Atypical El Tor Vibrio cholerae from the second major global seventh-pandemic cholera wave is endemic in Sabah, Malaysia
Jaeyres Jani, Jecelyn Leaslie John, Lia Natasha Amit, Deborah Yebon Kang, Marilyn Charlene Montini Maluda, Mohammad Jikal, Yann Felix Boucher, Kamruddin Ahmed

TL;DR
This study investigates the genomic traits of Vibrio cholerae in Sabah, Malaysia, revealing that atypical strains from a past cholera pandemic are still causing outbreaks.
Contribution
The study identifies atypical El Tor Vibrio cholerae strains in Sabah with unique genomic features linked to regional endemicity.
Findings
Sabah isolates have atypical El Tor biotype with Classical CTX prophage elements.
Strains carry tandem CTX prophage copies and RS1 sequences, indicating environmental adaptation.
Phylogenetic analysis shows Sabah strains belong to wave 2 of the seventh pandemic clade.
Abstract
Cholera, caused by Vibrio cholerae, remains a significant diarrheal disease, especially in coastal regions of developing countries. In Malaysia, cholera is largely non-endemic except in Sabah, which has had recurrent outbreaks accounting for ∼75% of national cases between 2004 and 2014. To understand the origin and transmission of the disease, we sequenced the genomes of clinical isolates of V. cholerae O1 collected during an outbreak in 2019 and 2020. Genotypic analyses revealed that all Sabah isolates were atypical El Tor biotype harboring Classical CTX prophage elements. In particular, the strains carried two tandem CTX prophage copies in chromosome 2 and