Asymmetric Total Syntheses of Eliglustat and C2-epi-Eliglustat
Miguel Mellado-Hidalgo, Anna M. Costa, Pedro Romea, Fèlix Urpí, Gabriel Aullón

TL;DR
This paper describes a method to create two drug-like molecules with high precision using a nickel-catalyzed reaction.
Contribution
A stereocontrolled nickel-catalyzed aldol-like reaction for synthesizing enantiomerically pure compounds.
Findings
A common key step using TMSOTf-mediated aldol-like reaction was developed for both syntheses.
Dibenzyl acetals significantly influence the stereochemical outcome of the reaction.
The method allows independent manipulation of amino and hydroxy groups.
Abstract
Parallel asymmetric total syntheses of eliglustat and C2-epi-eliglustat are reported. Both approaches feature a common key step, which involves a stereocontrolled TMSOTf-mediated aldol-like reaction of azidoacetyl thioimides with dibenzyl acetals of aromatic aldehydes catalyzed by chiral nickel(II) complexes. It is worth noting that the structure of dibenzyl acetals plays a key role in the stereochemical outcome of some of these reactions, as supported by theoretical calculations. Overall, this strategy enables the independent manipulation of the amino and hydroxy functional groups and favors the isolation of enantiomerically pure products.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1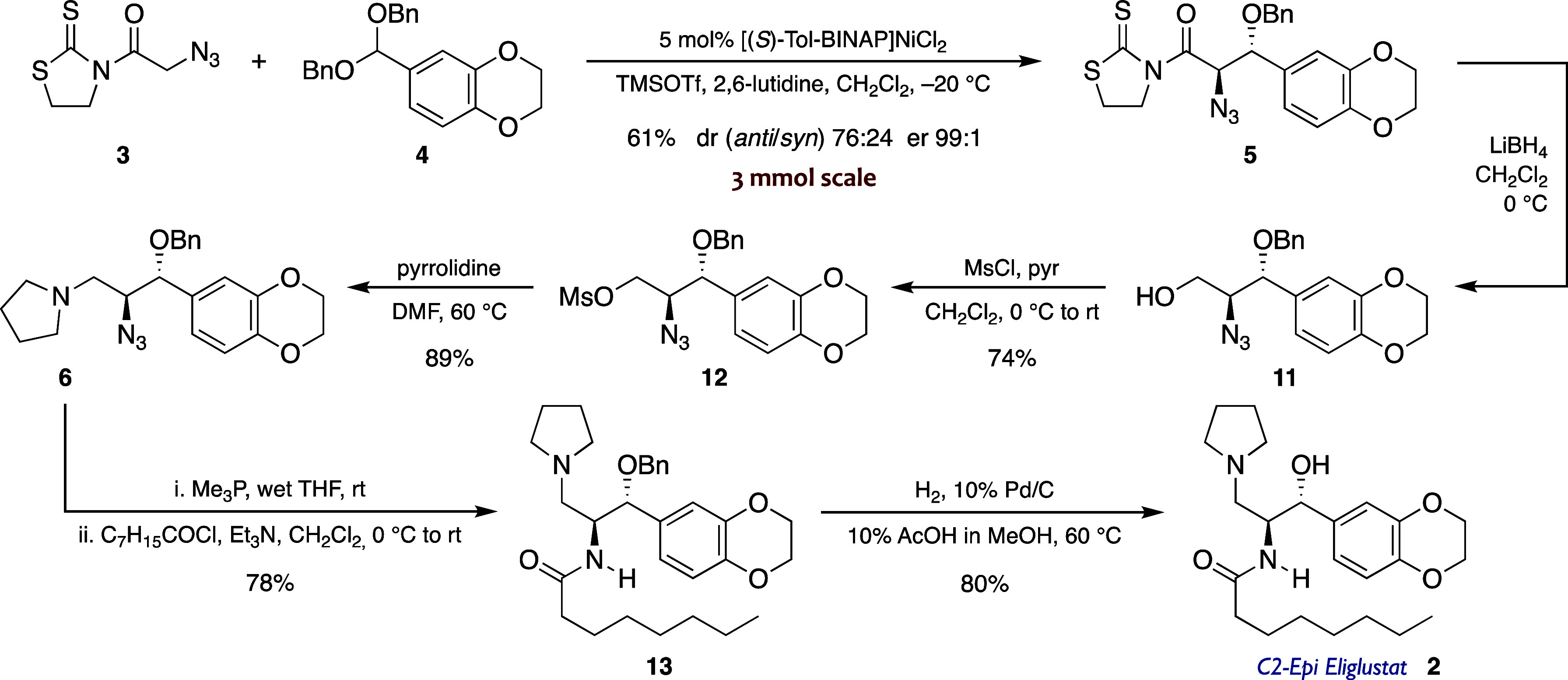 2
2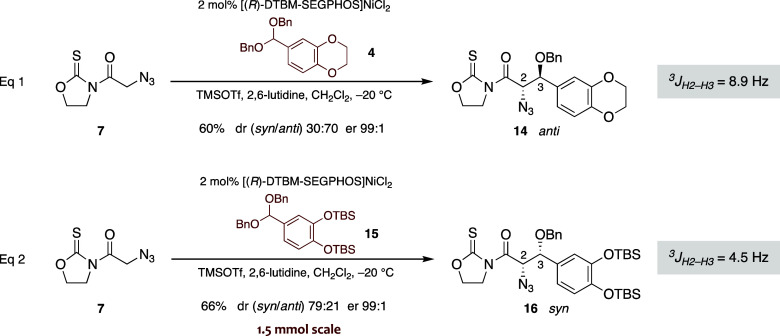 3
3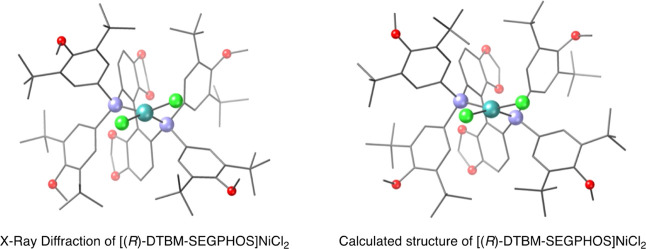 1
1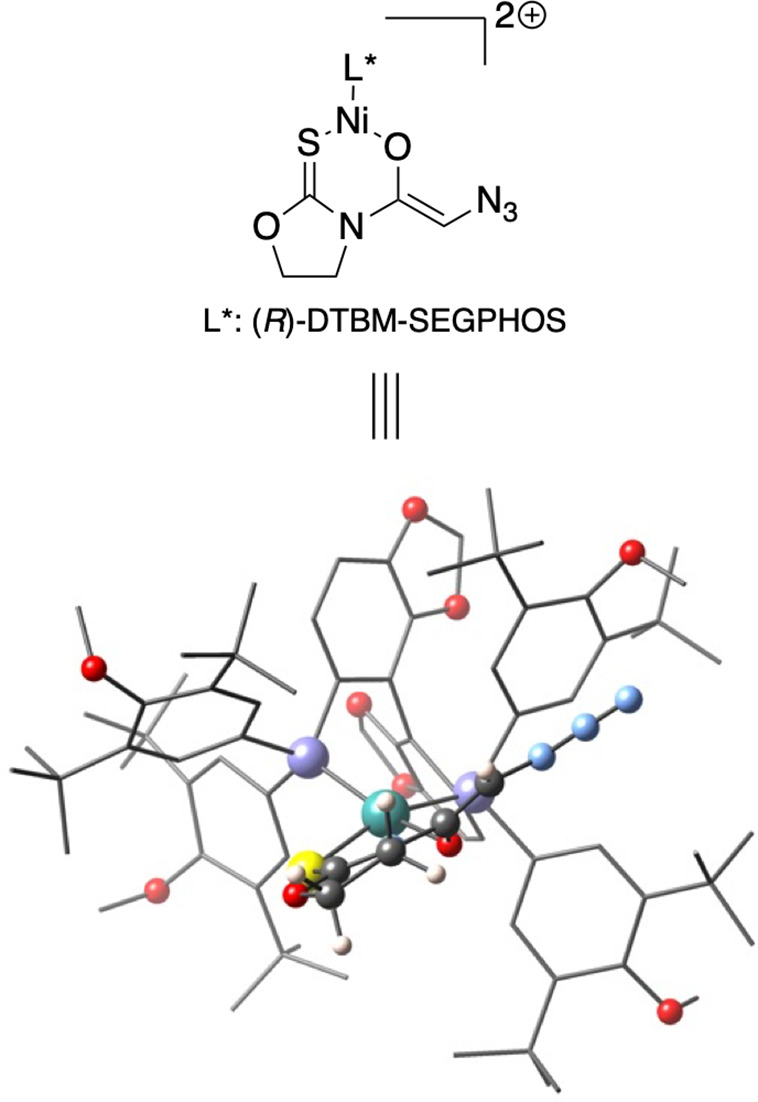 2
2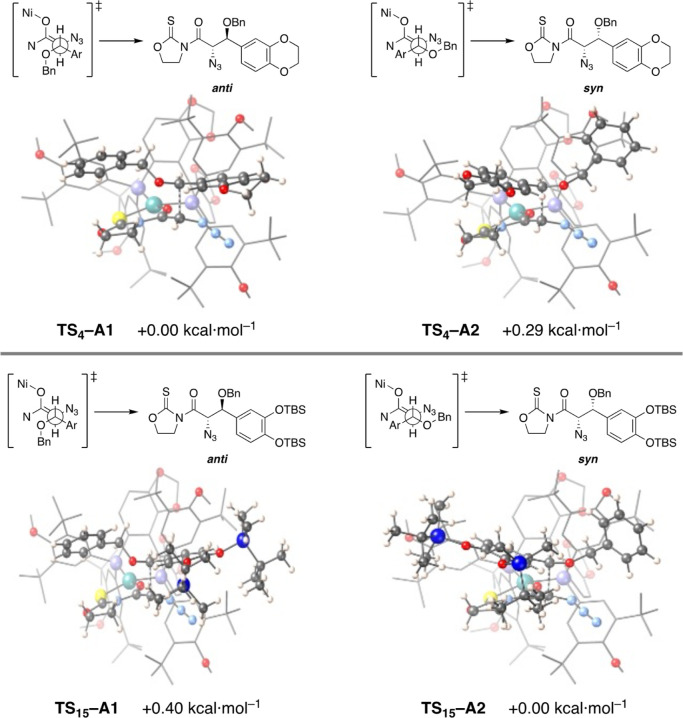 3
3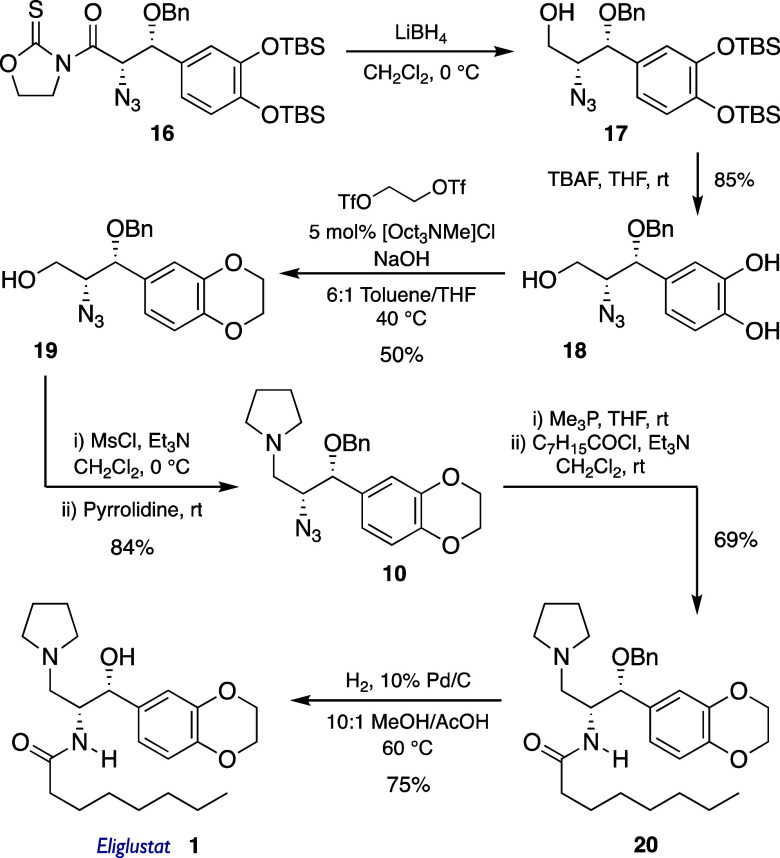 4
4- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Generalitat de Catalunya10.13039/501100002809
- —Generalitat de Catalunya10.13039/501100002809
- —European Regional Development Fund10.13039/501100008530
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAsymmetric Synthesis and Catalysis · Carbohydrate Chemistry and Synthesis · Synthetic Organic Chemistry Methods
Introduction
