Comparative phylogenetic, antimicrobial resistance, and clinical characterization of human spondylodiscitis-associated Staphylococcus pseudintermedius
Jakob Douan, Christian Kohler, Lyubomir Haralambiev, Evgeny A. Idelevich, Karsten Becker

TL;DR
A rare case of spinal infection caused by Staphylococcus pseudintermedius is analyzed using genome sequencing to track resistance genes and guide treatment.
Contribution
The study provides a detailed genomic and clinical analysis of a rare S. pseudintermedius infection case, revealing resistance gene profiles and phylogenetic context.
Findings
The isolate belonged to a rare sequence type ST2051 and carried multiple resistance genes but no mecA.
Genomic comparison revealed three AMR gene clusters, with the isolate in a multidrug-resistant intermediate group.
Phylogenetic analysis showed high global genomic diversity among S. pseudintermedius isolates.
Abstract
We report a case of spondylodiscitis caused by methicillin-susceptible Staphylococcus pseudintermedius (MSSP) in a 23-year-old male following lumbar spine stabilization. Despite initial recovery, the patient developed postoperative infection with elevated inflammatory markers and radiological signs of spondylodiscitis. Revision surgery revealed pus extending to the osteosynthesis device. S. pseudintermedius was identified from tissue and blood cultures by MALDI-TOF MS and molecular methods. Whole-genome sequencing (WGS) of three isolates collected at different time points revealed a single clonal strain carrying multiple chromosomal resistance genes [blaZ, cat, ermB, aph-Stph, ant6, aph(3″)-III, sat4A] and a 3.1 kb plasmid of unknown function, but no mecA. Phenotypically, the isolate was susceptible to all tested antibiotics except erythromycin and exhibited inducible clindamycin…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
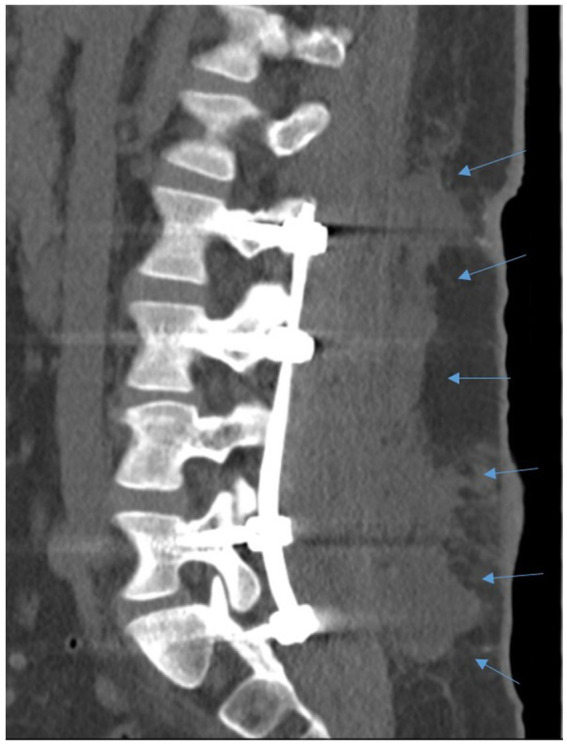 Figure 1
Figure 1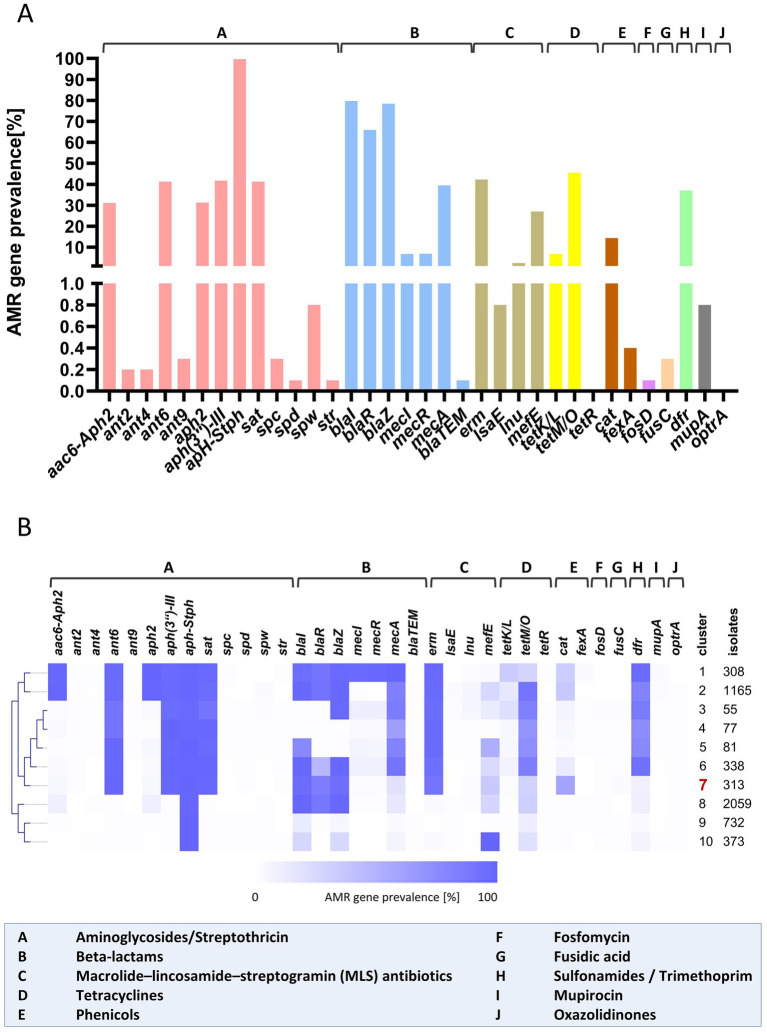 Figure 2
