A novel approach to combat Pseudomonas aeruginosa: repurposing pharmaceuticals for inhibition of phospholipase A
Matea Modric, Rocco Gentile, Raphael Moll, Ifey Alio, Wolfgang R. Streit, Karl-Erich Jaeger, Holger Gohlke, Filip Kovacic

TL;DR
This study shows that repurposing existing drugs to target bacterial enzymes like phospholipase A can help combat antibiotic-resistant Pseudomonas aeruginosa.
Contribution
The study introduces a novel approach of repurposing pharmaceuticals to inhibit bacterial phospholipase A as a strategy to combat antimicrobial resistance.
Findings
GW4869, darapladib, and rilapladib significantly inhibit Pseudomonas aeruginosa growth by more than 50%.
GW4869 compromises biofilm cell viability and enhances the efficacy of imipenem in combination treatments.
Rilapladib shows broad-spectrum antibacterial activity against Escherichia coli and Staphylococcus aureus.
Abstract
Phospholipase A (PLA) plays critical roles in cellular physiology, making human PLAs established drug targets. On the other hand, the potential of bacterial PLAs as targets for antimicrobial drug development remains underexplored. In this study, we curated a library of 23 approved and investigational pharmaceuticals, some of which inhibit human PLA-like enzymes, through a combination of ligand structure-based searches and textual mining in literature and compound databases. Experimental screening identified that compounds GW4869, darapladib, and rilapladib significantly inhibit Pseudomonas aeruginosa growth by more than 50%. While these compounds did not reduce biofilm formation, GW4869 increased the proportion of dead cells in established biofilms, suggesting its role in compromising biofilm cell viability. Biochemical assays revealed that all three compounds inhibited the enzymatic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
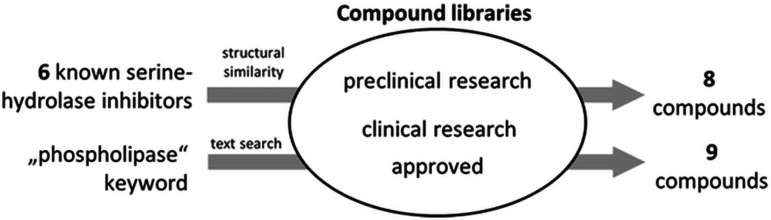 Fig 1
Fig 1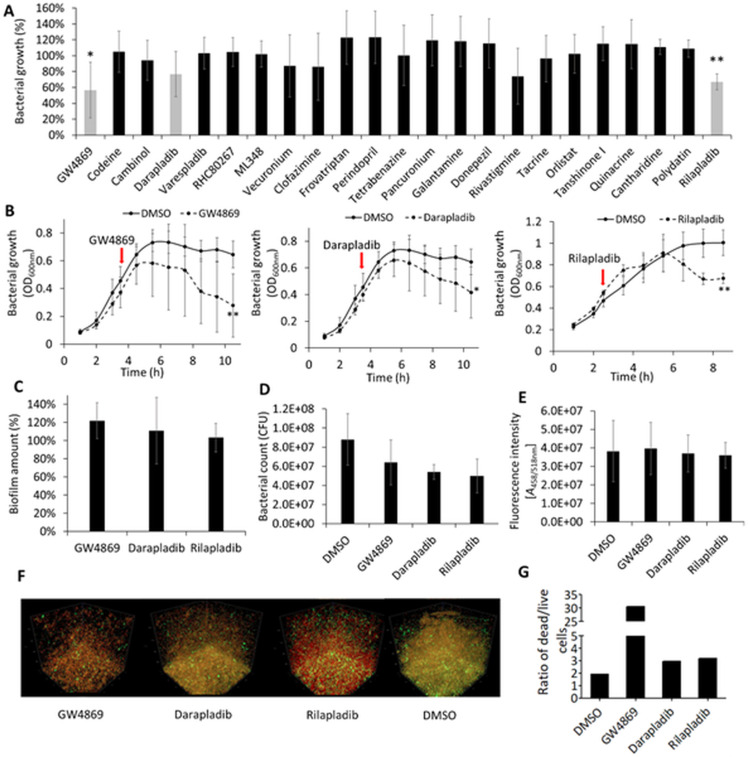 Fig 2
Fig 2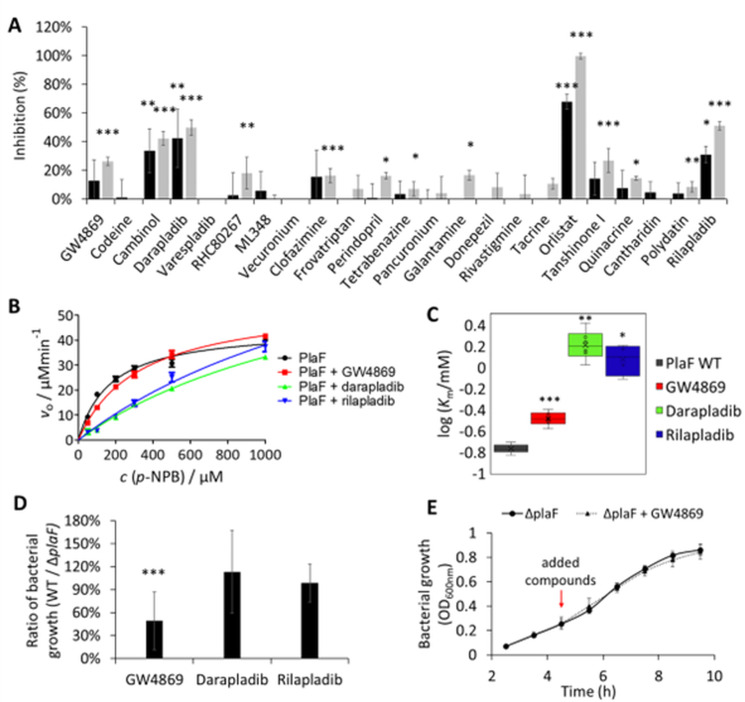 Fig 3
Fig 3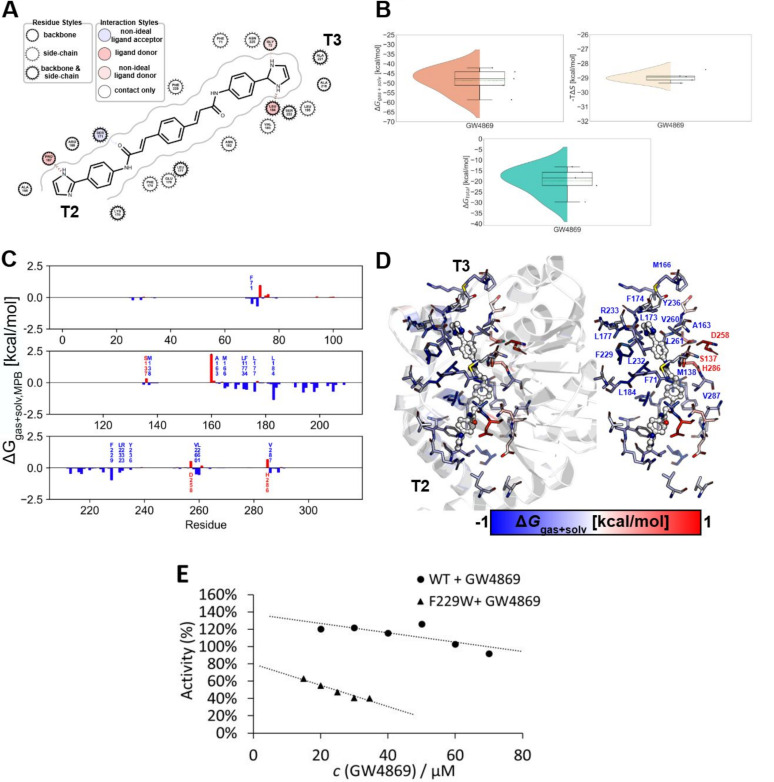 Fig 4
Fig 4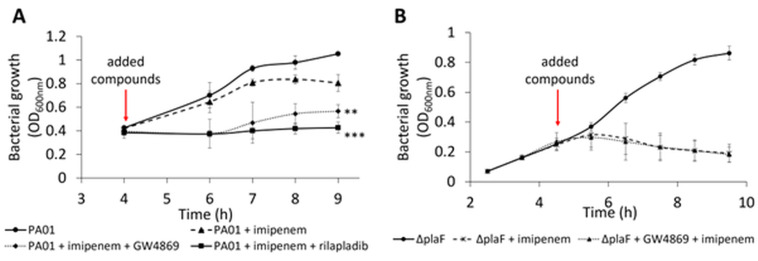 Fig 5
Fig 5| Compound | Clinical status | Human target | Mode of action | Literature | |
|---|---|---|---|---|---|
| Approved drugs | Rivastigmine | Approved by FDA and EMA | Acetylcholinesterase, butyrylcholinesterase | Pseudo-irreversible inhibitor | ( |
| Donepezil | Approved by FDA and EMA | Acetylcholinesterase | Reversible competitive inhibitor | ( | |
| Tacrine | Approved by FDA | Acetylcholinesterase, cholinesterase | Non-competitive reversible inhibitor | ( | |
| Clofazimine | Approved | Phospholipase A2 | Agonist modulator inhibitor | ( | |
| Galantamine | Approved by FDA and EMA | Acetylcholinesterase, cholinesterase, nicotinic acetylcholine receptor | Reversible competitive inhibitor | ( | |
| Orlistat | Approved by FDA and EMA | Triacylglycerol lipase | Competitive reversible inhibitor | ( | |
| DrugBank | Tetrabenazine | Approved | Synaptic vesicular amine transporter | Reversible inhibitor | ( |
| Codeine | Approved | Opioid receptors | Agonist, regulator | ( | |
| Frovatriptan | Approved | 5-Hydroxytryptamine receptor | Agonist | ( | |
| Perindopril | Approved | Angiotensin-converting enzyme | Inhibitor | ( | |
| Pancuronium | approved | Muscle nicotinic acetylcholine receptor | Competitive inhibitor | ( | |
| Vecuronium | Approved | Muscle nicotinic acetylcholine receptor | Competitive inhibitor | ( | |
| Cantharidin | Approved | Protein phosphatase | Inhibitor | ( | |
| Rilapladib | Investigational | Lipoprotein-associated phospholipase A2 | Inhibitor | ( | |
| Selleckchem | Darapladib | Investigational | Lipoprotein-associated phospholipase A2 | Competitive inhibitor | ( |
| Quinacrine | Investigational | Cytosolic phospholipase A2, DNA | Inhibitor intercalation | ( | |
| Varespladib | Investigational | Secretory phospholipase A2 | Inhibitor | ( | |
| Experimental | Diacylglycerol lipase, cholinesterase, cyclooxygenase, phospholipases C and A2 | Inhibitor | ( | ||
| ML348 | Experimental | Acyl protein thioesterase 1, lysophospholipase A1 | Reversible inhibitor | ( | |
| Cambinol | Experimental | Neutral sphingomyelinase 2, sirtuin 1/sirtuin 2 | Uncompetitive inhibitor | ( | |
| GW4869 | Investigational | Neutral sphingomyelinase 2 | Non-competitive inhibitor | ( | |
