Microbial landscape: composition and health associations of environmental microbiome in key functional spaces of premium elderly care facilities
Jianlou Yang, Xingsheng Qin, Dianbo Zhang, Chen Dong

TL;DR
This study shows that bacteria in elderly care facilities vary by room type and are influenced by factors like humidity, suggesting tailored cleaning and environmental controls could improve resident health.
Contribution
The study provides a detailed characterization of the environmental microbiome in premium elderly care facilities and identifies space-specific microbial patterns and health implications.
Findings
Dining areas and recreational rooms have higher microbial richness and human-associated taxa like Firmicutes.
Medical facilities and bathrooms have lower diversity but more opportunistic pathogens like Pseudomonas and Klebsiella.
Relative humidity and occupancy are key environmental drivers of microbial community structure.
Abstract
The environmental microbiome in elderly care facilities plays a crucial role in the health of aging populations with immunosenescence; however, its composition and health associations remain underexplored. This study characterizes the microbial ecology of premium elderly care facilities, focusing on key functional spaces, environmental drivers, and implications for resident health. We conducted 16S rRNA gene sequencing (V3–V4 regions) on 320 surface and air samples from six functional spaces (dining areas, medical facilities, bedrooms, bathrooms, recreational rooms, and corridors) across four premium elderly care facilities. Environmental parameters (temperature, humidity, CO₂, and occupancy) were measured concurrently. Bioinformatics analysis (QIIME 2, DADA2, and Silva database) and statistical modeling (permutational multivariate analysis of variance, distance-based redundancy…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
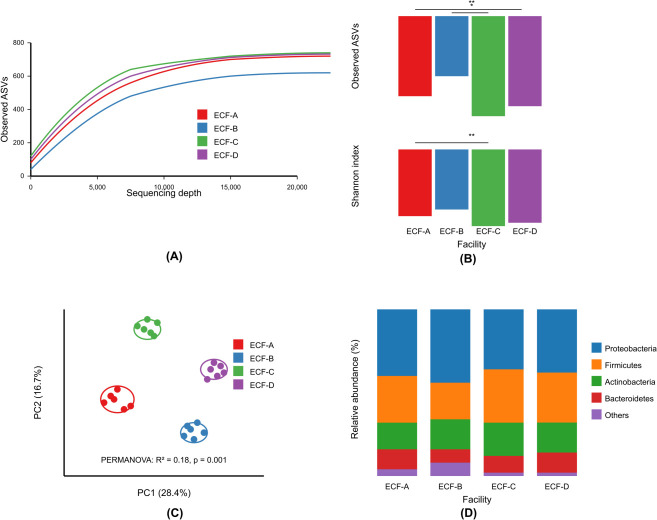 Fig 1
Fig 1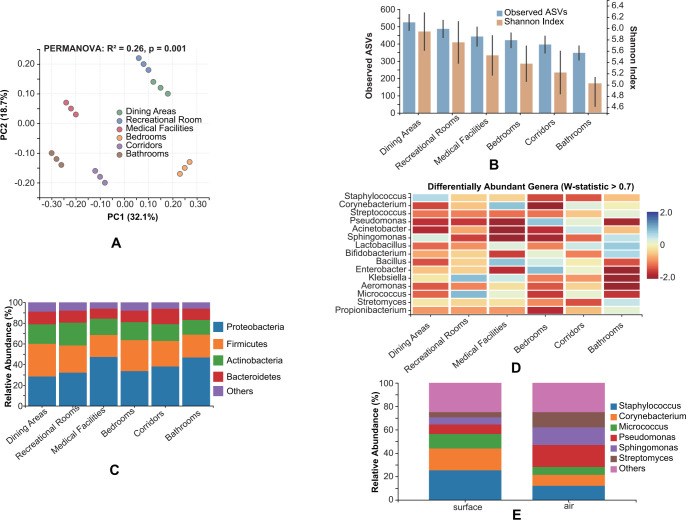 Fig 2
Fig 2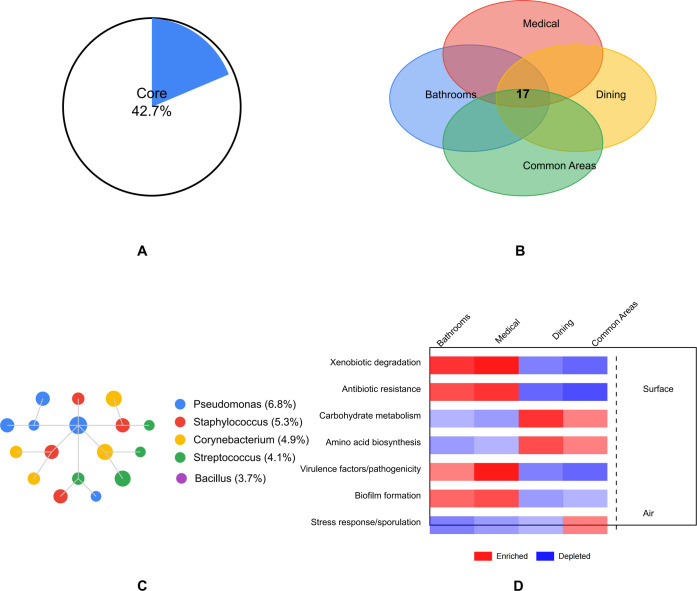 Fig 3
Fig 3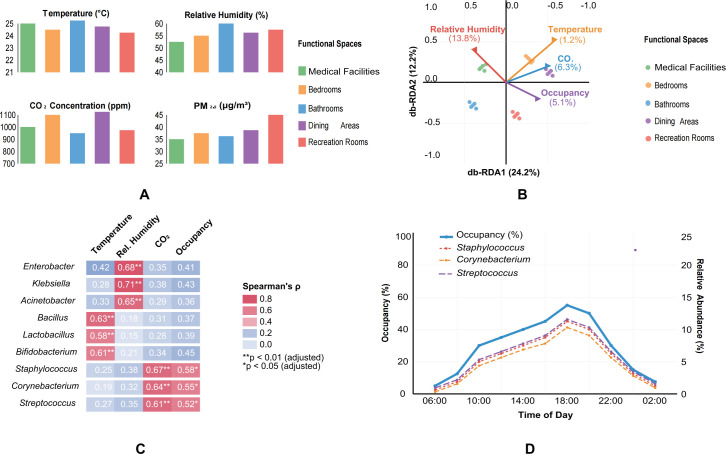 Fig 4
Fig 4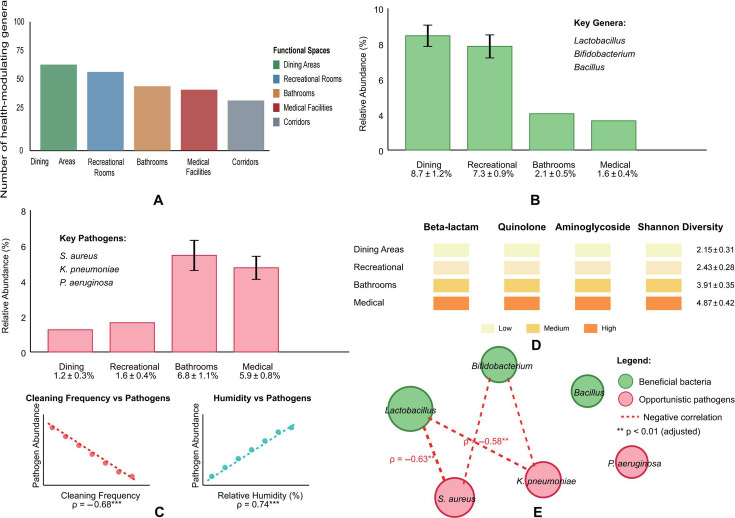 Fig 5
Fig 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIndoor Air Quality and Microbial Exposure · Infection Control in Healthcare · Urinary Tract Infections Management
INTRODUCTION
The global demographic shift toward aging populations has led to a substantial increase in elderly care facilities worldwide (1, 2). In China, the proportion of citizens aged 65 years and older has reached 14.9% in 2023, with projections indicating that this figure will exceed 30% by 2050 (3). Consequently, premium elderly care facilities have emerged as specialized living environments designed to optimize the quality of life for aging populations. These environments represent unique ecological niches where elderly residents, who often have compromised immune systems and specific health vulnerabilities, interact with environmental microbiomes for extended periods (4, 5).
