Unstable Hemoglobin, a Rare but Significant Cause of Hemolytic Anemia: Recognition of Peripheral Smear Findings Is Crucial for Diagnosis
Ryan C. Shean, Archana Agarwal, Anton V. Rets

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
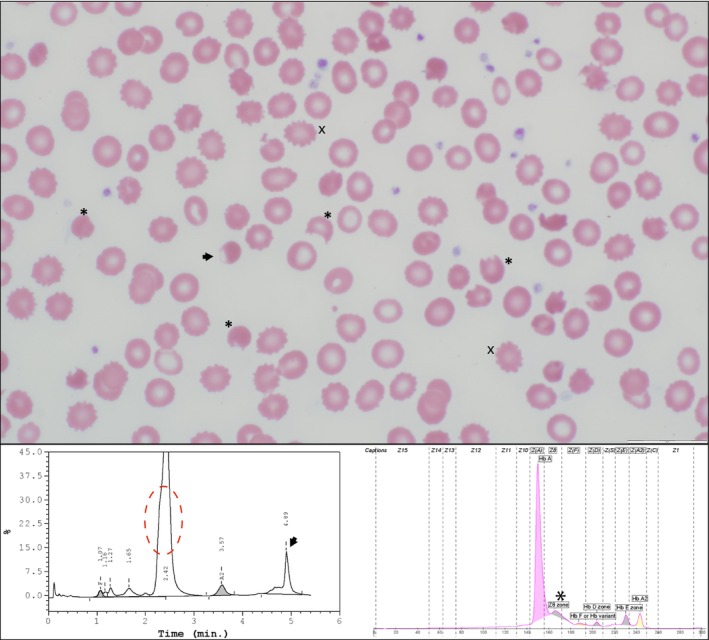 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHemoglobinopathies and Related Disorders · Blood groups and transfusion · Neonatal Health and Biochemistry
A 10‐year‐old male hospitalized for abdominal pain and jaundice was referred for hereditary hemolytic anemia workup. At admission, complete blood count (CBC) revealed severe normocytic normochromic anemia (hemoglobin (Hb) < 50 g/L, red blood cell (RBC) count < 2 × 10^12^/L), with normal mean corpuscular Hb concentration (MCHC) and mean corpuscular volume (MCV).
Pre‐transfusion peripheral smear (Figure 1, top panel) demonstrated normocytic normochromic erythrocytes with moderate anisopoikilocytosis, polychromasia, frequent bite cells (asterisk), echinocytes (x), and blister cells (arrow). Capillary electrophoresis (CE) and high‐performance liquid chromatography (HPLC) (Bio‐Rad) revealed abnormal patterns of Hb fractionation (bottom panel). CE showed mildly decreased Hb A (84%) with an unknown peak in the Z8 region (asterisk, 12%), suggesting a variant Hb. HPLC showed a slightly asymmetric “Hb A” peak (red dotted circle) of 78% and an abnormal peak of 12% at the retention time of 4.89 min (arrow). RBC osmotic fragility was increased, but Band 3 protein by flow cytometry and enzyme activities of pyruvate kinase and glucose‐6‐phosphate dehydrogenase (G6PD) were normal. Molecular testing demonstrated a heterozygous HBB variant c.295G>A; p.Val99Met, also known as Hb Köln.
Hb Köln is caused by a point mutation in HBB gene resulting in an abnormal beta‐globin chain with decreased stability [1]. The valine to methionine substitution in Hb Köln lies in a key binding region for beta‐globin folding and heme binding. The mutation leads to a hemoglobin tetramer with abnormally high oxygen affinity [2] and a tendency to denature.
When Hb Köln (and other unstable Hb) undergo oxidative damage, the denatured hemoglobin precipitates as intracellular inclusions called Heinz bodies. Heinz bodies are not visible on Wright‐Giemsa stains but may be viewed with supravital stains. Additionally, they can attach to the RBC membrane, forming “blister cells”. The precipitated globin in the Heinz bodies makes the RBCs less deformable, and thus the splenic macrophages remove these inclusions, causing the characteristic “bite cells” and ultimately extravascular hemolysis. These cells are not specific for unstable hemoglobin and can be seen in G6PD deficiency, oxidative drug‐induced hemolysis, and thalassemias.
The creation of bite and blister cells as well as extravascular hemolysis leads to increased variation in size and shape of erythrocytes (anisopoikilocytosis). Although polychromasia is not noted in this photomicrograph field, increased production of immature erythrocytes may be seen as a compensatory response to anemia.
Usually, because there is no charge or affinity change of the Hb Köln mutation, the variant hemoglobin elutes with Hb A and is frequently covered by the A peak, which can make diagnosis difficult. However, the asymmetric peak in the Hb A region (red circle) should raise suspicion. In our experience, in cases with Hb Köln and no normal Hb A, Hb Köln elutes on HPLC at 2.29 min. Additionally, the denatured Hb Köln can precipitate and be seen as an abnormal peak at ~5 min (black arrow).
Despite its overall rarity, Hb Köln is one of the most encountered unstable Hb variants. Recognizing the characteristic smear findings and biochemical profile of unstable hemoglobins like Hb Köln is crucial to avoid misdiagnosis and guide appropriate management [3].
Author Contributions
All authors created and edited the manuscript.
Funding
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1R. W. Carrell , H. Lehmann , and H. E. Hutchison , “Haemoglobin Köln (Beta‐98 Valine—Methionine): An Unstable Protein Causing Inclusion‐Body Anaemia,” Nature 210, no. 5039 (1966): 915–916, 10.1038/210915 a 0.5960324 · doi ↗ · pubmed ↗
- 2R. D. Woodson , J. D. Heywood , and C. Lenfant , “Oxygen Transport in Hemoglobin Köln: Effect of Increased Oxygen Affinity in Absence of Compensatory Erythrocytosis,” Archives of Internal Medicine 134, no. 4 (1974): 711–715, 10.1001/archinte.1974.00320220113014.4414970 · doi ↗ · pubmed ↗
- 3M. Risinger , M. Emberesh , and T. A. Kalfa , “Rare Hereditary Hemolytic Anemias: Diagnostic Approach and Considerations in Management,” Hematology/Oncology Clinics of North America 33, no. 3 (2019): 373–392, 10.1016/j.hoc.2019.01.002.31030808 · doi ↗ · pubmed ↗