| Tanshinone I | Investigational | Phospholipase A2, histone lysine methyltransferase, prostaglandin E2, tumor necrosis factor-α | Inhibitor | ( | |
| Polydatin | Investigational | Phospholipase A2, NF-κB and MAPK pathway, COX-2, iNOS, SIRT1 | Inhibitor modulator | ( |
- —German Research Foundationhttp://dx.doi.org/10.13039/501100001659
- —German Research Foundationhttp://dx.doi.org/10.13039/501100001659
- —German Research Foundationhttp://dx.doi.org/10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial biofilms and quorum sensing · Microbial Natural Products and Biosynthesis · Bacterial Genetics and Biotechnology
INTRODUCTION
Pseudomonas aeruginosa is a nosocomial gram-negative pathogen that poses a significant threat to public health, particularly among immunocompromised patients (1, 2). Its multidrug resistance has led the World Health Organization to classify it as a priority pathogen requiring the urgent development of effective antibiotics and alternative treatment strategies (3, 4). The overuse and misuse of antibiotics, inadequate waste management, and rapid environmental transmission have further exacerbated antibiotic resistance in P. aeruginosa and other ESKAPE pathogens (5, 6). To address the global resistance crisis that hinders the effectiveness of existing antibiotics against P. aeruginosa (7), recent advances have been made in the development of innovative antivirulence approaches against this human pathogen (5, 8, 9).
These antivirulence compounds target key virulence determinants, reducing pathogenicity or enhancing susceptibility to antibiotics, thereby improving infection treatment outcomes (9–13). Among them, the most promising approaches include the inhibition of quorum sensing (14, 15), surface-associated adhesins (16), and iron acquisition pathways (17) that collectively contribute to biofilm formation (18), inhibition of secretory systems (19), and direct neutralization of extracellular toxins such as phospholipase A (PLA) (20). However, monotherapy with these compounds has shown limited success in eradicating infections (18, 21). Combination therapies, such as quorum-sensing inhibitors and tobramycin or the iron chelator deferoxamine and tobramycin (22), have demonstrated enhanced efficacy in eliminating P. aeruginosa biofilms in infected hosts. These findings underscore the potential of adjuvant therapies targeting virulence factors to become integral components of clinical practice (8, 23, 24).
Among the virulence factors of P. aeruginosa, secreted PLAs play an important role in host cell invasion, membrane disruption, and immune evasion by releasing fatty acids from host phospholipids (25, 26). Notably, pseudolipasin A, a selective inhibitor of the ExoU phospholipase, has shown protective effects in vitro on hamsters, amoebae, and yeast cells against ExoU-mediated toxicity (20). Beyond secreted phospholipases, emerging evidence reveals the significance of intracellular bacterial phospholipases for virulence (27, 28) and antibiotic resistance (29). Their function is related to the adjustment of the bacterial membrane phospholipid composition to adapt to environmental stimuli. For example, Brucella melitensis PLA BveA increases the resistance to polymyxin B through the hydrolysis of membrane phosphatidylethanolamine (29). Similarly, the intracellular PLA PlaF of P. aeruginosa regulates membrane phospholipid composition, thereby influencing iron uptake, motility, biofilm formation, and signaling pathways (28). PlaF-deficient strains exhibit reduced virulence in mouse macrophage and Galleria mellonella infection models, underscoring their therapeutic potential (27, 30).
The crystal structure of PlaF reveals a transmembrane helix anchoring the catalytic domain, which contains a serine–hydrolase catalytic triad, to the cytoplasmic membrane (27). This configuration facilitates the interaction of PlaF with membrane phospholipid substrates and enables substrate extraction via one of three distinct active-site tunnels (31, 32). The crystallized complex of PlaF with the fatty acid product and molecular dynamics (MD) simulations identified putative product egress tunnels (31, 32). Detailed structural and mechanistic insights furthermore emphasize the drugability of PlaF.