Environmental microbiomes in built environments significantly impact human health, particularly for vulnerable populations such as the elderly (6, 7). Recent advances in next-generation sequencing technologies have enabled a comprehensive characterization of microbial communities in various built environments, including hospitals (8), schools (9), and residential buildings (10). However, there remains a notable knowledge gap regarding the microbial ecology of elderly care facilities, especially premium facilities with specialized functional spaces (11, 12). This gap is particularly significant considering that elderly individuals typically spend 80%–90% of their time indoors and may have altered microbiome interactions due to age-related immunosenescence (13, 14).
The functional spaces within elderly care facilities—such as dining areas, recreational rooms, medical facilities, and bedrooms—serve distinct purposes and potentially harbor unique microbial communities shaped by specific uses, occupancy patterns, and cleaning protocols (15). These spaces may act as reservoirs for various microorganisms, including beneficial commensals, opportunistic pathogens, and microbes with potential health-modulating effects (16, 17). Understanding the composition and dynamics of these microbial communities is crucial for developing evidence-based strategies to create healthier living environments for elderly populations (18).
Previous studies have primarily focused on healthcare-associated infections in nursing homes (19, 20) or specific pathogenic microorganisms in elderly care facilities (21). However, comprehensive characterization of the entire microbial community across different functional spaces in premium elderly care facilities remains unexplored. The transition from pathogen-centric approaches to ecosystem-based perspectives represents a paradigm shift in understanding environmental health in elderly care settings (22, 23).
In this study, we employed 16S rRNA gene sequencing to characterize the environmental microbiome across key functional spaces in four premium elderly care facilities in Jinan, Qingdao, Fuzhou, and Shanghai, China. Our research addresses the following three main questions. (i) What are the compositional characteristics and diversity patterns of microbial communities in different functional spaces of premium elderly care facilities? (ii) How do environmental factors influence the structure and diversity of these microbial communities? (iii) What are the potential implications of these microbial profiles for elderly health and facility management?
By addressing these questions, our study provides novel insights into the microbial ecology of premium elderly care environments, contributing to the development of microbially informed design and management practices that can potentially enhance the health and well-being of elderly residents.
MATERIALS AND METHODS
Study design and sampling sites
Four premium elderly care facilities (designated as ECF-A, ECF-B, ECF-C, and ECF-D) in Jinan, Qingdao, Fuzhou, and Shanghai, China, were selected for this study based on their service quality ratings, occupancy rates (>85%), and operational history (>3 years). For the purposes of this study, “premium” facilities were operationally defined by the following criteria: (i) monthly costs exceeding 5,000 renminbi per resident (approximately 2–3 times the regional average for standard nursing homes); (ii) infrastructure meeting or exceeding national Class A standards for elderly care facilities (24), including individual heating, ventilation, and air conditioning (HVAC) systems, mechanical ventilation with fresh air exchange rates ≥30 m³/h per person, and medical-grade air filtration systems; (iii) staffing ratios of at least 1:3 (staff to residents), compared to typical ratios of 1:6–1:10 in standard facilities; (iv) standardized cleaning protocols with daily disinfection of high-touch surfaces using hospital-grade disinfectants and weekly deep cleaning; and (v) on-site medical facilities staffed by licensed healthcare professionals. These characteristics distinguish premium facilities from standard nursing homes in terms of environmental control, hygiene management, and resource allocation, which may influence microbial community composition. Each facility accommodated between 120 and 180 residents, with an average age of 78.5 ± 6.2 years. The facilities provided comprehensive services including healthcare, rehabilitation, dietary management, and recreational activities. All facilities maintained similar environmental conditions (temperature: 22°C–26°C and relative humidity: 40%–60%) and implemented standardized cleaning protocols according to national elderly care standards (24).
Environmental sampling
Surface samples were collected using sterile nylon flocked swabs (Copan Diagnostics, Italy) pre-moistened with sterile 0.15 M NaCl solution with 0.1% Tween 20 (25). For each functional space, 10 sampling points were selected based on high-touch surfaces and areas with frequent human contact (26). Each sampling point covered approximately 100 cm² using the swabbing protocol described by Adams et al. (27). Swabs were immediately placed in sterile tubes containing 2 mL of RNAlater solution (Thermo Fisher Scientific, USA) and transported to the laboratory on ice within 4 h of collection.
Indoor air samples were collected using a BioSampler SKC (SKC Inc., USA) at a flow rate of 12.5 L/min for 30 min in each functional space (28). The sampler was placed at a height of 1.5 m to represent the breathing zone. Air samples were collected into 20 mL of phosphate-buffered saline (PBS) with 0.005% Tween 80. Upon completion of sampling, the liquid was transferred to sterile 50 mL centrifuge tubes and transported to the laboratory on ice.
Environmental parameters, including temperature, relative humidity, carbon dioxide (CO_2_) concentration, particulate matter (PM_2.5_ and PM_10_) levels, and occupancy counts, were measured simultaneously with microbial sampling using calibrated portable devices (TSI Q-Trak 7575, USA; Dusttrak DRX 8534, USA). These parameters were recorded at 5-min intervals throughout the 30-min sampling period (29).
DNA extraction and 16S rRNA gene sequencing
Sample processing prior to DNA extraction
For surface swab samples, the swabs stored in 2 mL RNAlater solution were first vortexed vigorously for 60 s to release microbial cells from the swab fibers into the liquid. The swab heads were then pressed against the inner wall of the tube while rotating to maximize cell recovery. The swabs were subsequently removed, and the liquid was transferred to sterile 2 mL microcentrifuge tubes. Samples were centrifuged at 10,000 × g for 10 min at 4°C to pellet microbial cells. The supernatant was carefully removed, leaving approximately 100 μL of liquid with the cell pellet to prevent sample loss. For air samples collected in PBS, the 20 mL liquid samples were first concentrated by centrifugation at 5,000 × g for 15 min at 4°C. The supernatant was discarded, retaining only the pellet and approximately 200 μL of residual liquid. If the pellet was not visible, the sample was subjected to a second centrifugation step at 10,000 × g for 10 min. The concentrated pellets from both surface and air samples were then resuspended in 200 μL of PBS by gentle pipetting before proceeding to DNA extraction.