The structure of biologically active compounds determines their pharmacological, physicochemical, and toxicological properties.? Notably, stereoisomers, whether enantiomers or diastereomers, often exhibit strikingly different interactions with biological targets and follow distinct metabolic pathways, despite sharing the same molecular formula and atomic connectivity.? This selective recognition of stereoisomers by chiral biomolecules, such as enzymes and receptors, underscores the importance of accessing not only both enantiomers but also all relevant diastereomers of a compound to fully characterize its biological profile. ?−? ?
Stereodivergent strategies,? which enable the synthesis of multiple stereoisomers from common precursors, provide a powerful platform for the systematic investigation of bioactive targets. By integrating modular synthetic routes that incorporate switchable stereocontrol elements, the access to molecules possessing different relative and absolute configurations becomes possible, which is key for the oriented synthesis of specific stereoisomers and for conducting comprehensive structure–activity relationship studies.?
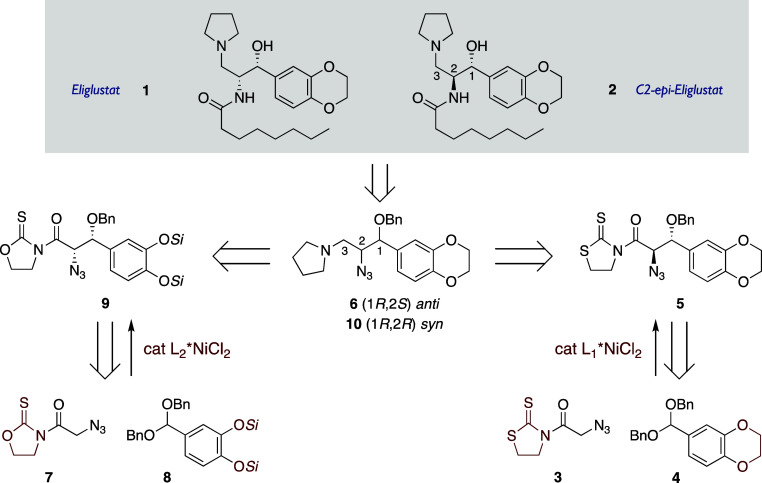
In this context, we envisioned that the parallel syntheses of both syn and anti stereoisomers of eliglustat would illustrate the benefits of this approach (Scheme). Eliglustat is a glucosylceramide synthase inhibitor that suppresses glycosphingolipid biosynthesis and has been identified as an effective long-term treatment for patients with Gaucher disease type 1.? Several synthetic routes have been developed for its preparation, mostly including chemical resolution of racemates, substrate-controlled, and chiral auxiliary-based methods. ?,? Conversely, only a few of the reported approaches rely on catalytic and enantioselective reactions to install the two stereocenters, such as a copper-mediated Henry reaction? or a ruthenium-mediated asymmetric reduction of an α-amino-β-keto ester.?
