SERPINB8-pEDD, MIM #617115), also known as peeling skin syndrome 5 (PSS5), which is characterized by skin peeling, superficial scales, hyperkeratotic plaques, and erythema on the palms and soles (Pigors et al. 2016). Its target substrate is furin, and study of cell culture models reveals a role in regulating desmosomal keratinocyte adhesion (Izaguirre et al. 2013; Pigors et al. 2016).
LOF pathogenic variants in SPINK5 (serine protease inhibitor of Kazal type 5) underlie the autosomal recessive skin condition termed Netherton syndrome [NS (SPINK5-sEDD), MIM #256500], which is clinically characterized by the triad of skin desquamation, atopic manifestation, and hair abnormalities (“bamboo-like” hair) (Fig. 1e; Netherton. 1958; Chavanas et al. 2000). Lympho-epithelial Kazal-type related inhibitor (LEKTI) is encoded by SPINK5 (Bitoun et al. 2003). Loss of LEKTI leads to over-activation of skin KLKs (KLK5, KLK7, KLK14) and subsequently increased targeting of DSG1, DSC1, and CDSN for breakdown (Pontone et al. 2022). In SPINK5-sEDD, upregulated KLKs can activate the PAR-2 pathway, driving Th2-mediated skin inflammation skin, while barrier disruption facilitates pathogen penetration and triggers a Th17 immune response (Zani et al. 2022).
Autosomal recessive ichthyosis-hypotrichosis syndrome [(ARIH) ST14-sEDD, MIM #602400], which is characterized by generalized congenital scaling, diffuse non-scarring hypotrichosis, and photophobia, has been associated with sequence variants in ST14 (type Ⅱ transmembrane serine protease matriptase) (Basel-Vanagaite et al. 2007; Paller et al. 2025). Matriptase deficiency leads to impaired degradation of corneodesmosomes within the stratum corneum, disrupted profilaggrin processing, abnormal hair follicle development, and increased apoptosis and marked depletion of thymocytes (List K et al. 2002; Alef et al. 2009). Matriptase has also been identified as an upstream activator of PAR-2, thereby triggering downstream signaling pathways that induce the expression of pro-inflammatory cytokines (Friis et al. 2017).
Cysteine proteases and cystatins
Cathepsins belong to the lysosomal cysteine proteases of the papain-like family and are primarily responsible for intra-lysosomal protein degradation as well as mediating cellular housekeeping functions (Turk et al. 2001; Zeeuwen et al. 2009). They are often inhibited by cystatins (Abrahamson et al. 2003). Genetic studies of rare inherited monogenic skin disorders have also shown cysteine proteases and their inhibitors (cystatins) to have key functions in epidermal homeostasis and barrier function (Toomes et al. 1999; Ngcungcu et al. 2017).
For example, LOF variants in CTSC encoding Cathepsin C (CTSC) underlie two similar autosomal recessive skin syndromes termed Papillon-Lefèvre syndrome [PLS (CTSC-pEDD); MIM #245000] and Haim-Munk syndrome [HMS (CTSC-pEDD-arachnodactyly), MIM #245010], which both share clinical features of PPK and periodontal inflammation (Fig. 1f, g; Toomes et al. 1999; Ghanei et al. 2021; Hart et al. 2000; Chitsamankhun et al. 2024). Also, inherited genomic duplications upstream of the Cathepsin B gene (CTSB) are associated with keratolytic winter erythema [KWE (CTSB-pEDD), MIM #148370], characterized by palmoplantar erythema and epidermal peeling (Ngcungcu et al. 2017).
Loss of cathepsin inhibitor activity has also been associated with skin disorders/syndromes in which superficial peeling of the epidermis is the primary clinical phenotype. This indicates a role for this class of protease inhibitors in regulating epidermal adhesion. For example, LOF variants in the CSTA gene, encoding cystatin A, underlie an autosomal recessive form of acral peeling skin syndrome [APSS (CSTA-nEDD), MIM #607936], also called peeling skin syndrome 4 (PSS4), characterized by superficial palmoplantar exfoliation and erythroderma (Blaydon et al. 2011a). Cystatin A (also known as stefin A) has been shown to regulate cathepsin L, cathepsin V, and dust mite cysteine proteases (Der f1, Der p1) (Muttardi et al. 2016). In addition, cystatin M/E has been found to inhibit both lysosomal cysteine proteases, such as cathepsin L and cathepsin V, as well as the asparaginyl endopeptidase legumain, and is essential for maintaining epidermal differentiation as well as for the development of the hair follicles and the corneal epithelium in mice (Cheng et al. 2006; Zeeuwen et al. 2010). Pathogenetic variants in CST6, encoding cystatin M/E, underlie ectodermal dysplasia 15, hypohidrotic/hair type [ECTD15(CST6-sEDD), MIM #618535] and keratosis follicularis spinulosa decalvans [KFSD (CST6-sEDD-cicatricial), MIM #612843], both of which are characterized by generalized scaling, hypotrichosis, and photophobia (van den Bogaard et al. 2019; Eckl et al. 2021; Paller et al. 2025).