In light of the challenges posed by traditional drug discovery, this study explores the repurposing of Food and Drug Administration (FDA)-approved or clinically tested compounds as potential inhibitors of PlaF. Some of the selected compounds have been described to target the human serine hydrolase family PLAs (33). From a library of 23 identified compounds, we experimentally tested their effects on P. aeruginosa PA01 growth, biofilm formation, and dispersal. Growth-inhibiting compounds were further assessed for PlaF inhibition using functional enzyme assays and a ΔplaF mutant strain. Enzyme kinetics and molecular dynamics simulations indicate the mode of action of the PlaF-targeting compound GW4869. Experiments to mimic combination therapy demonstrated synergism between GW4869 and the last-resort antibiotic imipenem, supporting the therapeutic potential of PlaF as an antivirulence target. This study underscores the inhibition of intracellular PLAs as a novel and previously underexplored strategy for developing effective treatments against P. aeruginosa.
MATERIALS AND METHODS
Protein expression and purification
Expression and purification of PlaF were performed as described previously (34). P. aeruginosa PA01 cells were transformed (35) with plasmid pBBR-plaF_H6_ or pBBR-plaF_H6_-F229W (36) and grown overnight at 37°C in lysogeny broth (LB) medium supplemented with tetracycline (100 µg/mL). These cultures were used to inoculate an expression culture in LB medium to an initial optical density at 600 nm (OD_600nm_) of 0.05. The cultures were grown at 37°C until an OD_600nm_ of ~2 was obtained and afterward harvested by centrifugation at 6,750 × g and at 4°C for 15 min. The total membrane fraction was solubilized with Triton X-100, and proteins were purified using Ni-NTA agarose (Qiagen, Hilden, Germany) with buffer supplemented with 0.25 mM n-dodecyl-β-D-maltoside. For biochemical analysis, proteins were transferred to Tris-HCl buffer (100 mM, pH 8) supplemented with the respective detergent. The purity of PlaF was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis under denaturation conditions on a 12% (wt/vol) gel (37).
Selection and preparation of compounds for screening library
The compound library was assembled based on the 2D structural similarity to six approved pharmaceuticals identified through a literature search, as indicated in Table 1. The DrugBank database was queried with these six selected compounds, and structures with more than 50% 2D structural similarity to the initial compounds were included in the library. Additionally, the Selleckchem database was searched using the keyword “phospholipase,” and the phospholipase inhibitors were added to the library. The stock solutions of selected compounds were prepared using dimethyl sulfoxide, methanol, or water as indicated in Table S1.
Growth curves
The growth of bacterial strains P. aeruginosa PA01 wild type, an isogenic mutant ΔplaF (27), Staphylococcus aureus ATCC 25923, and Escherichia coli ATCC 25922 was monitored by measuring the OD_600nm_ for 10 h in a 96-well plate shaken with agitation at 1,000 rpm. First, the OD_600nm_ of overnight cultures was diluted with lysogeny broth medium to 0.05. When the bacterial cultures reached an OD_600nm_ of approximately 0.4, 1.5 µL of inhibitor stock solutions (Table S1), 1.5 µL of antibiotic, or both were added in a total volume of 150 µL. The antibiotics were prepared in water at the following final concentrations: 0.5 mg/L (0.70 µM) for gentamicin, 2.0 mg/L (3.71 µM) for piperacillin, 1.0 mg/L (0.71 µM) for colistin, and 2.0 mg/L (6.30 µM) for imipenem. Cultures treated with respective solvents were used as no-compound controls.
Enzyme activity assay, inhibition, and enzyme kinetic studies
The esterase activity of the PlaF protein was determined spectrophotometrically with p-nitrophenyl butyrate (p-NPB) as a substrate, as described previously (84), using a 96-well microplate. Five microliters of purified protein (stock concentration 30 µg/mL) was mixed with 2 µL of potential inhibitor stock solution at concentrations of: 1.73 mM for GW4869, 3.34 mM for codeine, and 10.0 mM for all the remaining compounds, and 93 µL of freshly prepared 1 mM p-NPB solution was added. Esterase activity was monitored by an increase in absorbance at 420 nm for up to 2 h at 37°C. Kinetic parameters were determined by measuring activity with different substrate concentrations (0.05, 0.1, 0.2, 0.3, 0.5, 1.0, 1.3, and 1.5 mM) (27). Half-maximal inhibitory concentration (IC_50_) was determined by measuring enzyme activity with different compound concentrations and with 1 mM p-NPB substrate and was calculated from linear plots (85).
Biofilm assay
The microtiter dish biofilm assay was performed according to a modified protocol (86, 87). The respective number of single colonies of P. aeruginosa was inoculated in LB media and incubated for 24 h at 37°C with shaking. Overnight cultures were used to inoculate 150 µL of bacterial cultures of OD_600_ = 0.05 in LB medium in a plastic 96-well microtiter plate (MTP). In the case of compound-treated biofilm, 2 µL of the compound or respective solvent was added to a total volume of 150 µL. The inoculated microtiter plate was covered with air-permeable sealing film and incubated for 24 h at 37°C. Afterward, the OD_600nm_ was measured; the medium was removed; and each well was briefly washed two times with 200 µL LB media at 37°C. Biofilms were stained by adding 200 µL 0.1% (wt/vol) crystal violet solution, followed by 20 minutes of incubation at room temperature. The crystal violet solution was discarded, and the plate was washed three times with 200 µL water, followed by adding 200 µL 30% (vol/vol) acetic acid. The plate was incubated for 20 minutes at room temperature, and absorbance was measured at 585 nm using a plate reader (SpectraMax iD3; Molecular Devices GmbH, München, Germany).
Colony-forming unit count
Biofilm was grown as described above. After 24 h incubation, the supernatant was discarded; the biofilm was suspended in 200 µL LB medium, diluted 100,000-fold, and 100 µL of cell suspension was plated on LB medium agar plates. Plates were incubated overnight at 37°C.
Quantification of extracellular DNA in biofilm
Extracellular DNA was quantified according to a modified protocol (88). Biofilm was grown in a 96-well plate as described above. After washing two times with 200 µL LB medium, 100 µL of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8) was added, followed by adding 100 µL of SYBR Green I (Thermo Fisher Scientific, Germany). SYBR Green I was prepared freshly by diluting it 1:1,250 in TE buffer. The plate was incubated for 10 minutes in the dark at room temperature, and the fluorescence was measured using a fluorescence plate reader (SpectraMax iD3, Molecular Devices GmbH) at 485/518 nm excitation/emission filter setup.
Confocal laser scanning microscopy of biofilm
Confocal laser scanning microscopy (CLSM) of biofilm was performed as described previously (89). Briefly, bacterial biofilms were prepared under static conditions in an eight-well chamber µ-slide (ibidi GmbH, Gräfelfing, Germany) and treated with the compounds listed in Table S1. After 24 h incubation, the samples were incubated with live/dead staining solution (LIVE/DEAD BacLight Bacterial Viability Kit; Thermo Fisher Scientific, Waltham, USA) to assess the viability of the bacterial cells. The biofilms were visualized by Confocal Laser Scanning Microscope 800 (Axio observer.Z1/7; Carl Zeiss AG, Oberkochen, Germany) at settings listed in Table S2.
Preparation of starting structures for unbiased molecular dynamics simulations
The crystal structure of the PlaF dimer (PDB ID 6I8W) is available in the Protein Data Bank (90). The last five residues of the C-terminus of each monomer missing in the structure were added using MODELLER (91), and all small molecule ligands were removed. The dimer was oriented in the membrane using the PPM server (92). From that, the monomeric configuration of PlaF chain A was generated by removing chain B from the dimer orientation. Chain A was oriented again using the PPM server, resulting in the tilted configuration t-PlaF.