Figure 2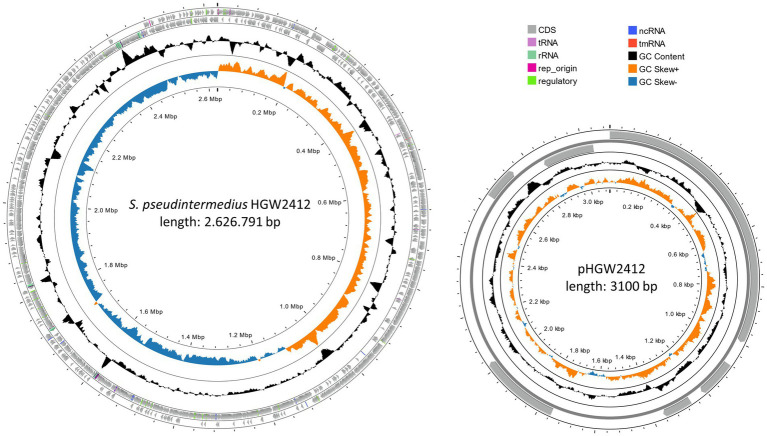 Figure 3
Figure 3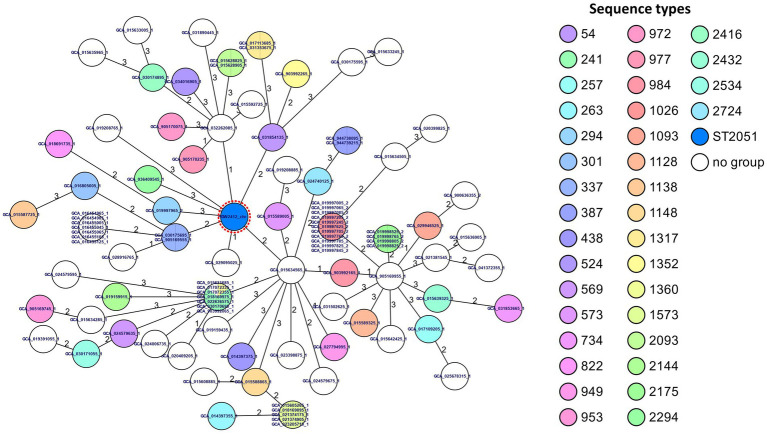 Figure 4
Figure 4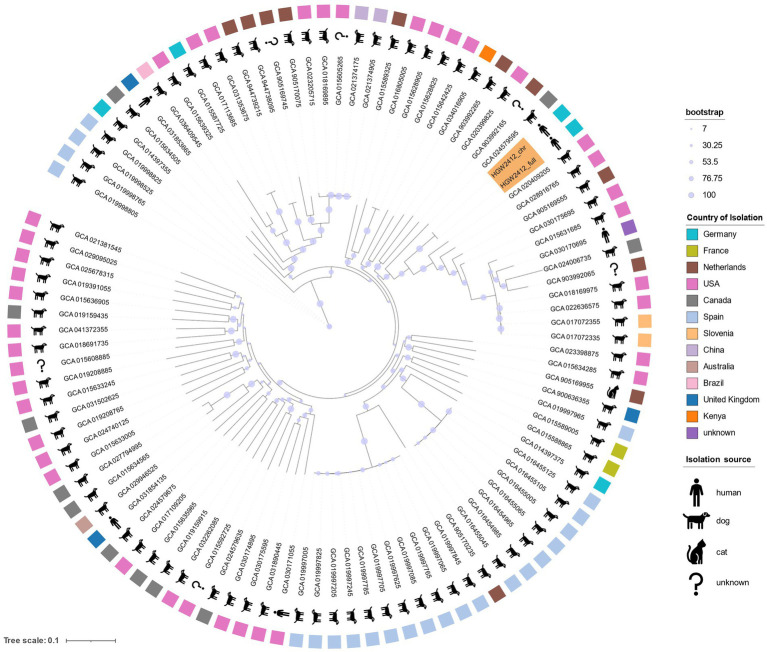 Figure 5
Figure 5| Antibiotics | Minimum inhibitory concentrations (MICs), mg/L | Interpretationa |
|---|---|---|
| Oxacillin | <=0.25 | Susceptible (S) |
| Levofloxacin | <=0.12 | No interpretation |
| Gentamicin | <=0.5 | No interpretation |
| Teicoplanin | 1 | No interpretation |
| Vancomycin | <=0.5 | No interpretation |
| Erythromycin | > = 8 | Resistance (R) |
| Clindamycin | 0.25 | Inducible clindamycin resistance (R) |
| Tetracycline | <=1 | Susceptible (S) |
| Tigecycline | <=0.12 | Susceptible (S) |
| Linezolid | 1 | Susceptible (S) |
| Daptomycin | 0.25 | Susceptible (S) |
| Fosfomycin | <=8 | Susceptible (S) |
| Fusidic acid | <=0.5 | Susceptible (S) |
| Trimethoprim sulfamethoxazole | <=10 | Susceptible (S) |
| Cluster | Total number of AMR gene combinations within cluster | Isolates of the predominant AMR gene combination | Proportion of the predominant AMR gene combination within the cluster [%] | Proportion of total ( | Predominant AMR gene combination | Theoretical resistance to some |
|---|---|---|---|---|---|---|
| Cluster 1 | 65 | 92 | 29.9 | 1.7 |
| Aminoglycosides, β-Lactams, Macrolides, Sulfonamides/Trimethoprim |
| Cluster 2 | 163 | 200 | 17.2 | 3.6 |
| Aminoglycosides, β-Lactams, Macrolides, Tetracyclines, Sulfonamides/Trimethoprim |
| Cluster 3 | 29 | 15 | 27.3 | 0.3 |
| Aminoglycosides, β-Lactams, Macrolides, Tetracyclines, Sulfonamides/Trimethoprim |
| Cluster 4 | 25 | 19 | 24.7 | 0.3 |
| Aminoglycosides, β-Lactams, Macrolides, Tetracyclines, Sulfonamides/Trimethoprim |
| Cluster 5 | 32 | 19 | 23.5 | 0.3 |
| Aminoglycosides, β-Lactams, Macrolides, Tetracyclines, Sulfonamides/Trimethoprim |
| Cluster 6 | 71 | 51 | 15.1 | 0.9 |
| Aminoglycosides, β-Lactams, Macrolides, Tetracyclines, Sulfonamides/Trimethoprim |
| Cluster 7 | 79 | 77 | 24.6 | 1.4 |