three tandem RS1 sequences on chromosome 1, including a Classical type rstR, which is atypical for canonical El Tor. Genome comparisons revealed conserved seventh-pandemic genomic islands (VSP1 and VSP2) and variably arranged…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
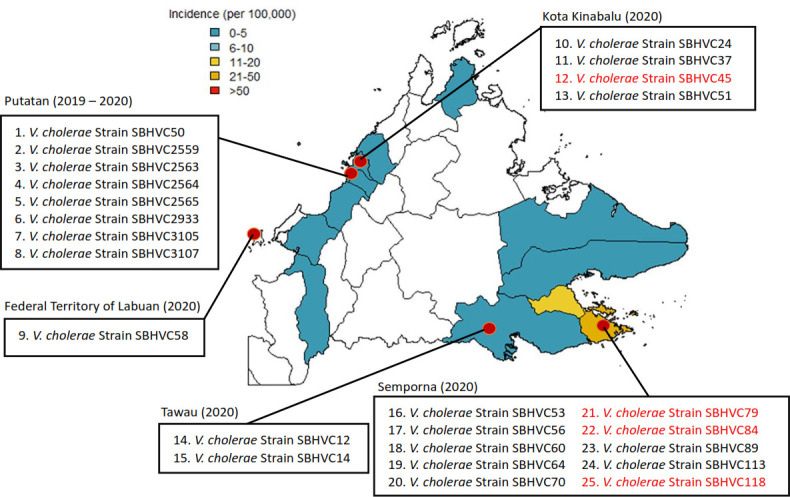 Fig 1
Fig 1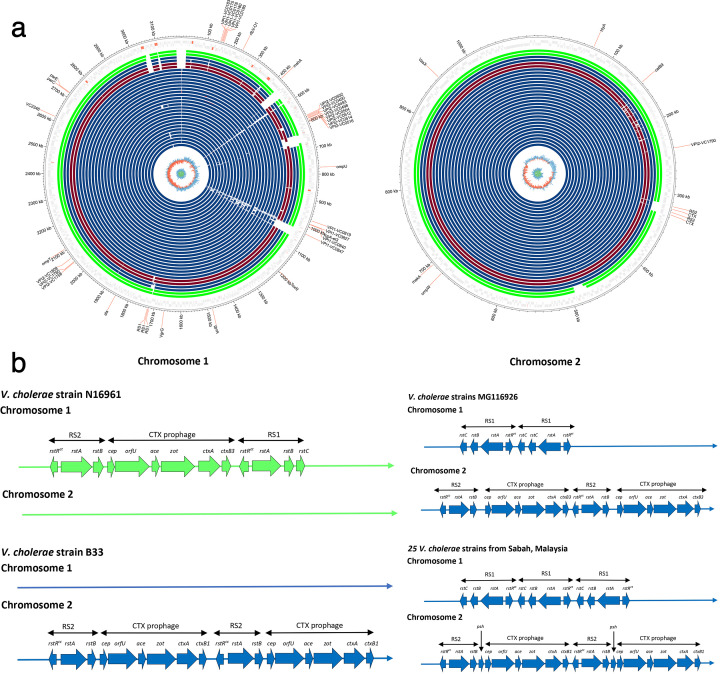 Fig 2
Fig 2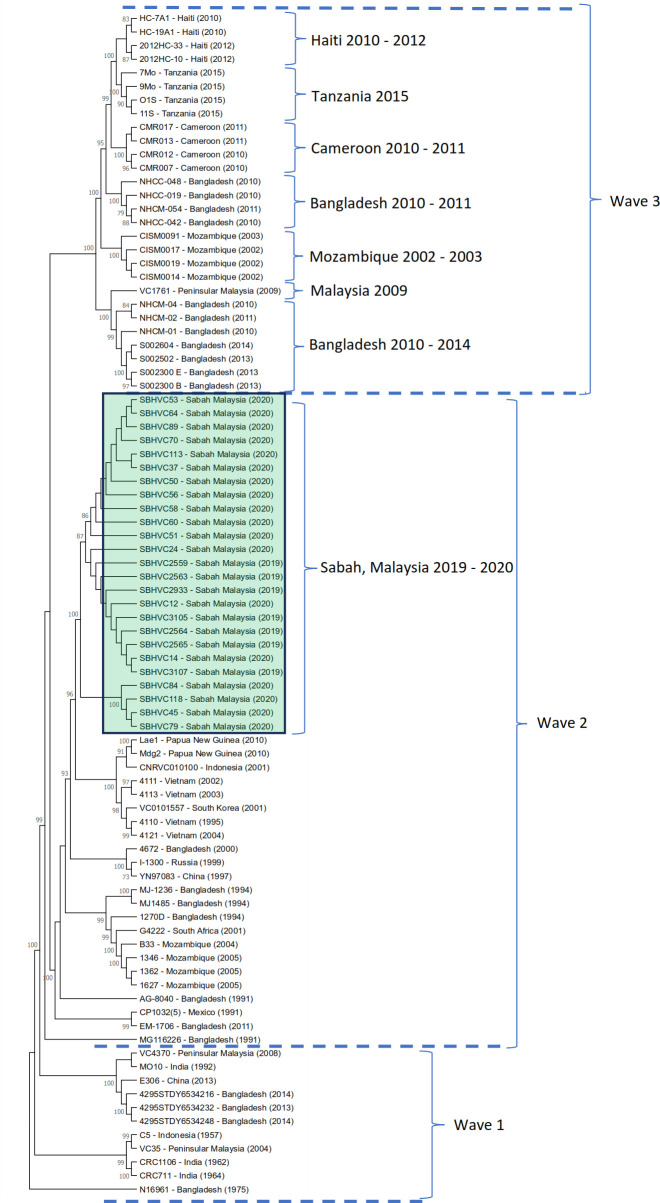 Fig 3
Fig 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVibrio bacteria research studies · Bacillus and Francisella bacterial research · Yersinia bacterium, plague, ectoparasites research
INTRODUCTION
Vibrio cholerae, the etiologic agent of cholera, remains a significant cause of bacterial diarrhea in developing countries, especially coastal areas. Among more than 200 V. cholerae serogroups that have been discovered, two serogroups, O1 and O139, are mainly linked with major epidemics (1). Serogroup O1 is further categorized into two biotypes, the Classical and El Tor. The Classical biotype was behind the first six pandemics between 1899 and 1923 (2, 3), while the El Tor biotype is known to have caused the seventh and ongoing pandemic, which was first reported in Indonesia in 1961. Serogroup O139 was identified in the Indian subcontinent in 1992 and then became widespread in Southeast Asia (2–4). V. cholerae notably has two circular chromosomes, with chromosome 1 carrying essential housekeeping genes and chromosome 2 containing genes involved in environmental adaptation, stress response, and pathogenicity (5).