Retrosynthetic Analysis
Considering the increasing importance of asymmetric and catalytic methodologies in modern organic synthesis and building upon recent advances in stereoselective carbon–carbon bond-forming reactions, we describe herein the syntheses of eliglustat and its C2 epimer (1 and 2, respectively, in Scheme). Our approach takes advantage of highly stereocontrolled silyl triflate-mediated additions of N-azidoacetyl thioimides to dibenzyl acetals of aromatic aldehydes catalyzed by chiral nickel(II) complexes, followed by appropriate transformations of the resulting enantiomerically pure adducts to enable the selective access to all the stereoisomers of eliglustat at will. ?−? ? ?
Results and Discussion
Preliminary Results
Exploratory studies established that the suitable selection of the catalyst, the thioimide, and the aromatic substituents of the dialkyl acetals directs the aldol-like addition toward either the syn or the anti aldol adducts with remarkable stereocontrol.?
As the anti adducts can be consistently obtained from the reaction of N-azidoacetyl thiazolidinethione 3 with dialkyl acetals, regardless of the steric bulk of the alkyl group or the substituents of the aromatic ring, we anticipated that the TMSOTf-mediated addition of thioimide 3 to dibenzyl acetal 4, catalyzed by [(S)-Tol-BINAP]NiCl_2_, would afford enantiomerically pure anti adduct 5 with the absolute configuration of C2-epi-eliglustat (2 in Scheme).? Following such a key carbon–carbon bond-forming step, reductive removal of the heterocycle and further manipulations of the resulting alcohol would yield the advanced intermediate 6, from which the azide could be chemoselectively converted into the desired octanamide without competing side reactions. Subsequent deprotection of the benzylic alcohol finally delivered the target C2-epi-eliglustat (2).
In a parallel approach, the reaction of oxazolidinethione 7 and dibenzyl acetal 8, derived from silyl-protected 3,4-dihydroxybenzaldehyde, could be catalyzed by [(R)-DTBM-SEGPHOS]NiCl_2_ to furnish the syn 9 adduct with the same absolute configuration as eliglustat (1 in Scheme).? As in the former sequence, removal of the heterocyclic scaffold would provide enantiomerically pure advanced intermediate 10, from which the synthesis of eliglustat could be smoothly attained.
Synthesis of C2-epi-Eliglustat
Based on the above considerations and with the aim of evaluating the feasibility of our strategy, we initially undertook the synthesis of C2-epi-eliglustat.? As expected, the TMSOTf-mediated addition of thiazolidinethione 3 to dibenzyl acetal 4 catalyzed by 5 mol % of [(S)-Tol-BINAP]NiCl_2_ at a 3 mmol scale led to a 76:24 mixture of anti/syn diastereomers from which the anti adduct 5 was isolated in an enantiomerically pure form (er 99:1) with a 61% yield (Scheme). Reductive removal of the heterocycle from 5 with LiBH_4_ gave alcohol 11 quantitatively, which was used in the next step without further purification. Treatment of 11 with mesyl chloride gave sulfonate 12 in 74% two-step yield, and the mesylate group was subsequently displaced with pyrrolidine to afford amine 6 in an 89% yield. The end game of the sequence involved the chemoselective Staudinger reduction of the azide,? followed by the acylation of the resulting primary amine with octanoyl chloride to give amide 13, and the final deprotection of the benzylic alcohol to deliver the desired C2-epi-eliglustat (2 in Scheme).? Therefore, the synthesis of 2 was achieved in seven steps and 25% overall yield from N-azidoacetyl thiazolidinethione 3.