The structure of GW4869 was prepared starting from its canonical SMILES using the fixpka option in OpenEye (93). The most favorable conformer was generated with Omega 4.1.1.1 (94) using the flag –maxconfs = 1. The charges of GW4869 were calculated following the RESP approach (95) using Gaussian 16 to compute electrostatic potentials at the HF/6-31G* level (96). GW4869 docking to t-PlaF was performed using AutoDock 3.0.5 (97) with the in-house developed scoring function DrugScore (98). The selected configuration represents the most populated cluster obtained.
The bound t-PlaF configuration was embedded into a DOPE:DOPG = 3:1 membrane (99) and solvated using PACKMOL-Memgen (100, 101). The membrane composition resembled that of the native inner membrane of gram-negative bacteria (99) and was already used for simulating PlaF in a membrane bilayer environment (27, 31, 32). A distance of at least 15 Å between the protein or membrane and the solvent box boundaries was kept. To obtain a neutral system, counter ions were added that replaced solvent molecules (KCl 0.15 M), resulting in the systems containing ~140,000 atoms.
Unbiased molecular dynamics simulations of selected inhibitors bound to t-PlaF
The GPU particle mesh Ewald implementation from the AMBER23 suite of molecular simulation programs (102) with the ff14SB (103), Lipid21 (104), and GAFF2 (105) force fields for the protein, membrane lipids, and ligands, respectively, was used; water molecules and ions were parametrized using the TIP3P model (106) and the Li and Merz 12-6 ion parameters (107, 108). For each t-Plaf-inhibitor complex, five independent replicas of 1 μs length were performed. Covalent bonds to hydrogens were constrained with the SHAKE algorithm (109) in all simulations, allowing the use of a time step of 2 fs. Details of the thermalization of the simulation systems are given below.
All unbiased MD simulations showed structurally invariant protein structures and membrane phases evidenced by electron density calculations (Fig. S1). The root mean square deviation (RMSD) of GW4869, after removal of the global motions of t-PlaF, showed structurally invariant binding configurations across five different replicas (Fig. S2).
Relaxation, thermalization, and production runs
An initial minimization step was performed with the CPU code of pmemd (110). Each minimization was organized in four steps of 1,000 cycles each, for a total of 4,000 cycles of minimization. Afterward, each minimized system was thermalized in one stage from 0 to 300 K over 25 ps using the NVT ensemble and the Langevin thermostat (111), and the density was adapted to 1.0 g/cm^3^ over 4,975 ps using the NPT ensemble with a semi-isotropic Berendsen barostat (112), with the pressure set to 1 bar. Thermalization and density adaptation were performed with the GPU code of pmemd (110).
For each replica, 1 μs of production run using the GPU code of pmemd was performed in the NPT ensemble at a temperature of 300 K using the Langevin thermostat (111) and a collision frequency of 1 ps^−1^. To avoid noticeable distortions in the simulation box size, semi-isotropic pressure scaling was employed using the Berendsen barostat (112) with a pressure relaxation time of 1 ps, coupling box size changes along the membrane plane (113).
Molecular mechanics–Poisson Boltzmann surface area calculations of GW4869 binding to t-PlaF
To pinpoint the most likely binding epitopes, we generated 3D density grids to map the location of GW4869 across multiple replicas. We considered stably bound conformations if a frame of the trajectory has a ligand RMSD of <1.5 Å to the previous frame (see supplementary results). These binding configurations were clustered using the hierarchical agglomerative (bottom-up) algorithm implemented in cpptraj (114), using the minimum distance ε between the clusters as the cluster criterion. Starting from ε = 2.0 Å, we gradually increased ε in 0.5 Å intervals until the population of the largest cluster remained unchanged (at ε = 4.0 Å). We calculated the 3D density maps of GW4869 considering all atoms using the grid function available in cpptraj (114) with a grid spacing of 1.5 Å. We applied a contour level of 1σ (one standard deviation above the mean value).
Subsequently, we conducted molecular mechanics–Poisson Boltzmann surface area (MM-PBSA) calculations with MMPBSA.py (115) and normal mode analysis computations to determine the binding effective energy and the binding entropy of GW4869 to t-PlaF, respectively. These calculations were performed across the different replicas, and the results per replica were averaged. Dielectric constants of 1 for the protein, 80 for the solvent, and 15 for the membrane were used, similar to our previous work (32). A heterogeneous dielectric model was used to represent the membrane; the implicit membrane model using spline fitting (memopt = 3) was employed for the binding effective energy calculations (116). This allowed us to compute ΔGsolv and ΔGgas+solv (equations 1 and 2) (117). The normal mode analysis calculation allowed us to calculate the loss of configurational entropy upon binding, −TΔStotal, considering the translational, rotational, and vibrational terms (equation 3) (117).
To compare the computed with experimentally determined binding affinities, we converted into the standard free energy of binding according to equation 4, as done previously (118, 119). This takes into account that translational entropy depends on solute concentration (120, 121), leading to the concentration dependence of chemical equilibria that do not conserve the number of molecules (such as binding reactions) (122, 123):
where R is the universal gas constant (R = 0.001987 kcal/K/mol); T = 298.15 K; C^0^ is the standard concentration of 1 mol/L; and C^ideal^ is the ligand concentration of 0.041 mol/L, derived from the general gas equation at a pressure of 101,325 Pa and a temperature of 298.15 K (118). is directly related to the computed dissociation constant according to equation 5:
The total standard error of the mean of the computations, denoted as , is estimated following the principles of Gaussian error propagation according to equation 6:
where and are the SEMs from MM-PBSA and NMA computations, respectively. The results from binding free energy calculations are reported as . The computations were converged, as evidenced by the comparison between the first and second halves of the trajectories (Fig. S3).
RESULTS
Identifying pharmaceuticals with potential inhibitory effects on bacterial PLA
This study aimed to identify approved drugs and pharmaceuticals from (pre)clinical studies that have potential inhibitory activity against the bacterial phospholipase PlaF to evaluate their suitability for repurposing as antimicrobials. In the following, we will jointly refer to these compounds as pharmaceuticals. According to the literature, the six FDA-approved drugs, rivastigmine (33, 38), donepezil (33, 38), galantamine (33, 38), tacrine (33, 38), orlistat (33, 44), and clofazimine (41, 42), target human PLAs or esterases/lipases.
Despite the generally low sequence identity between human and bacterial PLAs, both enzyme families share a conserved catalytic Ser–Asp dyad and a hydrophobic substrate-binding pocket that accommodates phospholipid acyl chains, enzymes known for their side PLA activity due to the shared serine hydrolase catalytic mechanism (27, 36, 124, 125). This conservation in the catalytic mechanism provides a mechanistic rationale for exploring human PLA or serine hydrolase inhibitors as potential scaffolds for bacterial PLA inhibition. At the same time, differences in the sequence similarity between PlaF, the human α/β hydrolase domain (ABHD6), and two epoxide hydrolases identified as the most similar targets in humans (Fig. S4) were considered favorable to identify selective bacterial PLA inhibition. Indeed, structural models of the target sequences superimposed with PlaF (Fig. S5) revealed, even when unaligned residues are not considered, a high RMSD for O60906; in all other cases, it is around 2 Å or below (Table S3). Still, when analyzing the binding site residues, we identified that a high proportion of residues were in alignment gaps (Table S3) and that the binding site cavity of PlaF slightly overlaps only with ABHD6 but not in the other structures, suggesting high structural diversity (Fig. S5). To accommodate this, we therefore enlarged the diversity in the screening pool by adding other compounds based on structural similarity.
Identified pharmaceuticals were used as templates to identify similar compounds among more than 500,000 investigational, clinically tested, or approved drugs in the DrugBank database (126, 127). This ligand-based search identified eight additional compounds showing 2D structural similarity with the initial six compounds, with similarity scores ranging from 54% to 72%. Furthermore, a targeted keyword search in the Selleckchem database, which includes over 120,000 bioactive small molecules, yielded nine additional compounds being investigated as human phospholipase, lysophospholipase, or lipase inhibitors (Fig. 1).