The El Tor biotype, first identified in 1905 at Egypt’s El Tor quarantine station (6), became epidemic in Indonesia before spreading across Asia, Africa, the Middle East, parts of Europe, Latin America, and lately Hispaniola in 2010 (3). Globally, cholera affects 1.3–4 million people annually, resulting in an estimated over 95,000 deaths (7). Previous studies have performed high-resolution single-nucleotide polymorphism (SNP) analyses of whole genomes, but these analyses represent only isolates from Peninsular Malaysia, which is approximately 640 km away from Sabah. In Malaysia, cholera has been documented since the 1950s, but it is now considered non-endemic, except in Sabah, which is located in East Malaysia (8). Outbreaks in Sabah, predominantly caused by the El Tor O1 serogroup, have been linked to contaminated food and water. Between 2004 and 2014, 3,481 cases and 32 deaths were reported nationwide, with Sabah accounting for 75.4% of the cases in Malaysia. Cholera outbreaks in Sabah peak every 3–5 years, with the highest incidence occurring along the east coast. In 2018, 163 cases were recorded across nine districts, with Semporna having the highest rate (51.5 per 100,000) (9). Given Sabah’s significant cholera burden, further research is crucial to identify risk factors and improve control measures. A detailed investigation into a 2014 cholera outbreak among Sea Gypsies (Bajau) in Kudat, with 44 symptomatic and 34 asymptomatic cases (10), revealed the main cause to be V. cholerae El Tor of the O1 Ogawa serotype. A retrospective study of strains isolated in 2015 across Sabah identified genetically diverse atypical El Tor strains among 65 clinical isolates and one environmental isolate through DNA fingerprinting (11). However, their origin and exact genotypes have remained a mystery, since whole-genome sequencing could not be performed at the time.
In May 2019, an outbreak of cholera occurred in the Putatan district of Sabah, and seven isolates could be obtained from patients. The following year, additional cases were reported across the state, and another 18 strains were isolated from clinical cases at local hospitals. Genome sequencing, followed by biotyping and phylogenetic analysis, revealed that the outbreak was caused by atypical V. cholerae El Tor O1 from the second wave of the current seventh pandemic.
MATERIALS AND METHODS
Strains and genome DNA preparation
Twenty-five V. cholerae strains were isolated from stool samples collected from patients with cholera-induced diarrhea across different districts in Sabah (Fig. 1; Table S1). The initial diagnosis involved culturing the stool samples on Thiosulfate-Citrate-Bile-Salts-Sucrose agar. Genomic DNA from the 25 V. cholerae strains was extracted from overnight cultures grown at 37°C using a genomic DNA purification kit (QIAGEN, Hilden, Germany), following the manufacturer’s instructions. The quality and quantity of the extracted DNA were assessed using a Nanodrop spectrophotometer.
Map of Sabah showing the locations of isolated V. cholerae strains, including Kota Kinabalu, Putatan, Tawau, Semporna districts, and the Federal Territory of Labuan. The red-colored strains belong to a specific monophyletic subclade of wave 2, including three strains from Semporna and one from Kota Kinabalu. The incidence rate per 100,000 is shown on the map for each district of Sabah, Malaysia, for the year 2020 (9).
Illumina and SMRTbell library preparation and sequencing
Strains of 25 V. cholerae were sequenced using the Illumina Novaseq 6000 system (Illumina, San Diego, CA, USA), generating 150 bp paired-end (PE) reads. All sequencing operations followed the protocols and reagents described in the manufacturing protocol. The genomic DNA library was prepared with the Nextera XT DNA Sample Prep Kit (Illumina). Briefly, 1 ng of input DNA was tagmented by the Nextera XT transposome at 55°C for 5 min, followed by end-repair, A-tailing, adaptor ligation, and library amplification, according to the manufacturer’s protocol. The DNA library was validated using the Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and the Qubit Fluorometer (Thermo Fisher Scientific) for quality control analysis. The library was then denatured, diluted to the optimal concentration, and used for cluster generation with the NovaSeq 6000 S1 Reagent Kit (Illumina) on the flow cell. Image analysis and base calling were performed using SCS2.8/RTA1.8 (Illumina). FASTQ file generation and the removal of failed reads were conducted using CASAVA ver.1.8.2 (Illumina).