DNA extraction protocol
Total genomic DNA was extracted from the concentrated swab and air samples using the DNeasy PowerSoil Pro Kit (Qiagen, Germany) following the manufacturer’s instructions with minor modifications as described below. Briefly, the 200 μL resuspended cell pellet was added to the PowerBead Pro Tube containing ceramic beads provided in the kit. An additional 550 μL of CD1 solution (lysis buffer) was added to each tube. Samples were subjected to bead-beating using a FastPrep-24 5G instrument (MP Biomedicals, USA) at 6.0 m/s for 45 s, followed by a 5-min incubation at room temperature. This bead-beating step was repeated once to ensure thorough cell lysis, particularly for gram-positive bacteria with robust cell walls.
Following bead-beating, the samples were briefly centrifuged at 15,000 × g for 1 min to pellet beads and debris. Up to 600 μL of the supernatant was transferred to a clean 2 mL microcentrifuge tube. Then, 200 μL of CD2 solution (inhibitor removal solution) was added, and the samples were vortexed for 5 s before incubation at 4°C for 10 min to precipitate inhibitors. Samples were centrifuged at 15,000 × g for 2 min, and up to 750 μL of supernatant was transferred to a new 2 mL microcentrifuge tube, avoiding the pellet.
Subsequently, 650 μL of CD3 solution (binding buffer) was added to the supernatant and mixed by vortexing for 5 s. The entire sample (approximately 1,400 μL) was loaded onto a MB Spin Column in two sequential applications, with centrifugation at 15,000 × g for 1 min between loadings. The flow-through was discarded after each centrifugation.
The MB Spin Column was washed twice: first with 500 μL of EA1 solution (wash buffer 1) and then with 500 μL of EA2 solution (wash buffer 2), with centrifugation at 15,000 × g for 1 min after each wash. After the final wash, the column was centrifuged for an additional 2 min at 15,000 × g to remove residual ethanol from the wash buffers.
Finally, the MB Spin Column was placed in a clean 2 mL microcentrifuge tube, and 50 μL of EB solution (elution buffer, prewarmed to 56°C) was applied directly to the center of the membrane. The column was incubated at room temperature for 5 min to maximize DNA yield and then centrifuged at 15,000 × g for 1 min to elute the DNA. This elution step was repeated once with an additional 50 μL of EB solution to maximize DNA recovery, resulting in a final elution volume of approximately 100 μL.
DNA concentration and purity were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) and Qubit 4.0 Fluorometer with the dsDNA HS Assay Kit (Invitrogen, USA). Only samples with DNA concentrations >5 ng/µL and A260/A280 ratios between 1.8 and 2.0 were used for subsequent analysis. Extracted DNA was stored at −80°C until PCR amplification. To minimize freeze-thaw cycles, DNA samples were aliquoted into multiple tubes (10 μL per tube) for PCR setup.
The V3–V4 hypervariable regions of the bacterial 16S rRNA gene were amplified using the universal primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) (30). It should be noted that while the 341F/805R primer pair provides broad coverage of bacterial diversity, certain taxa within Proteobacteria (e.g., some SAR11 clade members) and Firmicutes (e.g., certain Clostridiales) may be underrepresented due to mismatches in primer binding sites (30). This primer bias, common to all 16S rRNA-based approaches, may influence the completeness of taxonomic profiles and, consequently, the accuracy of downstream functional predictions based on these profiles. PCR amplification was performed in triplicate for each sample in a 25 μL reaction mixture containing 12.5 μL of 2× Phusion High-Fidelity PCR Master Mix (New England Biolabs, USA), 1 μL of each primer (10 μM), 2 μL of template DNA, and 8.5 μL of nuclease-free water. The thermal cycling conditions were as follows: initial denaturation at 98°C for 30 s; 30 cycles of denaturation at 98°C for 10 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s; and a final extension at 72°C for 5 min (31).
The triplicate PCR products were pooled, purified using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific, USA), and quantified using the Qubit 4.0 Fluorometer. Equimolar amounts of purified amplicons were pooled and paired-end sequenced (2 × 300 bp) on an Illumina MiSeq platform (Illumina Inc., USA) according to standard protocols (32).
Bioinformatic analysis
Raw sequencing data were processed using the QIIME 2 pipeline (version 2023.5) (33). After demultiplexing, the sequences were quality-filtered, denoised, merged, and chimera-filtered using the DADA2 plugin (34) with the following parameters: truncation length of 270 bp and 210 bp for forward and reverse reads, respectively; maximum expected error of 2.0; and truncation quality score threshold of 25. The resulting amplicon sequence variants (ASVs) were taxonomically classified using the Silva database (release 138.1) with a confidence threshold of 0.8 (35).
Alpha diversity metrics, including observed ASVs, Shannon diversity index, Faith’s phylogenetic diversity, and Pielou’s evenness, were calculated using the q2-diversity plugin in QIIME 2. Beta diversity was assessed using weighted and unweighted UniFrac distances (36) and visualized using principal coordinates analysis (PCoA). To identify differentially abundant taxa between functional spaces, analysis of composition of microbiomes (ANCOM) was implemented in QIIME 2 with a W-statistic cutoff of 0.7 (37).
Functional profiles of the microbial communities were predicted using PICRUSt2 (38) with default parameters. The predicted gene families were categorized into KEGG Orthology (KO) groups and pathways. Additionally, potential pathogenic bacteria were identified through taxonomic assignment and comparison with the Virulence Factor Database (VFDB) (39).
Statistical analysis
All statistical analyses were performed in R (version 4.2.1) (40) using the phyloseq (41), vegan (42), and DESeq2 (43) packages. Differences in alpha diversity metrics among facility types and functional spaces were evaluated using one-way ANOVA, followed by Tukey’s honestly significant difference (HSD) post-hoc test for normally distributed data or Kruskal-Wallis followed by Dunn’s test for non-normally distributed data. Normality was assessed using the Shapiro-Wilk test.
Permutational multivariate analysis of variance (PERMANOVA) with 999 permutations was used to test the significance of differences in microbial community composition between facility types and functional spaces (44). Distance-based redundancy analysis (db-RDA) was applied to examine the relationships between environmental parameters and microbial community structure (45). Spearman’s rank correlation analysis was used to assess associations between specific taxa and environmental parameters. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg procedure, with adjusted P < 0.05 considered statistically significant.
Network analysis was performed using the SPIEC-EASI (SParse InversE Covariance Estimation for Ecological Association Inference) method (46) to identify significant co-occurrence relationships between microbial taxa. Network properties, including modularity, connectivity, and centrality measures, were calculated using the igraph package in R (47).
RESULTS
Overall microbial diversity comparison among four elderly care facilities
The environmental microbiome sequencing analysis yielded a total of 3,842,156 high-quality sequences across all samples, resulting in 9,873 ASVs after quality filtering and chimera removal. Rarefaction curves reached saturation at approximately 15,000 sequences per sample, indicating sufficient sequencing depth to capture the microbial diversity (Fig. 1A).
Overall microbial diversity comparison among four elderly care facilities. (A) Rarefaction curves. (B) Alpha diversity metrics. (C) PCoA of weighted UniFrac distances. (D) Phylum-level composition.