| Aminoglycosides, β-Lactams, Macrolides, Phenicols |
| Cluster 8 | 140 | 724 | 35.2 | 13.2 |
| Aminoglycosides, β-Lactams |
| Cluster 9 | 36 | 512 | 69.9 | 9.3 |
| Aminoglycosides |
| Cluster 10 | 37 | 213 | 57.1 | 3.9 |
| Aminoglycosides, Macrolides |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInfectious Diseases and Tuberculosis · Antimicrobial Resistance in Staphylococcus · Orthopedic Infections and Treatments
Introduction
Spondylodiscitis encompasses vertebral osteomyelitis, spondylitis, and discitis as variations of a shared pathological process (Gouliouris et al., 2010). Among pyogenic causes, Staphylococcus aureus is most frequently isolated in humans (Mylona et al., 2009), whereas Staphylococcus pseudintermedius is the predominant agent in dogs (Barash et al., 2018; Pilkington et al., 2023). Staphylococci are known for antibiotic resistance and diverse virulence factors and are roughly classified for clinical purposes by coagulase production (Kloos and Bannerman, 1994). Importantly, S. pseudintermedius, a coagulase-positive species, is primarily part of the canine flora (Bannoehr and Guardabassi, 2012), yet it causes infections in companion but also wild animals (Smith et al., 2020; Mama et al., 2019). Though mainly an animal pathogen, it can act as a zoonotic agent with rare human cases mostly among those in close contact with animals (Darlow et al., 2017; Morris et al., 2010; Schwartz et al., 2021; Tugasworo et al., 2021). The first human case published with this species name was reported in 2005 involving an infected defibrillator implant (Van Hoovels et al., 2006), but, it can be assumed that earlier cases occurred under the designation S. intermedius.