V. cholerae strain SBHVC14 was also sequenced by Macrogen Inc. (Geumcheon-gu, Seoul, South Korea) using a PacBio RS II sequencer with one SMRT cell and P6-C4 chemistry at a 120-min movie length (Pacific Biosciences, Menlo Park, CA, USA). A 20-kb SMRTbell library was generated from sheared genomic DNA using a 20-kb template library preparation workflow, following the protocols and reagents provided by the manufacturer.
Hybrid genome assembly of V. cholerae strain SBHVC14
The genome of V. cholerae strain SBHVC14 was successfully assembled using a hybrid sequencing and assembly methodology. Long-read data generated from the PacBio RS II platform (P6-C4 chemistry) were first assembled using Canu, producing 16 contigs with a total length of 4,345,299 bp, an N50 of 1,053,043 bp, and a longest contig of 2,024,187 bp. While this initial assembly provided a comprehensive structural overview of the genome, it was further refined through error correction using high-accuracy Illumina short reads. These reads were aligned to the Canu V2.2 assembly contig using BWA-MEM2 V2.21, and the resulting alignments were polished with Pilon V1.24. This polishing process corrected sequencing errors and resolved local misassemblies, resulting in a significantly improved assembly with only two large scaffolds, with a total length of 4,293,028 bp (Table S2). The near-complete final assembly, with a consistent GC content of ~47.45%, demonstrates the effectiveness of integrating long-read and short-read sequencing platforms. Overall, this hybrid method not only enhanced genome continuity and accuracy but also provided a high-quality reference for downstream genomic and comparative analyses.
Genome mapping to V. cholerae strain SBHVC14
The reads of 24 V. cholerae strains sequenced using Illumina technology were first cleaned using trim BBDuk (bbduk.sh) version 38.90 to remove adapter sequences and trim reads with quality scores lower than Phred score Q20. Next, the cleaned reads were mapped to each chromosome of the V. cholerae strain SBHVC14 using BWA v0.7.13. A consensus sequence was then generated from the mapping results (Table S2).
Genome annotation and bioinformatics analyses
The genome annotation for each assembly was performed using the Prokka pipeline, which enables rapid and accurate annotation of bacterial genomes. Prodigal v3.3.6 was used to identify coding sequences, transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), and other genomic features. tRNAs were predicted using tRNAscan-SE v2.0 (12), a specialized tool for detecting tRNA genes with high sensitivity and accuracy. 5S, 16S, and 23S rRNAs were identified using barrnap v0.9, which uses hidden Markov models. The presence of potential virulence factors and antibiotic resistance genes was identified using CholeraFinder v1.0. CheckM v1.1.6 was used to estimate genome completeness and contamination levels based on lineage-specific marker genes.
Phylogenetic analysis and genome visualization
For comparative genomic analysis, SNPs were identified using the kSNP3 v3.1.2 tool. To accurately position the samples within a phylogenetic framework, the analysis included 62 previously published V. cholerae genomes retrieved from the NCBI database, as detailed in Table S3. A maximum likelihood phylogenetic tree was constructed using MEGA X based on the SNP-derived distance matrix, applying the general time reversible (GTR) model with 1,000 bootstrap replicates to assess the robustness of the inferred relationships. For genome-wide visualization, Circos (13) was used to generate circular genomic plots of 25 Sabah strains together with reference strains, providing an integrated view of the comparative genomic landscape.
RESULTS AND DISCUSSION
The 2019–2020 Sabah cholera outbreak occurred primarily in coastal areas but affected a geographically and demographically diverse population
The V. cholerae isolates collected during the 2019–2020 outbreak were obtained from patients residing in various districts across Sabah, including Semporna, Putatan, Kota Kinabalu, Tawau, and the Federal Territory of Labuan (Fig. 1). Among these, Semporna was the most affected district, with an incidence rate of 6.5 and 31.2 per 100,000 population in 2019 and 2020, respectively. This was followed closely by Putatan, with incidence rates of 11.4 and 4.3 per 100,000 population in those respective years. This geographic distribution suggests the presence of localized clusters of infection, possibly indicating environmental or community-based sources of transmission (9). In terms of gender distribution, approximately two-thirds of the total cases were female patients, whereas male patients comprised the remaining. This skew may reflect differences in exposure risks, health-seeking behavior, or report patterns between genders in the affected communities. The age of the patients varied widely, ranging from 4 to 83 years old, and a large proportion was young to middle-aged adults. This indicates that the outbreak affected a broad age range, including both pediatric and elderly populations (Table S1). Some patient records lacked complete demographic information, such as age or gender, which may limit demographic interpretation of the analysis. Nevertheless, the available data clearly depict a diverse demographic impact of cholera infections, affecting individuals across a broad spectrum of ages and both sexes, and concentrated in specific coastal and semi-urban districts of Sabah.