Alpha diversity metrics revealed significant differences in microbial community richness and diversity among the four elderly care facilities (ECF-A, ECF-B, ECF-C, and ECF-D) (Fig. 1B). ECF-C exhibited the highest microbial richness (observed ASVs: 482.3 ± 56.7) and Shannon diversity index (5.84 ± 0.41), followed by ECF-D (observed ASVs: 437.8 ± 62.3; Shannon index: 5.62 ± 0.38), ECF-A (observed ASVs: 398.5 ± 48.2; Shannon index: 5.43 ± 0.45), and ECF-B (observed ASVs: 356.9 ± 53.8; Shannon index: 5.21 ± 0.53) (ANOVA, P < 0.01 for both metrics). Faith’s phylogenetic diversity showed a similar pattern, with ECF-C displaying the highest values (42.7 ± 4.1), significantly different from ECF-B (35.8 ± 3.9) (Tukey’s HSD, adjusted P < 0.05).
Beta diversity analysis using weighted UniFrac distances revealed distinct clustering patterns among the four facilities (Fig. 1C). PERMANOVA analysis confirmed significant differences in microbial community composition between facilities (R^2^ = 0.18, P = 0.001). Pairwise comparisons revealed that all facilities differed significantly from each other (adjusted P < 0.05), with the greatest dissimilarity observed between ECF-B and ECF-C (R^2^ = 0.24, P = 0.001).
Despite these differences, all facilities shared a common core microbiome at the phylum level, dominated by Proteobacteria (38.4%–45.7%), Firmicutes (21.3%–28.5%), Actinobacteria (12.6%–18.1%), and Bacteroidetes (8.2%–12.3%), which collectively accounted for approximately 85% of the total microbial community (Fig. 1D).
Given that the four facilities were located in cities spanning different geographic and climatic zones (Jinan: temperate continental climate; Qingdao: temperate monsoon climate with maritime influence; Fuzhou: subtropical monsoon climate; and Shanghai: subtropical humid climate), we performed additional analyses to assess potential regional effects on microbial community composition. PERMANOVA analysis revealed significant but modest differences in microbial communities among cities (R² = 0.12, P = 0.002), which was considerably lower than the variation explained by functional space types (R² = 0.26, P = 0.001) described in subsequent sections. Pairwise comparisons showed that facilities in Fuzhou (subtropical) exhibited slightly higher relative abundance of thermophilic taxa compared to facilities in Qingdao and Jinan (temperate zones) (adjusted P < 0.05), likely reflecting regional temperature differences. However, the core microbiome composition at the phylum level remained consistent across all four cities, with no city-specific phyla detected. These findings suggest that while geographic location and outdoor climate exert some influence on indoor microbial communities, the functional characteristics of indoor spaces represent the dominant driver of microbial community structure in elderly care facilities, consistent with previous studies showing that building function and occupancy patterns override regional biogeography in shaping indoor microbiomes (15, 48, 49).
Microbial compositional characteristics and differences across functional spaces
Microbial community structure exhibited significant variations across the six functional spaces (dining areas, recreational rooms, medical facilities, bedrooms, corridors, and bathrooms) within the elderly care facilities (PERMANOVA, R² = 0.26, P = 0.001) (Fig. 2A). This spatial heterogeneity was more pronounced than the differences observed between facilities or geographic locations (city effect: R² = 0.12, P = 0.002), indicating that space functionality had a stronger influence on microbial composition than either facility-specific factors or regional biogeography.
Microbial compositional characteristics and differences across functional spaces. (A) Principal coordinate analysis (PCoA) of microbial communities. (B) Alpha diversity metrics across functional spaces. (C) Phylum-level taxonomic composition. (D) Differentially abundant genera (ANCOM analysis). (E) Surface vs air sample comparison.
The dining areas displayed the highest alpha diversity metrics (observed ASVs: 524.1 ± 48.9; Shannon index: 6.07 ± 0.37), followed by recreational rooms (observed ASVs: 486.3 ± 52.4; Shannon index: 5.86 ± 0.41) and medical facilities (observed ASVs: 441.7 ± 57.8; Shannon index: 5.61 ± 0.39). Bathrooms exhibited the lowest diversity (observed ASVs: 347.2 ± 43.6; Shannon index: 5.03 ± 0.47) (Fig. 2B). Significant differences were observed between dining areas and bathrooms for all alpha diversity metrics (Tukey’s HSD, adjusted P < 0.01).
Taxonomic analysis at the phylum level revealed distinct compositional patterns across functional spaces (Fig. 2C). Proteobacteria dominated in medical facilities (47.3% ± 5.1%) and bathrooms (46.8% ± 4.8%), while Firmicutes were more abundant in dining areas (31.5% ± 3.7%) and bedrooms (29.8% ± 4.2%). Actinobacteria showed the highest relative abundance in recreational rooms (22.1% ± 2.9%), and Bacteroidetes were most prevalent in corridors (14.8% ± 2.3%).
At the genus level, ANCOM analysis identified 28 differentially abundant taxa across functional spaces (W-statistic > 0.7, Fig. 2D). Notably, Staphylococcus, Corynebacterium, and Streptococcus were significantly enriched in bedrooms (adjusted P < 0.01), while Pseudomonas, Acinetobacter, and Sphingomonas dominated in medical facilities (adjusted P < 0.01). Dining areas showed higher abundance of Lactobacillus, Bifidobacterium, and Bacillus (adjusted P < 0.05), whereas bathrooms were characterized by elevated levels of Enterobacter, Klebsiella, and Aeromonas (adjusted P < 0.01).
Surface and air samples exhibited distinct microbial profiles within each functional space (Fig. 2E). Surface samples contained higher proportions of skin-associated genera such as Staphylococcus, Corynebacterium, and Micrococcus, while air samples were enriched with environmental genera including Pseudomonas, Sphingomonas, and Streptomyces. This pattern was consistent across all functional spaces, suggesting different sources and dispersal mechanisms for surface and airborne microbiota.
Core microbiome identification and functional prediction
Network analysis identified 83 ASVs as the core microbiome (present in >75% of all samples with relative abundance >0.1%), representing approximately 42.7% of the total microbial community (Fig. 3A). These core taxa formed a highly connected network with 376 significant co-occurrence relationships (Fig. 3B). The network exhibited a modular structure (modularity = 0.42) with five distinct clusters, suggesting potential ecological interactions and niche partitioning within the microbial community.
Core microbiome identification and functional prediction. (A) Core microbiome (83 ASVs). (B) Network of core taxa (376 connections). (C) Shared core taxa across functional spaces. (D) Predicted functional pathways.
The core microbiome was dominated by genera known to be widespread in built environments, including Pseudomonas (6.8%), Staphylococcus (5.3%), Corynebacterium (4.9%), Streptococcus (4.1%), and Bacillus (3.7%). Each functional space harbored a distinct subset of core taxa, with 17 ASVs shared across all spaces, representing a facility-wide core microbiome (Fig. 3C).
Functional prediction using PICRUSt2 revealed significant differences in the predicted metabolic potential across functional spaces (Fig. 3D). Based on taxonomic composition, the microbial communities in bathrooms and medical facilities were predicted to have higher relative abundance of genes related to xenobiotic degradation (KEGG pathway ko01220) and antibiotic resistance (KEGG pathway ko01501) (adjusted P < 0.01). Dining areas displayed higher abundance of genes involved in carbohydrate metabolism (KEGG pathway ko01200) and amino acid biosynthesis (KEGG pathway ko01230) (adjusted P < 0.05). Notably, medical facilities were predicted to harbor microbial communities with the highest proportion of genes associated with virulence factors and pathogenicity (KEGG pathway ko05111) (adjusted P < 0.01). It is important to note that these predictions represent the functional potential of the microbial communities based on reference genomes and do not confirm the actual expression or functional activity of these resistance genes.