Synthesis of C2-epi-Eliglustat
Studies toward the Synthesis of Eliglustat
Having completed the synthesis of anti-C2-epi eliglustat, we next focused on the diastereomeric syn eliglustat (1 in Scheme). Given the pronounced influence of aromatic substituents of dibenzyl acetals in dictating the diastereoselectivity of the key aldol-like reaction from N-azidoacetyl oxazolidinethione 7, we carried out exploratory studies to unequivocally determine the stereochemical outcome of the TMSOTf-mediated addition of 7 to dibenzyl acetals derived from 3,4-dihydroxybenzaldehyde, catalyzed by 2 mol % [(R)-DTBM–SEGPHOS]NiCl_2_ (Scheme). Acetal 4, featuring a sterically unhindered ethylene protecting group, delivered a 70:30 diastereomeric mixture, from which the major component 14 was obtained in an enantiomerically pure form (er 99:1) and 60% yield. However, analysis of the key coupling constant ^3^ J H2–H3 in the ^1^H NMR spectrum (^3^ J H2–H3 = 8.9 Hz) revealed that adduct 14 was the anti diastereomer rather than the required syn counterpart (eq 1 in Scheme). In contrast, use of dibenzyl acetal 15, incorporating bulky silyl protecting groups, shifted the reaction toward a mixture (dr 79:21) in which the major diastereomer exhibited a significantly lower ^3^ J H2–H3 coupling constant (^3^ J H2–H3 = 4.5 Hz), consistent with the syn relative configuration. To our pleasure, the desired syn diastereomer 16 was subsequently isolated as a single enantiomer (er 99:1) in 66% yield on a 1.5 mmol scale (eq 2 in Scheme).
Key Carbon–Carbon Bond-Forming Reactions toward Eliglustat
Computational Studies
The different stereochemical outcomes of the additions involving dibenzyl acetals 4 and 15 (Scheme) warranted further investigation. Accordingly, we conducted a detailed theoretical study of these reactions to elucidate the origin of the stereodivergent results.
Initially, we examined the structure of the putative nickel(II) enolate derived from N-azidoacetyl oxazolidinethione 7. Notably, the structure of the precatalyst [(R)-DTBM-SEGPHOS]NiCl_2_ had previously been determined by X-ray diffraction,? which provides a useful point of comparison for evaluating the structures arising from theoretical calculations (Figure). Indeed, the calculated Ni–Cl and Ni–P distances (2.23 and 2.19 Å, respectively) are in good agreement with those observed in the X-ray diffraction (2.22 and 2.15 Å, respectively). The experimental P–Ni–P angle is 94°, closely matching the calculated value of 95°, which is consistent with the constraints imposed by its chelating arrangement. Likewise, the Cl–Ni–Cl and Cl–Ni–P angles are 98° and ≈150°, respectively, whereas the calculated values are 95° and 158°. Taken together, the close correspondence between the experimental and the calculated geometric parameters underscores the strong agreement between the X-ray and the theoretical structures and lends support to our computational model. Furthermore, all these data indicate that the geometry around the nickel atom is more accurately described as distorted square-planar rather than tetrahedral, as is further supported by continuous shape measures (S SQ–4 = 7.0 and S TT–4 = 11.7).
X-ray diffraction and optimized geometries of [(R)-DTBM-SEGPHOS]NiCl2.
The substitution of both chlorides in precatalyst [(R)-DTBM-SEGPHOS]NiCl_2_ by the exocyclic sulfur and the oxygen atoms of the nickel(II) enolate generates four different conformers depending on the relative arrangement of the chelate and the oxazolidinethione ring. As these conformers exhibit very similar geometries, only the most stable one was considered for the analysis of their reactivity. From a structural point of view, the coordination environment around the nickel center became more planar (S SQ–4 = 2.8), with S–Ni–P and O–Ni–P angles of 160° and 167°, respectively, approaching ideal square-planar geometry (Figure). The reduced distortion around the nickel atom is associated with a small S–Ni–O angle of 89°, imposed by the chelated arrangement, while the P–Ni–P angle slightly increases to 99°. Finally, the metal–ligand bond lengths are consistent with expected values, including the absence of a trans effect for the two non-equivalent Ni–P bonds.
Structure of the nickel(II) enolate.
Having established the structure of the enolate and aiming to understand the stereochemical outcomes observed for dibenzyl acetals 4 and 15, we next carried out a comprehensive computational study of the reactions presented in Scheme. The calculations revealed that the corresponding oxocarbenium intermediates from 4 and 15 can approach only the Si π-face of the enolate, as access to the opposite Re π-face is sterically hindered by the bulky tert-butyl groups on the phosphine moieties. Accordingly, our analysis focused exclusively on the Si π-face approach using the most stable conformation of the oxazolidinethione ring. This approach generated two transition states, TS _ 4 _ –A1 and TS _ 4 _ –A2 for acetal 4 and TS _ 15 _ –A1 and TS _ 15 _ –A2 for acetal 15 in Figure, which differ in the relative disposition of the oxocarbenium substituents.