Selection of potential phospholipase PlaF inhibitors. A library of 23 pharmaceuticals to be tested as potential PlaF inhibitors (Table 1) was generated by screening compounds in preclinical and clinical research phases and approved drugs, from compound libraries of DrugBank and Selleckchem databases. The selection criteria involved a combination of structural similarity and text-based searches.
Of the resulting 23 pharmaceuticals, 13 have regulatory approval, 7 are in clinical research phases, and 3 are in preclinical studies, target esterases, lipases, and phospholipases. For the cases of tetrabenazine, codeine, frovatriptan, perindopril, pancuronium, vecuronium, and cantharidin, no related target has been described, despite an apparent molecular similarity to the template compounds or associated keywords. The advantage of this library is that the majority of the compounds have met safety and efficacy standards for use in humans (Table 1). In conclusion, this library of diverse potential phospholipase-inhibiting compounds provided a foundation for testing their repurposing as antimicrobial agents.
Effect of potential PlaF inhibitors on the growth and biofilm formation of P. aeruginosa
We investigated the effects of the 23 pharmaceutical compounds from the library on the planktonic growth of P. aeruginosa in a rich LB medium. In a screening experiment designed to identify the most potent pharmaceuticals, the compounds were added to exponentially growing bacterial cultures (OD_600nm_ ~0.5) in plastic MTPs, and optical density was measured after incubation at 37°C with agitation. In our study, relatively high concentrations of pharmaceuticals were used to overcome bioavailability issues due to poor aqueous solubility of many compounds. The results demonstrated that GW4869 and rilapladib significantly inhibited bacterial growth compared to the respective solvent-treated controls after 5 h incubation, while the effect of darapladib is significant after 7 h (Fig. 2A). Validation growth inhibition experiments under these screening conditions revealed that the effects of all three compounds were evident as early as 2–4 h post-treatment (Fig. 2B). Over 6–7 h, treated cultures exhibited a slower growth rate compared to the controls. After 6–7 h, darapladib and rilapladib reduced P. aeruginosa growth significantly by approximately 35%, while GW4869 exhibited a more pronounced effect, reducing growth by 57% (Fig. 2B).
*Potential PLA inhibitors impact the growth and viability of P. aeruginosa PA01. (A) Effect of 23 pharmaceuticals on the planktonic growth of P. aeruginosa after 5 h incubation with compounds. Relative growth values represent optical densities (OD600nm) of compound-treated cultures compared to the OD600nm of respective solvent-treated cultures set to 100%. Light gray bars represent pharmaceuticals that significantly impacted the growth of P. aeruginosa after 5 h or more (see panel B). Black bars represent compounds that showed no significant effect on the P. aeruginosa growth at any time point. Results are shown as the mean ± SD of six biological replicates (n = 6) from two independent experiments. t-test of normally distributed values; *P < 0.05, **P < 0.01. (B) Growth curves of P. aeruginosa treated with GW4869 (17.3 µM), darapladib (100.0 µM), and rilapladib (100.0 µM). Bacterial cultures were grown in LB medium at 37°C with shaking at 1,000 rpm in a plastic MTP. Red arrows indicate the beginning of treatment. Results are the mean ± SD of three independent experiments (n = 3). t-test of normally distributed values; *P < 0.05, *P < 0.01. (C) Biofilm amount of P. aeruginosa treated for 24 h with GW4869 (17.3 µM), darapladib (100.0 µM), and rilapladib (100.0 µM) was quantified by the crystal violet assay. The results are shown relative to the solvent-treated cultures, which were set to 100%. The results are the mean ± SD of >22 biological replicates from four independent experiments. t-test was calculated compared to the untreated samples. (D) Viable bacterial count determined as colony-forming units (CFUs) within biofilm treated for 24 h with GW4869 (17.3 µM), darapladib (100.0 µM), and rilapladib (100.0 µM) or the respective solvent (dimethyl sulfoxide [DMSO]). The y-axis represents colony-forming units per 1 mL of bacterial culture. Results are the mean ± SD of four biological replicates (n = 4); t-test showed no significant changes between treated and untreated cultures. (E) Quantification of eDNA in biofilm treated for 24 h with GW4869 (17.3 µM), darapladib (100.0 µM), and rilapladib (100.0 µM) or the respective solvent (DMSO) determined with SYBR Green I dye. Results are the mean ± SD of four biological replicates (n = 4); t-test showed no significant changes between treated and untreated cultures. (F) Representative confocal scanning microscopy figures of P. aeruginosa PA01 grown as static biofilms treated for 24 h with GW4869 (7.5 µM), darapladib (100.0 µM), rilapladib (100.0 µM), or DMSO as the solvent control. Biofilms were stained to visualize live (green) or dead/dying (red) cells. (G) The ratio of dead to live cells in biofilms was determined from red and green fluorescence intensity quantification from panel F using the BiofilmQ tool.
Additionally, evaluating the pharmaceutical compounds from our library, we found that rilapladib inhibited the growth of gram-negative enteropathogenic E. coli ATCC 25922 by 44% after 6 h of treatment (Fig. S6). Furthermore, the growth of pathogenic gram-positive S. aureus ATCC 25923 was reduced by 58% and 73% following treatment with rilapladib and darapladib, respectively (Fig. S7). Several other pharmaceuticals from the library also show specificity to inhibit the growth of E. coli and S. aureus in the range of 30%–55% (Fig. S6 and S7).
Biofilm formation represents a dominant lifestyle of P. aeruginosa during chronic infections (128, 129), functioning as a protective barrier against antibiotics (130, 131). Therefore, we next assessed the effects of GW4869, darapladib, and rilapladib on biofilm formation and cell viability in static cultures (without agitation) adhered to plastic MTP. Pharmaceuticals were introduced at the time of inoculation, followed by a 24-h incubation at 37°C under static conditions. Crystal violet staining of adherent cells revealed that these pharmaceuticals had no significant effect on biofilm quantity (Fig. 2C). Viable bacterial counts assessed by colony-forming unit assays indicated no significant differences between treated and untreated biofilms (Fig. 2D). Furthermore, quantification of extracellular DNA, a critical biofilm structural component linked to increased antibiotic resistance (132), showed no significant differences between the treated and untreated biofilms (Fig. 2E).
Given that the tested pharmaceuticals did not affect initial biofilm formation steps, we next investigated their impact on biofilm dispersal. Using CLSM, we examined biofilm architecture and cell viability. In this experiment, P. aeruginosa biofilms were preformed by incubating bacteria at 37°C under static conditions on microscopic glass slides for 24 h, followed by an additional 24 h treatment with GW4869, darapladib, or rilapladib. Biofilms were then stained with live/dead viability dyes: a green dye for live cells and a red dye for cells with compromised membranes, assigned as dead or dying cells. Results showed no discernible effect of the tested pharmaceuticals on biofilm dispersal (Fig. 2F). However, the ratio of dead/dying cells was much higher in biofilms treated with all three compounds compared to untreated controls (Fig. 2G). Quantitative analysis of CLSM images revealed that the dead-to-live cell ratio increased from 2:1 in untreated biofilms to approximately 3:1 in biofilms treated with darapladib or rilapladib. Notably, GW4869 had a strong effect, as only about 3% of the cells were estimated to be alive.
These findings highlight the potential of GW4869, darapladib, and rilapladib as inhibitors of planktonic growth and the viability of biofilm-forming P. aeruginosa cultures.