Significant differences were also observed in the predicted functional capacity between surface and air microbiomes. Surface microbiomes were predicted to show enrichment in genes related to biofilm formation (KEGG pathway ko05111) and antimicrobial resistance (KEGG pathway ko01501), while air microbiomes were predicted to be characterized by higher abundance of genes involved in stress response (KEGG pathway ko04010) and sporulation (KEGG pathway ko02020) (adjusted P < 0.01).
Correlation between environmental parameters and microbial composition
Environmental parameters varied significantly across functional spaces (Fig. 4A). Medical facilities maintained the lowest temperature (22.4°C ± 0.8°C) and relative humidity (42.3% ± 3.5%), while bathrooms exhibited the highest humidity levels (58.7% ± 4.2%). CO₂ concentrations were highest in bedrooms (972.6 ± 86.3 ppm) and dining areas during mealtimes (1,023.5 ± 112.8 ppm). PM₂.₅ levels were elevated in recreational rooms (38.6 ± 5.7 μg/m³) compared to other spaces (Kruskal-Wallis, P < 0.01).
Correlation between environmental parameters and microbial composition across functional spaces. (A) Environmental parameters by functional space. (B) Distance-based redundancy analysis (db-RDA). (C) Spearman’s correlation between environmental parameters and microbial taxa. (D) Temporal variations in occupancy and human-associated taxa.
db-RDA revealed that environmental parameters explained approximately 36.4% of the total variation in microbial community composition (Fig. 4B). Relative humidity and temperature emerged as the strongest predictors (13.8% and 11.2% of variation, respectively), followed by CO₂ concentration (6.3%) and occupancy (5.1%) (PERMANOVA, P < 0.01 for all parameters).
Spearman’s correlation analysis identified significant associations between specific environmental parameters and microbial taxa (Fig. 4C). Relative humidity showed significant positive correlations with the relative abundance of Enterobacter, Klebsiella, and Acinetobacter (Spearman’s ρ > 0.65, adjusted P < 0.01), while temperature was positively correlated with Bacillus, Lactobacillus, and Bifidobacterium (Spearman’s ρ > 0.58, adjusted P < 0.01). These associations suggest, but do not prove, that these environmental parameters influence the abundance of specific taxa. CO₂ concentration showed strong positive correlations with human-associated genera such as Staphylococcus, Corynebacterium, and Streptococcus (Spearman’s ρ > 0.61, adjusted P < 0.01).
Occupancy patterns also significantly influenced microbial community structure. Spaces with higher occupancy rates showed increased alpha diversity (Shannon index; Spearman’s ρ = 0.72, adjusted P < 0.001) and elevated relative abundance of human-associated taxa. Temporal variations in occupancy throughout the day correlated with fluctuations in the relative abundance of several human-associated genera, including Staphylococcus, Streptococcus, and Corynebacterium (Fig. 4D).
Distribution patterns of potentially beneficial bacteria and opportunistic pathogens
Analysis of the taxonomic profiles identified 37 genera with known health-modulating properties, including potentially beneficial bacteria and opportunistic pathogens (Fig. 5A). The relative abundance and distribution of these taxa varied significantly across functional spaces (PERMANOVA, R^2^ = 0.31, P = 0.001).
Distribution patterns of potentially beneficial bacteria and opportunistic pathogens. (A) Health-modulating genera distribution. (B) Potentially beneficial bacteria. (C) Opportunistic pathogens. (D) Antibiotic resistance gene (ARG) distribution. (E) Co-occurrence patterns between beneficial bacteria and pathogens.
Potentially beneficial bacteria, including Lactobacillus, Bifidobacterium, and Bacillus, were predominantly found in dining areas (collective relative abundance: 8.7% ± 1.2%) and recreational rooms (7.3% ± 0.9%) (Fig. 5B). These genera are known for their probiotic properties and potential health benefits for elderly populations. Lactobacillus species were particularly abundant on dining tables and food preparation surfaces (relative abundance: 4.2% ± 0.7%).
In contrast, opportunistic pathogens such as Staphylococcus aureus, Klebsiella pneumoniae, and Pseudomonas aeruginosa (identified through species-level classification and comparison with the VFDB) were more prevalent in bathrooms (collective relative abundance: 6.8% ± 1.1%) and medical facilities (5.9% ± 0.8%) (Fig. 5C). Notably, the relative abundance of these opportunistic pathogens showed significant negative correlation with cleaning frequency (Spearman’s ρ = −0.68, adjusted P < 0.001) and positively correlated with relative humidity (Spearman’s ρ = 0.74, adjusted P < 0.001). While these correlations are strong, the observational nature of our study precludes causal inference.
The predicted presence and diversity of antibiotic resistance genes (ARGs) inferred from PICRUSt2 analysis showed varying distribution patterns across functional spaces (Fig. 5D). Based on the taxonomic composition, medical facilities were predicted to harbor the highest diversity and abundance of ARGs (Shannon diversity of ARG profiles: 4.87 ± 0.42), particularly genes potentially conferring resistance to beta-lactams, quinolones, and aminoglycosides. It is important to note that these predictions represent the functional potential of the microbial communities based on reference genomes and do not confirm the actual expression or functional activity of these resistance genes. Bathrooms ranked second in ARG abundance, while dining areas exhibited the lowest levels.
The co-occurrence patterns between potentially beneficial bacteria and opportunistic pathogens revealed interesting ecological relationships (Fig. 5E). Significant negative correlations were observed between Lactobacillus and Bifidobacterium and S. aureus (Spearman’s ρ = −0.63, adjusted P < 0.01) as well as K. pneumoniae (Spearman’s ρ = −0.58, adjusted P < 0.01). While these negative correlations are consistent with potential competitive or antagonistic interactions between beneficial and pathogenic bacteria within the built environment of elderly care facilities, it is important to note that such patterns in relative abundance data may also arise from compositional effects rather than genuine biological interactions and would require validation with absolute quantification methods.
DISCUSSION
This study provides a comprehensive characterization of the environmental microbiome across different functional spaces in premium elderly care facilities, revealing distinct microbial communities shaped by space functionality, environmental parameters, and human occupancy patterns. Our findings contribute to the growing body of knowledge on microbial ecology in built environments and offer insights into potential implications for elderly health and facility management.
Spatial heterogeneity of microbial communities
The observed spatial heterogeneity in microbial communities across different functional spaces aligns with previous studies on built environments (48, 49). However, our finding that space functionality exerts a stronger influence on microbial composition than facility-specific factors represents a novel contribution to understanding microbial ecology in elderly care settings. This pattern suggests that the activities and purposes associated with each space create unique microbial niches, potentially through differences in human behavior, materials, cleaning protocols, and environmental conditions (50). An important finding of our study is that space functionality exerted a substantially stronger influence on microbial composition (R² = 0.26) than geographic location/city (R² = 0.12), despite the four facilities being located in cities with distinct climatic zones ranging from temperate continental to subtropical climates. This pattern indicates that the microbial ecology of indoor environments in elderly care facilities is primarily shaped by anthropogenic factors—such as space use, occupancy, and management practices—rather than by regional biogeography or outdoor microbial sources (15, 48). The observed consistency in core microbiome composition across geographic regions, combined with pronounced differences across functional spaces within facilities, supports the concept that built environments create unique selective pressures that homogenize microbial communities across geographic scales while differentiating them at functional scales (49, 51). This finding has important implications for facility management, suggesting that standardized, space-specific protocols may be broadly applicable across different geographic regions, although local climate factors (e.g., humidity influenced by regional weather patterns) should still be considered in implementation (52).