Formally delimited from S. intermedius in 2005 (Devriese et al., 2005), S. pseudintermedius is phylogenetically part of the S. intermedius group (SIG) and was in the pre-MALDI-TOF mass spectrometry era frequently confused with other SIG species or misidentified as S. aureus due to coagulase activity (Becker et al., 2005; Fitzgerald, 2009; Sasaki et al., 2007). Misidentification may affect the interpretation of susceptibility testing and treatment decisions (Borjesson et al., 2015). S. pseudintermedius produces various virulence factors and, like other staphylococci, presents challenges through methicillin resistance (Bannoehr et al., 2011; Piriz et al., 1996; Pitchenin et al., 2018; Roberts et al., 2024; Teixeira et al., 2024). Further, methicillin-resistant S. pseudintermedius (MRSP) isolates are often multidrug-resistant and pose significant zoonotic risks (Kadlec and Schwarz, 2012; Paul et al., 2011).
Infections caused by S. pseudintermedius are increasingly recognized as clinically similar to those caused by S. aureus, particularly in vulnerable populations such as the elderly and immunocompromised (Somayaji et al., 2016). A 2017 review identified MRSP as one of the most common antimicrobial-resistant (AMR) bacteria transmitted from companion animals to humans (Pomba et al., 2017). In Germany, individuals aged 60 and older represent 25% of pet owners (Schirmer, 2018), and their close contact with pets may elevate the risk of zoonotic transmission of pathogens like S. pseudintermedius. This underscores the necessity for targeted surveillance and preventive measures within this demographic and beyond. Further emphasizing its public health relevance, the European Food Safety Authority’s Panel on Animal Health and Welfare named S. pseudintermedius among significant AMR pathogens isolated from dogs in the EU, alongside Escherichia coli and Pseudomonas aeruginosa (Nielsen et al., 2021). The misuse of antimicrobials shared between companion animals and humans exacerbates this issue (Chow et al., 2024). These findings align with global health research advocating for integrated surveillance systems to monitor AMR in zoonotic bacteria, including Staphylococcus species, under the One Health framework (Dafale et al., 2020).