Inhibitory potential of selected compounds against PlaF
Next, we investigated whether PlaF, an intracellular PLA recently discovered by our group, serves as a potential target of GW4869, darapladib, and rilapladib. PlaF plays a critical role in maintaining phospholipid homeostasis and impacts the pathogenicity of P. aeruginosa (27, 28). To address this, we first performed biochemical assays using purified PlaF reconstituted into phospholipid liposomes to ensure a near-native conformation (31). To assess the inhibition of its hydrolytic activity in the presence of the three selected pharmaceuticals, a spectrophotometric assay based on the hydrolysis of the pro-chromogenic p-NPB substrate revealed that all three compounds inhibited PlaF activity. Darapladib and rilapladib at 400 µM reduced PlaF activity by ~50%, while GW4869 (69.2 µM) inhibited PlaF activity by ~30% (Fig. 3A). Although some of the remaining compounds showed an inhibitory effect on the PlaF activity, we did not consider them further as they did not impact the growth of P. aeruginosa.
*Inhibitory effect of potential PLA targeting pharmaceuticals on PlaF. (A) Purified PlaF was treated with compounds (34.6 and 69.2 µM for GW4869, 66.8 and 133.6 µM for codeine, and 200 and 400 µM for the remaining compounds) in the presence of the p-NPB substrate, and the released product was quantified spectrophotometrically during 1 h. Black and gray bars indicate lower and higher concentrations of pharmaceuticals, respectively. Results are shown as relative inhibition compared to samples treated with the respective solvent. Results are the mean ± SD of three experiments, each measured with three replicates (n = 9). t-test was calculated by comparing pharmaceutical- and solvent-treated samples; *P < 0.05, **P < 0.01, ***P < 0.001. (B) Michaelis–Menten curve showing enzyme kinetic studies with purified PlaF treated with GW4869 (34.6 µM), darapladib (200.0 µM), or rilapladib (200.0 µM) measured spectrophotometrically using p-NPB assay. The curves are calculated with GraphPad software from two independent experiments, each measured three times (n = 6). (C) Log-transformed Michaelis–Menten constants calculated from curves in panel B. The average value is indicated with a cross, while the center line represents the median. The box limits indicate the interquartile range, and the whiskers indicate the minimum and maximum values. t-test of normally distributed values is calculated compared to the non-treated sample; *P < 0.05, **P < 0.01, ***P < 0.001. (D) The ratio of P. aeruginosa WT/ΔplaF bacterial growth after 6 h incubation with GW4869 (17.3 µM), darapladib (100.0 µM), and rilapladib (100.0 µM). Bacterial cultures were grown in LB medium at 37°C with shaking at 1,000 rpm. Results are shown as the ratio of the mean values ± SD of two independent experiments, each measured three times (n = 6). t-test of normally distributed values, **P < 0.001. (E) Growth curves of PA01 ΔplaF treated with GW4869 (17.3 µM). Bacterial cultures were grown in LB medium at 37°C with shaking at 1,000 rpm in a plastic MTP. Red arrows indicate time points of compound addition. Results are the mean ± SD of four biological replicates (n = 4).
Next, we employed enzyme kinetic studies to determine whether these compounds inhibit PlaF competitively. Using the p-NPB assay, we measured PlaF activity at various substrate concentrations (0.05–1.0 mM) and calculated the initial velocity (v₀) and Michaelis–Menten constant (Km). Kinetic plots showed that darapladib and rilapladib strongly, and GW4869 less prominently, affected the catalytic properties of PlaF (Fig. 3B) without altering vmax but increasing the Km (Fig. 3C). These results indicate that PlaF treated with these compounds exhibits reduced substrate affinity, thus suggesting competitive inhibition.
To further explore whether these pharmaceuticals target PlaF in bacteria, we compared the growth of P. aeruginosa wild-type and an isogenic ΔplaF mutant strain in the presence of the compounds (Fig. 3D). Results revealed that darapladib and rilapladib showed no differential effects on the growth of the ΔplaF strains, indicating that their mode of action is likely independent of PlaF. In contrast, after 6 h of incubation with GW4869, the ΔplaF mutant grew to a higher cell density than the wild-type strain (Fig. 3D), whereas the growth curves of GW4869-treated and GW4869-untreated ΔplaF did not differ (Fig. 3E). These results suggest that the action of GW4869 may be directly or indirectly related to PlaF.
Overall, our findings suggest that GW4869, darapladib, and rilapladib modulate PlaF activity in vitro, although a distinct inhibitory effect potentially mediated by inhibition of PlaF in bacterial culture was only observed with GW4869.
Mechanism of inhibition of PlaF by GW4869 involves binding to the substrate-binding tunnel
To obtain atomistic insights into the inhibition of PlaF by GW4869, we conducted unbiased MD simulations. Results from five independent 1 μs long MD simulations indicated that the t-PlaF:GW4869 complex, where the ligand was initially docked at the active site, remains structurally invariant, as indicated by a low overall RMSD of <1.5 Å (Fig. S2). Hierarchical clustering of the simulated configurations sampled every 50 ps across the five replicas revealed that GW4869 preferentially binds to tunnels T2 and T3, which are structural elements previously identified as essential for substrate hydrolysis (31, 32). The most abundant cluster (C1), where GW4869 occupies parts of T2 and T3, accounted for 25.7% ± 4.2% of the analyzed frames (Fig. 4A).
Molecular mechanism of GW4869 binding to t-PlaF. (A) Binding of GW4869 to t-PlaF in the most populated cluster from MD simulations represented as a 2D interaction map. Residues interact with their side chain, backbone, or both, as detailed by the legend. The entrances of tunnels T2 and T3 are indicated. (B) Effective energy (top left) and configurational entropy (top right) contributions to binding and total binding energy (bottom) of GW4869. The violin plots indicate the distributions of the data points; the inner box of a box plot represents the interquartile range, with the horizontal black line indicating the median. The dotted green line represents the mean, and the whiskers show the rest of the distribution, excluding points determined to be outliers when they fall outside 1.5 times the interquartile range. (C) Per-residue binding effective energy of tunnel residues. The energies were averaged across five independent replicas (n = 5). Error bars denote the SEM. (D) Per-residue binding effective energy mapped at the structural level. Interacting residues are depicted as sticks, colored according to the effective binding energy. The entrances of tunnels T2 and T3 are indicated. The interacting residues of T3 are labeled. The GW4869 inhibitor is depicted as red, blue, and gray spheres. (E) Determination of half-maximal inhibitory concentration for inhibition of phospholipid liposome-reconstituted PlaFWT or PlaFF229W with GW4869 using p-NPB assay. Graphs show linear plots of relative activities compared to the untreated sample set to 100%. The result represents the mean ± SD of six measurements (n = 6).
To quantify the binding energy of GW4869 in cluster C1, the unbiased MD simulations were used for MM-PBSA calculations, incorporating an implicit membrane model (115, 116). The computations were converged, as evidenced by the comparison between the first and the second halves of the trajectories (Fig. S3). These trajectories yielded a favorable binding effective energy (ΔGgas+solv) of −48.8 ± 2.9 kcal/mol and an unfavorable entropic contribution to binding (TΔS) of −28.9 ± 0.2 kcal/mol. The resulting binding free energy (ΔGbind) estimate is −19.8 ± 2.9 kcal/mol (Fig. 4B). The corrected standard free energy of binding ( ) is −21.7 ± 2.9 kcal/mol, indicating that binding of GW4869 to T2 and T3 is exergonic.
The residue-specific decomposition of the binding effective energy revealed key residues in T2 and T3 that contribute significantly to GW4869 binding. Residues in T3 that were proposed to be part of the product egress pathway (31) contributed more to GW4869 binding (62.2% of the total favorable interacting residues) than residues in T2 (37.8%). This resulted in a more favorable interaction energy with T3 (ΔGgas+solv, T3 = −30.4 ± 2.2 kcal/mol) than T2 (ΔGgas+solv, T2 = −15.0 ± 3.2 kcal/mol). Notably, residue F229 in T3, previously shown to be critical for product release, exhibited one of the strongest binding interactions. Other residues near F229, including F71, L177, L184, and L232, also contributed markedly to GW4869 binding (Fig. 4C and D).