Calculated transition states for the reaction of oxocarbenium intermediates from acetals 4 (top) and 15 (bottom).
The geometries of TS–A1 and TS–A2 involve a distorted planar environment for the metal center (S SQ–4 = 3.0–3.4), with angles for the opposite atoms and the diphosphine close to 162° and 96°, respectively. Furthermore, distances for the new C···C bond in TS–A1 are slightly longer than those in TS–A2, while the dihedral angle for the two methine groups ranges from 157° to 167°. Nevertheless, differences between approaches TS–A1 and TS–A2 became clear when analyzing their relative energies. The transition state TS _ 4 _ –A1 of the oxocarbenium intermediate from acetal 4 containing a dioxanyl moiety at the aromatic ring is more stable than the TS _ 4 _ –A2 counterpart by 0.29 kcal mol^–1^ (Figure top). By application of the Boltzmann distribution, the predicted diastereomeric syn/anti ratio of 36:64 is consistent with the 30:70 value obtained experimentally. In comparison, the oxocarbenium arising from acetal 15, which contains two TBS groups that dramatically increase the steric hindrance of the aromatic moiety, prefers to approach A2, so TS _ 15 _ –A2 turns out to be more stable than TS _ 15 _ –A1 by 0.40 kcal mol^–1^ (Figure bottom). This reversal gives a diastereomeric syn/anti ratio of 69:31, in line with the 79:21 value obtained experimentally.
These calculations indicate that tert-butyl groups of the phosphine moieties in the chiral ligand determine the π-face of the enolate to which electrophilic oxocarbenium intermediates approach, while the observed diastereoselectivity (syn versus anti) arises from the optimal accommodation of the aryl and the benzyloxy substituents at the electrophilic center, minimizing steric interactions with the chiral environment.
Synthesis of Eliglustat
Two main lessons were learned from former studies. First, dibenzyl acetal 4, bearing the 1,4-dioxane motif, proved suitable exclusively for the synthesis of C2-epi eliglustat 2, irrespective of the thioimide or catalyst employed. Second, successful synthesis of eliglustat required the stringent combination of oxazolidinethione 7, [DTBM-SEGPHOS]NiCl_2_ catalyst, and a dibenzyl acetal with bulky silyl-protected phenol groups on the aromatic ring, such as 15. As a result, reconstruction of the characteristic oxygenated heterocycle? became essential once the C2 and C3 stereocenters had been properly installed through the reaction of oxazolidinethione 7 with dibenzyl acetal 15, in which both phenol groups are protected as silyl ethers.
Based on these findings, the synthesis of eliglustat was pursued, as outlined in Scheme. Reduction of syn adduct 16 and subsequent treatment of resulting alcohol 17 with TBAF afforded trihydroxy derivative 18 in an 85% two-step yield. Then, reconnection of both phenol groups with an ethylene handle was attempted. Initial efforts employing ethylene dibromide as the alkylating agent under different conditions consistently resulted in complex mixtures.? The use of the more reactive ethylene ditosylate did not improve these results either. Finally, much more active ethylene bistriflate under phase transfer conditions enabled the formation of the desired dioxane 19 in 50% yield.? Transformation of the hydroxyl group in 19 with mesyl chloride and displacement of the resulting mesylate with pyrrolidine provided advanced intermediate 10 in an 84% yield. Comparison of the spectra from amines 6 and 10 (Scheme) confirmed that they were indeed diastereomers, thereby validating the use of acetal 15 in the synthesis of eliglustat. As in the case of C2-epi-eliglustat (Scheme), the final steps involved Staudinger reduction of the azide, followed by acylation of the resulting primary amine with octanoyl chloride to give amide 20 in 69% two-step yield. Eventually, deprotection of the benzylic alcohol delivered the targeted eliglustat (1) in 75% yield (Scheme), with spectroscopic data matching those reported in the literature.? Overall, the synthesis of eliglustat (1 in Scheme) was accomplished in nine steps and 12% yield from N-azidoacetyl oxazolidinethione 7.