Comparative genomics reveals atypical hybrid El Tor O1 characteristics in Sabah V. cholerae
A visualization of whole-genome alignment of 24 draft V. cholerae genomes and the complete genome of strain SBHVC14 is shown in Fig. 3a. This comparative genomic map offers a circular overview of both chromosomes, providing detailed insights into the genomic architecture and conservation across strains. The alignment reveals a high degree of genome similarity among the strains, with most regions aligning closely to the reference genome. The high degree of genome synteny suggests evolution of all Sabah outbreak strains from a common ancestor, consistent with their phylogeny (Fig. 2). The Sabah strains display all key genetic elements consistent with the seventh pandemic, including the El Tor variant of the toxin coregulated pilus tcpA gene, the Vibrio seventh-pandemic islands (VPI1 and VPI2), and the El Tor CTX prophage carrying the cholera toxin ctxA and ctxB genes (14). This analysis also highlights the presence of VPI1, specifically the VC2346 gene in Sabah strains (Table S4), which is one of the most acquired genes associated with the seventh-pandemic strain (15). In addition, two other groups of VPI1-associated genes were identified. The first group includes VC0183, VC0175, VC0178, VC0180, and VC0185, located approximately between 100 and 200 kb on chromosome 1. The second group comprises VC0819, VC0827, VC0840, and VC0847, located at around 1,000 kb on chromosome 1.
Comparative genomic analysis of pandemic V. cholerae strains generated using Circos. The genome visualization highlights key loci, including VPI1, VPI2, biotype-specific genes, and other important genetic regions. (a) From the outermost track: reference V. cholerae strains N16961 and C5 from wave 1 (green track), reference V. cholerae strains B33 and MG116226 from wave 2 (blue track), and reference V. cholerae strains 7Mo and S002604 from wave 3 (red track). The subsequent blue tracks represent the 25 V. cholerae strains from Sabah, Malaysia, with the complete V. cholerae SBHVC14 genome used as a template for the comparison at the center (wave 2). (b) Schematic representation of the arrangement of the CTX prophage and RS1 element in V. cholerae O1 strains. For comparison, the genetic organization of the CTX prophage, along with the RS1 and RS2 elements, is shown for the El Tor reference strain V. cholerae N16961 (AE003852) from wave 1, strain B33 (ACHZ00000000) and the Bangladesh strain MG116926 (GQ485646) from wave 2, alongside the genetic arrangement of the 25 V. cholerae strains from Sabah, Malaysia.
VPI2 plays an important role in enhancing the bacterium’s ability to survive and thrive within the host (16). It contains genes involved in sialic acid metabolism and neuraminidase production, allowing V. cholerae to degrade mucin in the intestinal lining and use sialic acid as a nutrient source (17). VPI2 was identified within the region spanning approximately 550–600 kb on chromosome 1 (VC0490, VC0493, VC0498, VC0502, VC0504, VC0512, VC0514, and VC0516) and between 2,000 and 2,100 kb (VC1758, VC1760, and VC1809). Additionally, VC1790 encoding transposase was identified on chromosome 2. All these genetic features, including VPI1, VPI2, CTX prophage, VPI1, VPI2, and RS1, represent horizontally acquired DNA segments that have transformed V. cholerae into a highly virulent and human-adapted pathogen (18). These elements form the foundation for the bacterium’s ability to persist in environmental reservoirs and spread effectively during epidemics. The genomic map highlighted the distribution of biotype-specific genes distinguishing Classical and El Tor strains (Fig. 3a; Table S2), supporting their classification and potential lineage-specific adaptations. It also revealed structural variations such as insertions, deletions, and rearrangements, which may involve mobile elements or phage regions contributing to virulence and environmental adaptation. This overview complements phylogenetic analysis and provides a basis for further functional studies.