The highest microbial diversity observed in dining areas and recreational rooms likely reflects the convergence of diverse microbial sources, including food-associated microorganisms, human-associated taxa from multiple occupants, and environmental species (53). These spaces function as community hubs with high occupancy rates and diverse activities, fostering rich microbial ecosystems. Conversely, the lower diversity in bathrooms and medical facilities may result from more stringent cleaning protocols, antimicrobial surface materials, and environmental conditions that select for specific microbial taxa (54, 55).
The taxonomic composition across functional spaces revealed noteworthy patterns. The predominance of Proteobacteria in medical facilities and bathrooms aligns with previous studies reporting high abundance of this phylum in healthcare environments and moisture-rich settings (56, 57). These environments may select for gram-negative bacteria with higher tolerance to disinfectants and antimicrobial agents commonly used in these spaces (58). The enrichment of Firmicutes in dining areas and bedrooms likely reflects human influence, as this phylum includes many human-associated genera such as Staphylococcus and Streptococcus (59). The higher abundance of Actinobacteria in recreational rooms may be related to the diverse materials present in these spaces, as members of this phylum are known to degrade complex organic compounds (60).
Environmental drivers of microbial community structure
The significant influence of environmental parameters on microbial community composition underscores the importance of indoor environmental quality in shaping microbial ecosystems. Relative humidity emerged as the strongest predictor of microbial community structure, consistent with previous research demonstrating humidity as a critical factor affecting microbial growth and survival in indoor environments (61, 62). This finding aligns with a continental-scale study of hotel rooms across China, which identified relative humidity and temperature as primary environmental determinants of microbial richness and composition, with humidity showing particularly strong effects on community structure (61). The consistency of humidity effects across different building types (hotels and elderly care facilities) and geographic scales suggests universal principles governing indoor microbial ecology. The positive correlation between relative humidity and opportunistic pathogens such as Enterobacter, Klebsiella, and Acinetobacter is particularly notable, suggesting that moisture control may be an effective strategy for reducing the prevalence of these potentially harmful microorganisms (63). However, it is important to note that our cross-sectional, observational study design does not establish causation, and experimental or intervention studies would be necessary to confirm whether humidity control directly reduces pathogen abundance.
Temperature also significantly influenced microbial community composition, with thermophilic genera like Bacillus showing positive associations with higher temperatures. This finding aligns with the known temperature preferences of these microorganisms and suggests the potential for temperature regulation as a microbial management strategy (64), although controlled intervention studies would be needed to establish causal relationships. The correlations between CO₂ concentration and human-associated genera further demonstrate the impact of occupancy on microbial communities, supporting the concept that humans serve as important sources and vectors for microorganisms in built environments (15, 65).
The temporal variations in microbial community structure associated with occupancy patterns reveal the dynamic nature of indoor microbiomes. These findings suggest that microbial communities in elderly care facilities undergo daily fluctuations in response to human activities, a phenomenon previously observed in other built environments (9, 66). This temporal dimension adds complexity to environmental microbiome management and highlights the need for adaptive strategies that account for occupancy patterns.
Implications of core microbiome and functional potential
The identification of a core microbiome across all functional spaces suggests the existence of a stable microbial community adapted to the general conditions of elderly care facilities. This core community, dominated by widespread environmental and human-associated genera such as Pseudomonas, Staphylococcus, and Corynebacterium, may represent key microbial taxa that persist despite variations in space functionality and environmental conditions (51). The modular structure of the co-occurrence network indicates potential ecological interactions and niche partitioning within this core community, contributing to its stability (67).
The predicted functional profiles revealed significant differences in metabolic potential across functional spaces, providing insights into the ecological roles of these microbial communities. The enrichment of genes related to xenobiotic degradation and antibiotic resistance in bathrooms and medical facilities suggests adaptation to the frequent use of cleaning agents and antimicrobials in these spaces (68). This finding raises concerns about these environments potentially serving as reservoirs for antimicrobial resistance genes, a significant public health issue, particularly in healthcare settings (69).
The higher abundance of genes involved in carbohydrate metabolism and amino acid biosynthesis in dining areas likely reflects adaptation to nutrient-rich conditions from food residues (70). Similarly, the enrichment of biofilm formation genes in surface microbiomes compared to air microbiomes demonstrates adaptation to different lifestyle strategies based on habitat (71). These functional differences highlight the specialized roles that microorganisms play in different microhabitats within elderly care facilities. However, it is crucial to emphasize that these functional predictions are based on taxonomic composition and reference genomes and do not represent direct measurements of gene presence or expression. The predicted presence of antibiotic resistance genes, in particular, does not necessarily indicate functional resistance phenotypes, as gene expression, regulation, and horizontal gene transfer dynamics are not captured by 16S rRNA-based predictions (38, 72). Therefore, while these predictions provide valuable hypotheses about the functional potential of microbial communities, they should be interpreted cautiously and validated through metagenomics, metatranscriptomics, or culture-based approaches in future studies.
Health implications of environmental microbiome
The distribution patterns of potentially beneficial bacteria and opportunistic pathogens across functional spaces have important implications for elderly health. The higher abundance of potentially beneficial bacteria such as Lactobacillus and Bifidobacterium in dining areas may contribute to a healthier microbiome environment in spaces where food consumption occurs (73). These bacteria are known for their probiotic properties and may help maintain a balanced microbial ecosystem through competitive exclusion of pathogenic species (74).
Conversely, the higher prevalence of opportunistic pathogens in bathrooms and medical facilities highlights these spaces as potential hotspots for harmful microorganisms (23). The negative correlation between cleaning frequency and the relative abundance of these pathogens is consistent with the hypothesis that regular cleaning may reduce potential health risks. Similarly, the positive correlation with relative humidity suggests that moisture control may complement cleaning protocols to help manage pathogen levels (75). However, these correlational observations do not prove causation, and controlled intervention studies are needed to establish the actual effectiveness of these management strategies in reducing pathogen abundance and associated health risks.
The negative correlations observed between beneficial bacteria and opportunistic pathogens are suggestive of potential ecological interventions to promote healthier indoor microbiomes. For instance, if these negative correlations reflect true competitive or antagonistic interactions, encouraging the growth of beneficial bacteria through environmental management may potentially help suppress pathogen abundance through competitive interactions (76, 77). However, it is critical to acknowledge that our study employed 16S rRNA gene sequencing, which provides relative abundance data rather than absolute cell counts or biomass measurements. Negative correlations in compositional data can arise from mathematical constraints of relative abundances (i.e., the closed sum problem) rather than genuine biological antagonism (67). For example, a dramatic increase in the absolute abundance of one taxon will necessarily decrease the relative abundances of all other taxa, even if their absolute abundances remain unchanged or increase modestly. Therefore, while the observed negative correlations between Lactobacillus/Bifidobacterium and S. aureus/K. pneumoniae are consistent with competitive exclusion or antagonistic interactions, they could also represent compositional artifacts. Definitive establishment of antagonistic relationships would require absolute quantification approaches, such as quantitative PCR (qPCR) targeting specific taxa, flow cytometry with microbial cell counts, or culture-based enumeration methods (67). Despite this limitation, the ecological hypothesis that beneficial bacteria may competitively exclude pathogens remains plausible and warrants investigation through experimental validation studies. This approach represents a shift from conventional hygiene practices focused solely on reducing microbial load to strategies that promote balanced microbial communities (78).