Calpains are calcium-dependent cysteine proteases that are highly expressed in stratified squamous epithelia and are inhibited by calpastatin, encoded by the CAST gene. LOF variants in CAST are associated with PLACK syndrome (CAST-pEDD, MIM #616295), which presents with peeling skin, leukonychia, acral punctate keratosis, cheilitis, and knuckle pads (Lin et al. 2015). In CAST-pEDD, the subsequent upregulated epidermal calpain activity seems to increase proteolysis of epidermal desmosomal components, induce keratinocyte apoptosis, and impair keratinocyte adhesion (Nguyen et al. 2024).
Metalloproteinases
The metzincin superfamily of metalloproteinase primarily includes matrix metalloproteinases (MMPs), a disintegrin and metalloproteinases (ADAMs), ADAM with thrombospondin motifs (ADAMTS), and their endogenous inhibitors, known as tissue inhibitors of metalloproteases (TIMPs) (Rivera et al. 2019; Zhu 2021). These metalloproteinases play crucial roles in processing various extracellular matrix molecules and mediating cell signaling within the skin (Kümper et al. 2022).
ADAM17 (a disintegrin and metalloproteinase 17) is essential for processing numerous membrane-bound substrate proteins, including activating epidermal growth factor receptor (EGFR) ligands and releasing pro-inflammatory cytokines such as TNF-α (Rabinowitsch et al. 2023). Loss-of-function pathogenic variants in ADAM17 are associated with inflammatory skin and bowel syndrome (ADAM17-sEDD, MIM #614328) (Blaydon et al. 2011b; Imoto et al. 2021; Chang et al. 2025). Gain-of-function variants in RHBDF2 (encoding iRhom2 inactive rhomboid-like protein 2) underlie tylosis with oesophageal cancer [TOC (RHBDF2-pEDD), MIM #148500], characterized by PPK, oral and oesophageal leukoplakia, and a significantly high lifetime risk of oesophageal squamous cell carcinoma (Blaydon et al. 2012). Interestingly, ADAM17 protease activity, including EGFR signaling, is dependent on the “inactive” rhomboid protease iRhom2 (Adrain et al. 2012; Maretzky et al. 2013; Brooke et al. 2014).
MBTPS2 (membrane-bound transcription factor protease, site 2), also known as S2P (site-2 protease), is a membrane-embedded zinc metalloproteinase associated with lipogenesis and the endoplasmic reticulum stress response. S2P cleaves sterol regulatory element-binding proteins (SREBPs), which act as transcriptional factors involved in cholesterol metabolism (Haghighi et al. 2013). Variants in MBTPS2 are linked to three X-linked recessive skin disorders: Olmsted syndrome [OS (MBTPS2-pEDD), MIM #300918], ichthyosis follicularis with atrichia and photophobia (IFAP) syndrome (with or without BRESHECK syndrome) (MBTPS2-sEDD-IFAP, MIM #308205), and keratosis follicularis spinulosa decalvans (KFSD (MBTPS2-sEDD-cicatricial alopecia), MIM #308800) (Haghighi et al. 2013; Oeffner et al. 2009; Naiki et al. 2012; Aten et al. 2010). These syndromes share the common clinical manifestation of keratotic skin, suggesting that pathogenic variants in MBTPS2 reduce protease activity and impair sterol responsiveness in keratinocytes, thereby disrupting epidermal barrier function.
It should be noted that most cases of OS arise from pathogenic variants in the TRPV3 (transient receptor potential vanilloid-3) gene, which can manifest in either autosomal dominant (gain-of-function) or recessive forms (Lin Z et al. 2012; Eytan et al. 2014).
Concluding remarks
Proteases and their inhibitors are pivotal in skin barrier function, inflammation, and immune regulation. Dysregulated proteolytic activity contributes to a series of monogenic skin disorders with overlapping clinical and cellular phenotypes, often with impaired desmosomal function and inflammation. To date, the management of these disorders has remained largely supportive and symptomatic, underscoring the lack of disease-specific therapies. However, some clinical strategies have been tried in SPINK5-sEDD, including repurposing therapies linked to immune dysregulation, and used to treat more prevalent skin conditions such as psoriasis and atopic eczema. Monoclonal antibodies targeting IL-12/IL-23, IL-17, IL-4/IL-13, TNF-α, and IL1β have been trialed in SPINK5-sEDD (Samuelov et al. 2023; Gan et al. 2022; Blunder et al. 2025; Zingkou et al. 2022; Ragamin et al. 2023). In addition, JAK inhibitors have shown therapeutic benefit in some SPINK5-sEDD cases by dampening cytokine-mediated signaling and downstream inflammatory responses (Tang et al. 2025). Intravenous immunoglobulin has also demonstrated efficacy in SPINK5-sEDD through immunoglobulin replacement (Neema et al. 2023). Recently, GSK951, a KLK5 inhibitor, has been proposed as a promising therapeutic candidate (Liddle et al. 2021). Nevertheless, the current evidence for these therapeutic strategies remains largely limited to a small number of participants. Further multi-centre trials are warranted to establish the long-term efficacy of these therapies in SPINK5-sEDD.
Beyond recent advances in these targeted biologics and small-molecular immunomodulators, protease replacement strategies and protease inhibitor analogs represent emerging therapeutic avenues for conditions with this pathogenesis basis. Although steps have been made in elucidating protease-related mechanisms in genetic skin diseases, further research is needed for in-depth understanding of the disease pathogenesis and to facilitate the development of effective targeted treatments.