Synthesis of Eliglustat
At this point, considering both the experimental and theoretical results reported in this and earlier studies,? it is reasonable to assert that the stereochemical outcome of aldol-like additions of thioimides to oxocarbenium intermediates generated from aromatic acetals catalyzed by chiral [DTBM-SEGPHOS]NiCl_2_ complexes is strongly governed by the steric demands of the aromatic moiety. Accordingly, a rigorous analysis is necessary to delineate the synthetic route best suited to the desired syn diastereomers.
Conclusions
In summary, we have successfully completed the asymmetric total syntheses of eliglustat (1) and its C2-epimer (2) in nine and seven steps, respectively, with overall yields of 10–25% using a common approach. The key step involves the forging of the C1–C2 carbon bond through Lewis acid-mediated, stereocontrolled additions of azidoacetyl thioimides to dibenzyl acetals catalyzed by chiral nickel(II) complexes. Both experimental and theoretical studies indicate that the nature of the substituents on the aromatic moiety is crucial for determining the stereochemical outcome of the carbon–carbon bond-forming step; indeed, a dioxanyl-like substituent consistently leads to the anti relative configuration found in C2-epi-eliglustat, whereas bulky TBS protecting groups of phenols favor the syn configuration required for eliglustat. Importantly, this approach enables the chemoselective acylation of the amine at C2 without competition from the C1-hydroxyl counterpart in the final steps of the syntheses. Therefore, the appropriate choice of the starting thioimide, acetal, and catalyst gives access to any of the potential stereoisomers of eliglustat at will, providing a valuable platform for the accurate evaluation of their biological activity.
From a broader perspective, this approach complements previously reported strategies based on the asymmetric and catalytic construction of key carbon–carbon bonds ?,? while also enabling the enantioselective synthesis of all potential stereoisomers of targeted eliglustat through the use of robust nickel(II) complexes containing commercially available ligands, rather than bespoke ligands as required in earlier approaches. ?,?
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Eliel, E. L. ; Wilen, S. H. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, 1994.
- 2Lovering F.Bikker J.Humblet C.Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success J. Med. Chem.2009526752675610.1021/jm 901241 e 19827778 · doi ↗ · pubmed ↗
- 3Silvestri I. P.Colbon P. J. J.The Growing Importance of Chirality in 3D Chemical Space Exploration and Modern Drug Discovery Approaches for Hit-IDACS Med. Chem. Lett.2021121220122910.1021/acsmedchemlett.1c 0025134413951 PMC 8366003 · doi ↗ · pubmed ↗
- 4Scott K. A.Ropek N.Melillo B.Schreiber S. L.Cravatt B. F.Vinogradova E. V.Stereochemical diversity as a source of discovery in chemical biology Curr. Res. Chem. Biol.2022210002810.1016/j.crchbi.2022.100028 · doi ↗
- 5a Schreiber S. L.Target–Oriented and Diversity–Oriented Organic Synthesis in Drug Discovery Science 20002871964196910.1126/science.287.5460.196410720315 · doi ↗ · pubmed ↗
- 6a Krautwald S.Carreira E. M.Stereodivergence in Asymmetric Catalysis J. Am. Chem. Soc.20171395627563910.1021/jacs.6b 1334028384402 · doi ↗ · pubmed ↗
- 7For a recent example in our group, see Galeote O.Kennington S. C. D.Benedito G.Fraedrich L.Davies-Howe E.Costa A. M.Romea P.UrpíF.Aullón G.Font-Bardia M.Puigjaner C.Direct, Stereodivergent, and Catalytic Michael Additions of Thioimides to α,β-Unsaturated Aldehydes – Total Synthesis of Tapentadol Angew. Chem., Int. Ed.202463 e 20231930810.1002/anie.20231930838231568 · doi ↗ · pubmed ↗
- 8Lee L.Abe A.Shayman J. A.Improved Inhibitors of Glucosylceramide Synthase J. Biol. Chem.1999274146621466910.1074/jbc.274.21.1466210329660 · doi ↗ · pubmed ↗