SNP-based phylogenetic tree of 87 seventh-pandemic V. cholerae El Tor strains from wave 1, wave 2, and wave 3 outbreaks. Strains from the Sabah 2019–2020 outbreak are highlighted in green. Phylogenetic analysis was performed using the general time reversible (GTR) model with 1,000 bootstrap replicates in the MEGA X software.
To further investigate the biotypes of outbreak strains in Sabah, representative biotype-specific genetic markers, including ctxB1, rstR^cc^, rstR^et^, and tcpA^et^ were analyzed (19, 20). The rstR^et^ and tcpA^et^ genes were highly similar to those of the El Tor V. cholerae O1 El Tor strain N16961 (AE003852 and AF325734). On chromosome 2, ctxB1 resembled the sequence found in O1 V. cholerae (CP001235), whereas rstR^cc^ was similar to the sequence from O139 V. cholerae (KJ023707). Additionally, RS1 elements were detected in chromosome 1, where three tandem copies of RS1 were arranged as rstC, rstB, rstA, and rstR^et^ in the studied V. cholerae strains. This genetic arrangement is characteristic of the RS1 structure observed in the El Tor variant strains from Bangladesh, which were reported in 2002 (21, 22).
All epidemic and pandemic V. cholerae strains are lysogens of the CTX prophage. In Sabah isolates, the 6.5 kb CTX prophage present in chromosome 2 was found to be organized into two repeating units: a 2.4 kb Repeat Sequence 2 (RS2) and a 4.6 kb CTX prophage (Fig. 2b) (21, 22). RS2 contains three genes (rstR^cc^, rstA, and rstB), which are essential for regulation, replication, and integration, respectively. The lysogeny of CTX is primarily regulated by the phage repressor protein rstR^cc^, which binds to the rstA promoter region and represses the transcription of rstA and rstB, ultimately affecting CTX production (21, 22).
The observed genetic variability in the CTX prophage and RS1/RS2 elements among V. cholerae strains highlights the dynamic nature of its genome architecture. The presence of duplicated CTX prophage regions in the Mozambique V. cholerae strain B33, along with strain-specific arrangements of virulence genes (cep, orfU, ace, zot, ctxA, ctxB1, and R2 elements) (23) suggests potential mechanisms for enhanced toxin production or transmission efficiency. Such duplications may arise from recombination events or prophage integration, contributing to the evolutionary success of certain lineages. The high variability in RS1 elements, particularly the repetitive occurrences of rstC, rstB, and rstR^et^, underscores the role of mobile genetic elements in shaping V. cholerae diversity (24). These rearrangements could facilitate adaptive advantages, such as antibiotic resistance or immune evasion, by enabling rapid genomic changes. The hybrid architectures observed in some strains, including integrations of RS2 and colonization factors, further support the idea that horizontal gene transfer plays a key role in this pathogen’s evolution.
While this study provides insights into the structural diversity of CTX-related elements, the functional consequences of these genetic variations remain to be fully elucidated (20). Future work should investigate whether specific arrangements correlate with clinical outcomes, environmental persistence, or epidemic potential. Additionally, experimental studies could clarify how these genomic features influence virulence regulation and host-pathogen interactions. In summary, the plasticity of CTX prophage and RS1/RS2 elements in V. cholerae reflects its capacity for rapid adaptation. Understanding these genetic dynamics is critical for tracking emerging strains and developing targeted public health interventions.