To experimentally validate the role of F229 in GW4869 binding, we aimed to determine the IC_50_ of GW4869 for wild-type PlaF (PlaF_WT_) and the F229W variant (PlaF_F229W_). Both variants were purified and reconstituted into phospholipid liposomes to ensure near-native PlaF conformations (31). Inhibition curves obtained by varying the GW4869 concentration at constant protein and substrate (p-NPB) concentrations displayed a linear relationship within the tested concentration range without reaching a plateau, likely due to the low solubility of the hydrophobic GW4869 in water and its adsorption to phospholipid liposomes. Although precise IC_50_ values could not be determined, the observed linear relationship suggests that GW4869 inhibits PlaF_F229W_ activity more efficiently compared to PlaF_WT_ within the tested concentration range (Fig. 4E). We speculate that the increased indolyl π-system of Trp and/or the presence of a hydrogen bond donor in the Trp ring provides more favorable interactions with GW4869 compared to the smaller phenyl ring of phenylalanine.
In summary, these findings suggest that GW4869 binds noncovalently to the active site of PlaF, which might lead to interference with substrate binding and product release.
Compounds inhibiting P. aeruginosa growth potentiate the effect of common antibiotics
To evaluate the potential synergistic effects of P. aeruginosa growth-inhibiting pharmaceuticals GW4869, darapladib, and rilapladib (Fig. S6) with common antibiotics against P. aeruginosa, we conducted combinatorial treatments with antibiotics of differing modes of action (133). Synergy with antibiotics over time was assessed using growth curve analysis rather than checkerboard fractional inhibitory concentration indices (134) to keep the conditions identical to growth studies with pharmaceuticals only.
The selected last-resort antibiotics included gentamicin, which targets ribosomal function (135); colistin, which disrupts cytoplasmic membrane integrity (136); and piperacillin and imipenem, which target cell wall synthesis (137, 138). To assess synergistic effects, these bactericidal antibiotics were used at sub-inhibitory concentrations for P. aeruginosa. The concentrations used were as follows: gentamicin at 0.5 mg/L (0.70 µM), piperacillin at 2 mg/L (3.71 µM), colistin at 1 mg/L (0.71 µM), and imipenem at 2 mg/L (6.30 µM) (133).
Treatment of planktonic P. aeruginosa PA01 cultures in the exponential growth phase with combinations of pharmaceutical and antibiotic revealed that darapladib did not enhance the activity of any of the four antibiotics tested (Fig. S7). Similarly, GW4869 and rilapladib did not improve the bactericidal effects of gentamicin, colistin, or piperacillin (Fig. S7). However, combinations of GW4869 or rilapladib with imipenem showed significant synergy (Fig. 5A). Imipenem at a concentration fourfold lower than its minimum inhibitory concentration had only a minimal impact on bacterial growth. In contrast, co-treatment with GW4869 (17.3 µM) or rilapladib (100 µM) abolished P. aeruginosa growth within 2 h and significantly decreased the optical density after an additional 3 h (Fig. 5A).
*PlaF inhibitors potentiate the antibiotic activity of imipenem on P. aeruginosa. (A) The combination of imipenem (6.3 µM) and GW4869 (17.3 µM) or rilapladib (100.0 µM) resulted in reduced growth of P. aeruginosa. Bacterial cultures were grown in LB medium at 37°C with shaking at 1,000 rpm. Results are shown as the mean ± SD of four biological replicates (n = 4). t-test of normally distributed values; **P < 0.01, **P < 0.001. (B) Growth of P. aeruginosa ΔplaF mutant in the presence of a combination of imipenem (6.3 µM) and GW4869 (17.3 µM). Bacterial cultures were grown in LB medium at 37°C with shaking at 1,000 rpm. Results are shown as the mean ± SD of four biological replicates (n = 4).
To explore the mechanism of action of GW4869, presumed to inhibit PlaF, we examined its effect in combination with imipenem on the ΔplaF mutant strain (Fig. 5B). Growth analyses indicated that the GW4869-imipenem combination had no different effect than imipenem alone. In both cases, growth was slowed within 1 h of treatment, with no subsequent growth over the next 4 h. These findings suggest that GW4869’s synergism with imipenem is dependent on the presence of PlaF, further supporting the hypothesis that GW4869 acts on P. aeruginosa by inhibiting PlaF.
DISCUSSION
Inhibition of intracellular PLA is a novel strategy to combat P. aeruginosa and other pathogens
Various virulence factors of P. aeruginosa play a crucial role in host infections by facilitating bacterial adhesion, invasion, immune suppression, tissue damage, and nutrient acquisition (139). These mechanisms highlight the potential of antivirulence strategies as innovative therapeutic approaches (8). This study investigates whether inhibiting intracellular phospholipase PlaF, a virulence factor of P. aeruginosa, can suppress the growth of bacterial culture in vitro, providing a foundation for developing antivirulence therapies targeting intracellular PLAs. We acknowledge that P. aeruginosa encodes several PLA enzymes, including ExoU, PldA, and OMPLA, which could potentially interact with the tested compounds. However, an overlay of ExoU (RCSB_ID: 3TU3), PldA (RCSB_ID: 7V53), and OMPLA-PlpD (RCSB_ID: 5FQU, full sequence AlphaFold model AF-Q9HYQ6-F1) with PlaF reveals that all structures are dissimilar from PlaF (Fig. S8). Moreover, we have explored the possibility of translating these results to pathogenic E. coli and S. aureus strains.
Intracellular PLAs are essential for remodeling the bacterial membrane phospholipid composition, a vital adaptation that enhances virulence and antibiotic resistance in P. aeruginosa (27, 28) and other pathogens (29). Using a target-based drug repurposing strategy, 23 potential PLA inhibitors were selected from compounds already approved or undergoing (pre)clinical trials for human diseases. This approach could leverage the safety profiles of these compounds, including data on toxicity, pharmacokinetics, and reproductive and carcinogenic effects (140). While cross-reactivity with human pathways could pose challenges, it may also offer dual benefits by targeting pathogen factors and host pathways critical for infection resolution or damage mitigation (141).
Limited aqueous solubility and poor bacterial membrane permeability likely had a negative effect on bioavailability inside bacteria, which is required for drugs acting on intracellular targets in P. aeruginosa. Hence, many of the tested compounds were applied at concentrations above clinically achievable levels in humans or mice, with concentrations of codeine, GW4869, cambinol, RHC80267, ML348, and varespladib being closest to a therapeutically relevant range. Therefore, to elicit antibacterial effects in vitro, future investigations should focus on structure–activity relationship-guided modification of the core scaffolds (142), including reducing lipophilicity to improve aqueous solubility, optimizing substituents to enhance affinity for the PlaF active-site tunnels, and incorporating polar groups to improve bacterial membrane permeability. These pathogen-directed optimization strategies aim to generate derivatives with markedly improved potency and activity at therapeutically relevant concentrations. Moreover, optimizing drug formulation by using nanocarrier-based delivery, lipid or polymeric nanoparticles, and prodrug strategies (143) could significantly enhance solubility and intracellular uptake, thereby improving effective bioavailability of poorly soluble compounds.
We have not performed cytotoxicity tests for the pharmaceuticals examined here, as these assessments had already been carried out during their respective (pre)clinical testing. However, the feasibility of repurposing these pharmaceuticals as antibacterial scaffolds remains to be elucidated through future assessment of their cytotoxicity and selectivity toward bacterial versus mammalian PLA targets. Furthermore, for clinical application, it is necessary to establish pharmacological safety profiles of optimized derivatives even if they are very similar to an approved drug.