The predicted enrichment of antibiotic resistance genes across different functional spaces, with highest inferred abundance in medical facilities, aligns with patterns observed in hospital environments (22). However, it is crucial to emphasize that these functional predictions are based on taxonomic composition and reference genomes and do not represent direct measurements of gene presence or expression. The predicted presence of antibiotic resistance genes, in particular, does not necessarily indicate functional resistance phenotypes, as gene expression, regulation, and horizontal gene transfer dynamics are not captured by 16S rRNA-based predictions (38, 72). Therefore, while these predictions provide valuable hypotheses about the functional potential of microbial communities, they should be interpreted cautiously and validated through metagenomics, metatranscriptomics, or culture-based approaches in future studies. Our findings are further supported by a global urban microbiome study that identified environmental factors (particularly humidity and antimicrobial use) and demographic characteristics as key drivers of ARG distribution in built environments across multiple continents (69). That study’s demonstration of significant spatial heterogeneity in ARG profiles across different urban settings parallels our observation of space-specific ARG enrichment patterns within elderly care facilities. While these predictions suggest potential ARG reservoirs, direct metagenomic sequencing would be necessary to confirm the actual presence and diversity of resistance genes (72). While these predictions suggest potential ARG reservoirs, direct metagenomic sequencing would be necessary to confirm the actual presence and diversity of resistance genes (72). This finding underscores the need for antimicrobial stewardship and targeted cleaning protocols in these areas to minimize the spread of resistance genes. The co-occurrence patterns between antibiotic resistance genes and specific taxa provide insights into potential reservoirs and vectors for these genes within the built environment (79).
Practical implications for elderly care facility management
Our findings have several practical implications for the design and management of elderly care facilities. The distinct microbial communities associated with different functional spaces suggest that cleaning protocols and environmental management strategies should be space-specific rather than facility-wide (18). For example, based on the strong positive correlations we observed, more stringent humidity control in bathrooms and medical facilities may help reduce the prevalence of moisture-loving opportunistic pathogens, while alternative approaches focusing on microbial balance might be more appropriate for dining areas and recreational rooms. However, intervention studies are needed to confirm the causal effects of these management strategies.
The significant influence of environmental parameters on microbial community structure highlights the importance of indoor environmental quality management. Given the significant correlations between environmental parameters and microbial community structure, maintaining optimal temperature and humidity levels, ensuring adequate ventilation to control CO₂ concentrations, and implementing occupancy-based ventilation strategies may help create healthier microbial environments (52, 80), although these recommendations should be validated through controlled interventions. These approaches align with the concept of "bioinformed design," which incorporates microbial ecology principles into building design and management (81).
The temporal dynamics of microbial communities in response to occupancy patterns suggest that cleaning schedules should be aligned with periods of high human activity (82). Additionally, the use of real-time monitoring of environmental parameters may inform adaptive management strategies that respond to changing conditions throughout the day (83), although the effectiveness of such approaches in modulating microbial communities requires empirical testing. Such approaches would be particularly valuable in spaces with fluctuating occupancy, such as dining areas and recreational rooms.
The potential health implications of environmental microbiomes underscore the need for a balanced approach to hygiene in elderly care facilities. While controlling potentially harmful microorganisms remains important, especially for immunocompromised elderly residents, maintaining diverse and balanced microbial communities may also contribute to health benefits (84, 85). This perspective aligns with recent theoretical advances in indoor microbiome research, which emphasize prevention strategies that promote beneficial microbial communities rather than solely focusing on pathogen elimination (75). Such strategies, including targeted probiotic applications and microbiome-informed ventilation protocols, represent promising approaches for elderly care environments where residents may be particularly susceptible to microbiome-related health effects. This perspective represents a paradigm shift from viewing all microorganisms as threats to recognizing their ecological roles and potential contributions to human health (86).
Limitations and future directions
Despite the comprehensive nature of this study, several limitations should be acknowledged. First, while 16S rRNA gene sequencing provides valuable information about bacterial community composition, it does not capture other microorganisms such as fungi, viruses, and archaea, which are also important components of indoor microbiomes (87). Future studies should employ metagenomic approaches to characterize the complete microbial community, including non-bacterial members and their functional potential (88).
Furthermore, our reliance on 16S rRNA gene sequencing for microbial community characterization means that all abundance data are compositional (relative abundances) rather than absolute. This creates inherent challenges in interpreting correlations between taxa, particularly negative correlations that we interpreted as potentially indicating antagonistic relationships. The compositional nature of sequencing data imposes a mathematical constraint, whereby all relative abundances must sum to 100%, meaning that changes in one taxon necessarily affect the relative abundances of all others, even in the absence of biological interactions. This “closed sum” problem can generate spurious negative correlations that mimic competitive or antagonistic relationships (67). To definitively establish whether the negative correlations we observed between beneficial bacteria (e.g., Lactobacillus) and opportunistic pathogens (e.g., S. aureus) represent genuine biological antagonism or compositional artifacts, future studies should incorporate absolute quantification methods such as quantitative PCR, flow cytometry, or spike-in standards during sequencing library preparation. Such approaches would enable differentiation between scenarios where both taxa decline in absolute abundance versus scenarios where one increases while the other remains constant or decreases, thereby clarifying the nature of their ecological relationships (46).
Meanwhile, we acknowledge a methodological limitation regarding the absence of negative controls (e.g., blank swabs, DNA extraction kit blanks, and PCR-negative controls) in our sampling and laboratory procedures. In low-biomass environmental microbiome studies, contamination from reagents, consumables, and laboratory environments represents a significant concern that can introduce exogenous DNA and potentially confound taxonomic assignments (89). Without negative controls, we cannot definitively quantify or exclude potential contamination signals from our data set. This limitation is particularly relevant for low-abundance taxa detected in our samples, as certain genera commonly found in reagent contamination may overlap with genuine environmental microorganisms (15, 27). Future studies should incorporate comprehensive negative controls throughout the sampling, extraction, and sequencing workflow to distinguish genuine environmental signals from potential contamination. Despite this limitation, our focus on high-abundance, ecologically relevant taxa and the observation of consistent patterns across multiple facilities and sample types provide confidence in our major findings regarding spatial heterogeneity and environmental drivers of microbial communities (48, 49).
Second, our study provides a snapshot of microbial communities at specific time points but does not capture long-term temporal dynamics across seasons or years. Longitudinal studies would provide insights into temporal stability and succession patterns of microbial communities in elderly care environments (90). Additionally, incorporating resident health data would enable direct assessment of associations between environmental microbiomes and health outcomes, strengthening the clinical relevance of these findings (91).
Third, while PICRUSt2 provides useful predictions of functional potential, these predictions are based on reference genomes and may not accurately represent the actual functional capabilities of the microbial communities. Several factors further limit the accuracy of functional inference in built environment microbiomes: (i) the underrepresentation of environmental microbial taxa in reference databases, as most reference genomes derive from cultured clinical or soil isolates rather than built environment-adapted strains (72); (ii) the inability of 16S rRNA-based methods to resolve strain-level functional variation, which is particularly relevant for antibiotic resistance genes that often show strain-specific distribution patterns (92); and (iii) primer bias inherent in the 341F/805R primer set, which may underrepresent certain Proteobacteria and Firmicutes lineages, potentially skewing functional predictions (30). These limitations are especially important when interpreting our ARG predictions, as the actual presence, copy number, and expression of resistance genes cannot be confirmed without direct metagenomic sequencing or culture-based phenotypic testing. Future studies should incorporate metatranscriptomic or metaproteomic approaches to directly measure functional activity rather than inferring it from taxonomic composition (72), alongside metagenomics to validate the presence and diversity of predicted functional genes.