Wave 2 El Tor strains are responsible for recent outbreaks in Sabah
To determine the phylogenetic relationship of V. cholerae strains from Sabah and put them in the context of the seventh pandemic, we added 62 reference genomes from three major waves of global cholera spread to reconstruct their phylogeny (Fig. 3). These 62 genomes were carefully selected to capture genetic diversity across different geographic regions and time periods, ensuring the inclusion of well-characterized strains previously used in global phylogenetic frameworks. This approach balanced computational efficiency and phylogenetic clarity, allowing the Sabah isolates to be accurately positioned within the established wave 1, wave 2, and wave 3 lineages without introducing redundancy or analytical noise from thousands of highly similar genomes (8, 25, 26). The SNP-based phylogenetic tree revealed distinct clustering patterns corresponding to the known cholera pandemic waves. Strains from Haiti (2010–2012), Cameroon (2010–2011), Bangladesh (2010–2011), Tanzania (2015), and Mozambique (2002–2010) showed close phylogenetic relationships, forming clusters within a larger wave 2 clade. These groupings support earlier studies suggesting transcontinental transmission routes during this period. Surprisingly, despite their more recent isolation, V. cholerae strains isolated from the Sabah 2019−2020 outbreak were also found to cluster within wave 2, suggesting that they share a common ancestor with older strains from South Asia and Africa. This finding highlights the ongoing global circulation of wave 2 V. cholerae. Furthermore, the monophyly of all Sabah strains suggests a single introduction into the state and subsequent spread. Two subclades were identified among the Sabah isolates, with the major cluster comprising 21 strains that appeared first during the 2019–2020 outbreak, and the minor cluster containing four strains, indicating subsequent genetic diversification following the initial introduction into Sabah.
The study of V. cholerae strain MG116926, reported by Nair et al. (21) and Safa et al. (22), shows that strain MG116926 belongs to the Matlab variant III hybrid strain. This strain contains a tandem repeat of the classical CTX prophage on chromosome 2 and a tandem repeat of the RS1 element on the larger chromosome 1, as shown in Fig. 2b. This genetic structure is similar to variants identified in Bangladesh (South Asia) and is classified as a new variant of V. cholerae O1. Among the 25 clinical strains, the presence of a tandem repeat of the RS1 element on chromosome I was observed, which is characteristic of the Atypical El Tor biotype of the El Tor lineage (21, 22). The most significant difference compared to the 25 clinical strains is the presence of a tandem repeat of RS1 on chromosome 1, where three copies are present (Fig. 2b). This feature indicates a new variant of V. cholerae O1 Biotype El Tor as an Atypical El Tor responsible for the outbreak in Sabah. Another unique characteristic is the presence of the V2364 marker. Additionally, the 25 isolate strains belonged to the wave 2 V. cholerae clades, retaining the ctxB1 classical cholera toxin B subunit allele while exhibiting an El Tor tcpA allele (23, 27). Phylogenetic analysis based on whole-genome SNPs also showed and confirmed that the 25 isolate strains clustered within wave 2 strains.
Genome mapping and SNP-based phylogenetic analysis placed the Sabah strains within the seventh-pandemic lineage, confirming their global pandemic lineage status. Despite their classification, these isolates exhibited sufficient genomic divergence to suggest regional adaptation, supported by the clustering of Sabah isolates in a distinct phylogenetic clade. Genome mapping and SNP-based phylogenetic analysis placed the Sabah strains within the second wave of the seventh-pandemic lineage, confirming their global pandemic lineage status. In addition, the Lae1, Mdg2, and CNRVC010100 strains, which belong to wave 2, were reported to lack the SXT or GI-15 elements, and their antimicrobial resistance genes were not clustered together, as shown in Fig. 3 (26). This finding indicates the absence of genetic relatedness and the lack of SXT, GI-15, or antimicrobial resistance genes among the 25 V. cholerae strains isolated from Sabah.
Whole-genome assemblies of 25 V. cholerae strains isolated during the 2019–2020 cholera outbreak in Sabah were successfully obtained, revealing comprehensive genomic features across all samples (Table S2). The number of contigs per assembly ranged from 2 to 92 contigs. The total genome lengths were consistent across all strains, ranging between 4,259,716 and 4,293,028 bp. GC content was remarkably consistent among all strains, with each displaying a GC percentage of approximately 45.47%, which is typical for V. cholerae. These findings confirm the high quality and consistency of the genome assemblies suitable for downstream comparative and phylogenomic analyses.
Conclusion
This investigation represents the first complete genomic characterization of V. cholerae in Sabah, revealing hybrid biotype traits associated with the second global wave of cholera when compared with isolates from Peninsular Malaysia. Interestingly, wave 2 has been largely replaced by its wave 3 descendants in most parts of the world, making its continued presence and dominance in recent outbreaks in West Malaysia unexpected. This persistence may reflect limited genomic surveillance in Southeast Asia or indicate specific ecological adaptations that confer competitive advantages to wave 2 strains in the coastal environment of Sabah. Such adaptation may reduce the introduction and establishment of newer strains due to local environmental resilience and restricted population movement.