Efficacy of PLA inhibitors in inhibiting P. aeruginosa, E. coli, and S. aureus growth
Our results reveal that GW4869, darapladib, and rilapladib significantly inhibit the in vitro growth of planktonic P. aeruginosa and decrease the viability of biofilm-forming cells. Interestingly, rilapladib showed a broad inhibitory effect as it also inhibited the growth of E. coli and S. aureus. Application of putative PLA inhibitors from our library of pharmaceuticals may show potential for monotherapies, as several other inhibitors selectively inhibited the growth of pathogenic E. coli and S. aureus. The inhibitory effects of rilapladib on species that lack PlaF homologs indicate that this pharmaceutical may also target other lipid-metabolizing enzymes or membrane-associated processes. This cross-species activity suggests potential as broad-spectrum membrane disruptors and that PlaF inhibition may represent one of several contributing mechanisms.
While for rilapladib and darapladib—targeting human lipoprotein-associated phospholipase A2 (56, 57)—no clearly defined bacterial target could be identified, GW4869 demonstrated potent inhibition of the intracellular PLA PlaF. This was supported by the resistance observed in P. aeruginosa ΔplaF mutants and the inhibition of purified PlaF. PlaF is a non-essential P. aeruginosa gene, as deletion of plaF does not impair bacterial growth under standard laboratory conditions (27), suggesting that P. aeruginosa encodes several PlaF homologs. Such predicted single-transmembrane helix proteins (Fig. S8) could compensate for PlaF’s biological function in PL remodeling. These homologs likely reveal structural differences in their active sites, potentially preventing interactions with GW4869.
The observed competitive inhibition kinetics, consistent docking poses within the catalytic pocket, and resistance of the ΔplaF variant together provide indirect yet convergent evidence that GW4869 may target PlaF. However, it is likely that PlaF inhibition alone may not fully account for the observed antibacterial effects. Instead, PlaF inhibition may sensitize cells to metabolic or envelope stress, consistent with its role in membrane homeostasis. Our results suggest that GW4869’s specificity, while evident for PlaF, may not extend uniformly to all phospholipase homologs, warranting further investigation into their individual susceptibilities. While we cannot exclude contributions from other PLA-active proteins, the consistent correlation between PlaF inhibition in vitro and reduced bacterial growth supports PlaF as a plausible, although not exclusive, target. Although direct biophysical characterization of PlaF-ligand binding is challenging due to the high hydrophobicity of the interacting molecules, such experiments would be valuable for elucidating the mechanism of PlaF targeting by GW4869. GW4869 at 17 µM concentration, which is close to the concentration used for mice treatment (~10 µM, 5 mg/kg) (144), reduced planktonic P. aeruginosa growth to more than 50% within 7 h and drastically reduced biofilm-cell viability to 3% post-treatment. These findings align with prior studies showing impaired biofilm formation in plaF knockout strains (27). Interestingly, GW4869 had no significant impact on Legionella pneumophila replication or outer membrane vesicle secretion in infected human THP-1 monocytes (145), consistent with the absence of PlaF homologs in L. pneumophila (25).
Synergy with antibiotics
GW4869 also demonstrated notable adjuvant activity when combined with imipenem, even at sub-minimum inhibitory concentrations. The synergy might result from reduced production of the imipenem-neutralizing β-lactamase AmpC (146) in the ΔplaF mutant (27). Imipenem disrupts peptidoglycan synthesis by inhibiting penicillin-binding proteins, destabilizing the cell wall (146). Thus, a dual assault on the bacterial envelope due to a putative membrane dysfunction caused by GW4869-mediated PlaF inhibition might enhance bactericidal activity.
Potential alteration of host–pathogen interactions
Rilapladib and darapladib inhibit host lipoprotein-associated PLA2, which releases precursors of inflammatory lipid mediators (56–59). Rilapladib or darapladib would help dampen systemic inflammation and simultaneously slow bacterial proliferation, thus helping clear infections.
GW4869 is a non-competitive inhibitor of neutral sphingomyelinase 2, which regulates the conversion of sphingomyelin to ceramide (74, 75, 147). This mechanism has been shown to inhibit exosome release, which results in impaired pro-tumorigenic macrophage differentiation (148) and suppression of systemic inflammation in the murine sepsis model and LPS treatment (148). These immunomodulatory effects, coupled with its direct activity against P. aeruginosa, suggest that GW4869 may bolster host defenses against severe bacterial infections.
Additionally, it remains to be determined whether GW4869 mediates ceramide reduction during P. aeruginosa infection, as ceramide was shown to promote infection and immune evasion (149, 150).
Future directions
Further preclinical and clinical research is required to determine whether GW4869 could be effectively used alongside antibiotics to improve outcomes in bacterial infections. GW4869 holds promise as an antivirulence compound targeting intracellular bacterial PLAs and host lipid pathways critical to host–pathogen interactions. These findings open novel avenues for the development of innovative therapies targeting intracellular PLAs to combat bacterial infections.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Reynolds D, Kollef M. 2021. The epidemiology and pathogenesis and treatment of Pseudomonas aeruginosa infections: an update. Drugs (Abingdon Engl) 81:2117–2131. doi:10.1007/s 40265-021-01635-6PMC 857214534743315 · doi ↗ · pubmed ↗
- 2Jesudason T. 2024. Antibacterial agents in preclinical and clinical development. The Lancet Microbe 5:100962. doi:10.1016/j.lanmic.2024.10096239102836 · doi ↗ · pubmed ↗
- 3Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, Han C, Bisignano C, Rao P, Wool E, et al.. 2022. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet 399:629–655. doi:10.1016/S 0140-6736(21)02724-0PMC 884163735065702 · doi ↗ · pubmed ↗
- 4Melchiorri D, Rocke T, Alm RA, Cameron AM, Gigante V. 2025. Addressing urgent priorities in antibiotic development: insights from WHO 2023 antibacterial clinical pipeline analyses. The Lancet Microbe 6:100992. doi:10.1016/j.lanmic.2024.10099239454608 PMC 11876093 · doi ↗ · pubmed ↗
- 5Qin S, Xiao W, Zhou C, Pu Q, Deng X, Lan L, Liang H, Song X, Wu M. 2022. Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Sig Transduct Target Ther 7. doi:10.1038/s 41392-022-01056-1PMC 923367135752612 · doi ↗ · pubmed ↗
- 6Elfadadny A, Ragab RF, Al Harbi M, Badshah F, Ibáñez-Arancibia E, Farag A, Hendawy AO, De Los Ríos-Escalante PR, Aboubakr M, Zakai SA, Nageeb WM. 2024. Antimicrobial resistance of Pseudomonas aeruginosa: navigating clinical impacts, current resistance trends, and innovations in breaking therapies. Front Microbiol 15:1374466. doi:10.3389/fmicb.2024.137446638646632 PMC 11026690 · doi ↗ · pubmed ↗
- 7Miethke M, Pieroni M, Weber T, Brönstrup M, Hammann P, Halby L, Arimondo PB, Glaser P, Aigle B, Bode HB, et al.. 2021. Towards the sustainable discovery and development of new antibiotics. Nat Rev Chem 5:726–749. doi:10.1038/s 41570-021-00313-1PMC 837442534426795 · doi ↗ · pubmed ↗
- 8Veesenmeyer JL, Hauser AR, Lisboa T, Rello J. 2009. Pseudomonas aeruginosa virulence and therapy: evolving translational strategies. Crit Care Med 37:1777–1786. doi:10.1097/CCM.0b 013e 31819 ff 13719325463 PMC 2749241 · doi ↗ · pubmed ↗