An additional limitation of our study is the exclusive focus on resident-centered functional spaces, without sampling visitor reception areas, staff break rooms, or administrative zones. While our sampling design prioritized spaces where elderly residents spend the majority of their time (80%–90% of daily activities), visitor and staff areas may serve as critical entry points for externally transmitted microbes and could significantly influence the overall microbial ecology of the facilities. High-traffic zones such as main entrances, visitor waiting areas, and staff changing rooms warrant investigation in future studies, as these spaces may harbor distinct microbial communities shaped by outdoor air exchange, transient human traffic, and different cleaning protocols. Understanding the microbial transmission pathways from these external contact points to resident spaces would provide a more comprehensive picture of how environmental microbiomes are established and maintained in elderly care facilities. Future research should incorporate the sampling of these transitional zones, coupled with visitor/staff microbiome profiling and tracking studies, to elucidate the dynamics of microbial introduction and dissemination throughout the facility environment.
Finally, our study was conducted in a specific geographic region and cultural context, which may limit the generalizability of findings to elderly care facilities in other regions with different building designs, management practices, and cultural norms. Comparative studies across different geographic and cultural contexts would enhance our understanding of universal versus context-specific patterns in elderly care facility microbiomes (93).
Future research should explore interventions aimed at promoting healthier indoor microbiomes in elderly care facilities. These could include probiotic cleaning approaches, environmental modification strategies to encourage beneficial microorganisms, and personalized microbial management based on resident health status (94, 95). Additionally, investigating the interactions between the environmental microbiome and the human microbiome in elderly populations would provide valuable insights into microbial transmission dynamics and health implications (92).
In conclusion, our study reveals the complex microbial ecology of premium elderly care facilities, demonstrating how space functionality, environmental parameters, and human factors shape microbial communities across different indoor environments. These findings contribute to our understanding of microbial ecology in built environments and provide a foundation for developing evidence-based strategies to create healthier living environments for elderly populations. By recognizing the ecological complexity of indoor microbiomes and their potential health implications, we can move beyond conventional hygiene approaches toward more nuanced, ecologically informed management strategies that promote both microbial and human health.
Conclusion
This study provides the first comprehensive analysis of the environmental microbiome across key functional spaces in premium elderly care facilities, revealing critical insights into microbial ecology, environmental drivers, and potential health implications. Our findings demonstrate that microbial community composition and diversity are strongly shaped by space functionality, with dining areas and recreational rooms harboring higher microbial diversity dominated by human-associated and food-related taxa, while medical facilities and bathrooms exhibit lower diversity but higher relative abundance of opportunistic pathogens. Importantly, the effect of functional space (R² = 0.26) exceeded that of geographic location (R² = 0.12) across four cities spanning different climatic zones, indicating that anthropogenic factors associated with space use represent the primary drivers of indoor microbial ecology in elderly care facilities. Environmental parameters, particularly relative humidity and occupancy patterns, emerged as key determinants of microbial community structure, highlighting the importance of indoor environmental management in modulating microbial ecosystems.
The identification of a core microbiome across facilities underscores the existence of stable microbial consortia adapted to elderly care environments, while functional predictions revealed space-specific metabolic potentials, including predicted enrichment of antibiotic resistance genes in medical facilities and carbohydrate metabolism pathways in dining areas. Notably, negative correlations between beneficial bacteria (e.g., Lactobacillus) and opportunistic pathogens (e.g., S. aureus) observed in relative abundance data are consistent with potential antagonistic interactions, although absolute quantification would be necessary to confirm these ecological relationships and suggest ecological strategies to promote healthier microbial equilibria.
These findings have direct implications for facility management: space-specific hygiene protocols, humidity control, and ventilation strategies may help mitigate pathogen risks while preserving beneficial microbial communities. Furthermore, our results support the potential value of a paradigm shift from pathogen-centric disinfection to ecologically informed microbial stewardship, balancing pathogen reduction with microbiome resilience.
Limitations, including the exclusion of non-bacterial taxa and snapshot sampling, underscore the need for longitudinal, multiomics studies to explore microbial dynamics and host-microbiome interactions. Future research should integrate resident health data to directly link environmental microbiomes to clinical outcomes and test interventions such as probiotic cleaning or bioinformed design. By bridging microbial ecology and geriatric care, this work lays the foundation for creating healthier built environments tailored to the unique needs of aging populations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1United Nations. 2023. Department of economic and social affairs, population division. World population ageing 2023: highlights. UN, New York.
- 2Fang EF, Xie C, Schenkel JA, Wu C, Long Q, Cui H, Aman Y, Frank J, Liao J, Zou H, et al.. 2020. A research agenda for ageing in China in the 21st century (2nd edition): focusing on basic and translational research, long-term care, policy and social networks. Ageing Res Rev 64:101174. doi:10.1016/j.arr.2020.10117432971255 PMC 7505078 · doi ↗ · pubmed ↗
- 3National Bureau of Statistics of China. 2024. Statistical communiqué of the People’s Republic of China on the 2023 National Economic and Social Development. Beijing, China
- 4Salazar N, Valdés-Varela L, González S, Gueimonde M, de Los Reyes-Gavilán CG. 2017. Nutrition and the gut microbiome in the elderly. Gut Microbes 8:82–97. doi:10.1080/19490976.2016.125652527808595 PMC 5390822 · doi ↗ · pubmed ↗
- 5Santoro A, Zhao J, Wu L, Carru C, Biagi E, Franceschi C. 2020. Microbiomes other than the gut: inflammaging and age-related diseases. Semin Immunopathol 42:589–605. doi:10.1007/s 00281-020-00814-z 32997224 PMC 7666274 · doi ↗ · pubmed ↗
- 6Gilbert JA, Stephens B. 2018. Microbiology of the built environment. Nat Rev Microbiol 16:661–670. doi:10.1038/s 41579-018-0065-530127345 · doi ↗ · pubmed ↗
- 7Jayaprakash B, Adams RI, Kirjavainen P, Karvonen A, Vepsäläinen A, Valkonen M, Järvi K, Sulyok M, Pekkanen J, Hyvärinen A, Täubel M. 2017. Indoor microbiota in severely moisture damaged homes and the impact of interventions. Microbiome 5:138. doi:10.1186/s 40168-017-0356-529029638 PMC 5640920 · doi ↗ · pubmed ↗
- 8Lax S, Sangwan N, Smith D, Larsen P, Handley KM, Richardson M, Guyton K, Krezalek M, Shogan BD, Defazio J, Flemming I, Shakhsheer B, Weber S, Landon E, Garcia-Houchins S, Siegel J, Alverdy J, Knight R, Stephens B, Gilbert JA. 2017. Bacterial colonization and succession in a newly opened hospital. Sci Transl Med 9:eaah 6500. doi:10.1126/scitranslmed.aah 650028539477 PMC 5706123 · doi ↗ · pubmed ↗