The use of both PacBio and Illumina sequencing platforms proved essential for resolving repetitive and biotype-specific genomic structures, particularly the CTX prophage arrays. The hybrid sequencing strategy enhanced genomic resolution and accuracy, demonstrating its value in molecular epidemiology for tracking pathogen evolution and transmission. As sequencing technologies become more affordable and portable, their integration into regional laboratories will enable faster, evidence-based public health responses.
From a public health perspective, the detection of hybrid and potentially more virulent V. cholerae strains underscores the urgent need to strengthen environmental and clinical surveillance systems. Diagnostic protocols should be updated to account for genomic variability, including hybrid biotype markers, to improve detection accuracy. Intervention strategies should prioritize environmental monitoring of coastal reservoirs, rapid molecular diagnostics for outbreak detection, and genomic-based tracking of emerging variants. Furthermore, genomic data can inform vaccine efficacy assessments, guide antimicrobial use policies, and support the development of early warning systems for future cholera outbreaks in Malaysia and neighboring regions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jaskólska M, Adams DW, Blokesch M. 2022. Two defence systems eliminate plasmids from seventh pandemic Vibrio cholerae. Nature 604:323–329. doi:10.1038/s 41586-022-04546-y 35388218 PMC 7613841 · doi ↗ · pubmed ↗
- 2Bhandari M, Jennison AV, Rathnayake IU, Huygens F. 2021. Evolution, distribution and genetics of atypical Vibrio cholerae - a review. Infect Genet Evol 89:104726. doi:10.1016/j.meegid.2021.10472633482361 · doi ↗ · pubmed ↗
- 3Montero DA, Vidal RM, Velasco J, George S, Lucero Y, Gómez LA, Carreño LJ, García-Betancourt R, O’Ryan M. 2023. Vibrio cholerae, classification, pathogenesis, immune response, and trends in vaccine development. Front Med (Lausanne) 10:1155751. doi:10.3389/fmed.2023.115575137215733 PMC 10196187 · doi ↗ · pubmed ↗
- 4Parvin I, Shahid A, Das S, Shahrin L, Ackhter MM, Alam T, Khan SH, Chisti MJ, Clemens JD, Ahmed T, Sack DA, Faruque ASG. 2021. Vibrio cholerae O 139 persists in Dhaka, Bangladesh since 1993. P Lo S Negl Trop Dis 15:e 0009721. doi:10.1371/journal.pntd.000972134473699 PMC 8443037 · doi ↗ · pubmed ↗
- 5Lukjancenko O, Ussery DW. 2014. Vibrio chromosome-specific families. Front Microbiol 5:73. doi:10.3389/fmicb.2014.0007324672511 PMC 3957060 · doi ↗ · pubmed ↗
- 6Clemens JD, Nair GB, Ahmed T, Qadri F, Holmgren J. 2017. Cholera. The Lancet 390:1539–1549. doi:10.1016/S 0140-6736(17)30559-728302312 · doi ↗ · pubmed ↗
- 7Mutale LS, Winstead AV, Sakubita P, Kapaya F, Nyimbili S, Mulambya NL, Nanzaluka FH, Gama A, Mwale V, Kim S, Ngosa W, Yard E, Sinyange N, Mintz E, Brunkard J, Mukonka V. 2020. Risk and protective factors for cholera deaths during an urban outbreak-Lusaka, Zambia, 2017-2018. Am J Trop Med Hyg 102:534–540. doi:10.4269/ajtmh.19-067831933465 PMC 7056416 · doi ↗ · pubmed ↗
- 8Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, Croucher NJ, Choi SY, Harris SR, Lebens M, Niyogi SK, Kim EJ, Ramamurthy T, Chun J, Wood JLN, Clemens JD, Czerkinsky C, Nair GB, Holmgren J, Parkhill J, Dougan G. 2011. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477:462–465. doi:10.1038/nature 1039221866102 PMC 3736323 · doi ↗ · pubmed ↗