Here, we report a rare case of spondylodiscitis caused by S. pseudintermedius in a young male with no known animal contact following spinal trauma and surgery. The isolates recovered underwent whole-genome sequencing (WGS) and genetic characterization to enable phylogenetic classification and to identify virulence factors and resistance genes. This case highlights the importance of recognizing zoonotic pathogens within a “One Health” framework and emphasizes the need for precise and advanced diagnostic techniques.
Materials and methods
Patient history
A 23-year-old male patient with a history of Tourette syndrome and a documented penicillin allergy presented to the emergency department after sustaining a traumatic fall from a second-story balcony. Upon admission, the patient reported severe pain in the cervical, thoracic, and lumbar regions, with additional discomfort in the thorax and both ankles. The initial examination revealed an unstable compression fracture of the fourth lumbar vertebra without evidence of an open wound or external communication. Imaging confirmed the lumbar fracture without additional injuries to the head, thorax, or abdomen. The patient was admitted for surgical treatment of the spinal fracture and underwent transpedicular dorsal stabilization of L2 to L5 at the end of September 2024. The postoperative course was initially uneventful, and the patient demonstrated adequate recovery. However, despite a sudden deterioration in the patient’s infection markers, he was discharged against medical advice 11 days after admission, without undergoing any antibiotic treatment. Three days after being discharged, the patient presented at a nearby hospital with worsening general condition and progressive pain. As a result, the patient was readmitted to our clinic for further treatment. Upon readmission, the patient exhibited signs of infection, including fever and elevated inflammatory markers, with a C-reactive protein (CRP) level of 282 mg/L and leukocytosis with a white blood cell count 11.9 × 10^9^/L. A series of blood cultures were collected for microbiological examination. Further, a computed tomography (CT) scan was performed, revealing signs of spondylodiscitis at the L4 vertebral level (Figure 1). Consequently, the patient underwent revision surgery, which revealed pus in the wound extending to the osteosynthesis. Following meticulous surgical debridement and lavage, five tissue samples were obtained from different areas. The transpedicular osteosynthesis was retained as the fracture had not yet consolidated.
CT scan of spondylodiscitis in a 23-year-old patient following surgical stabilization of L2–L5. The CT image reveals substantial fluid accumulation surrounding the internal transpedicular osteosynthesis of the lumbar spine, signifying early postoperative infection. The arrows mark the soft tissue involvement. The imaging findings are consistent with the patient’s septic clinical presentation, thereby confirming the suspected diagnosis of a peri-implant abscess.
Microbiological analysis
Bacterial growth was observed after overnight cultivation of the tissue samples, with colonies appearing white and haemolytic on Columbia agar plates with 5% sheep blood (BD Diagnostics, Heidelberg, Germany). All tested colonies were positive in the catalase test, while the clumping factor test using the Pastorex Staph-Plus (Bio-Rad, Marnes-la-Coquette, France) was negative. Identification of the bacterial colonies from these samples was performed using MALDI-TOF MS utilizing the MALDI Biotyper® sirius system (Bruker Daltonics, Bremen, Germany) with MBT Biotargets 96 (Bruker Daltonics). Identification was initially performed using two protocols, direct transfer (DT) and extended direct transfer (eDT), following the manufacturer’s instructions and a previous study (Letunic and Bork, 2024). Some spots showed no peaks or identification scores below 2.00 and therefor the median identification score was 1.70 (range = 1.60–2.12). To improve sensitivity and accuracy, a protein extraction (PE) procedure was applied before repeating MALDI-TOF MS measurements (Idelevich et al., 2023). This resulted in a marked increase in the identification scores, with a median value of 2.22 (range = 2.08–2.37), and all isolates were unequivocally identified as S. pseudintermedius. For quality control and standardization, a 1 μL aliquot of IVD Bacterial Test Standard (IVD BTS, Bruker) was included on the MBT Biotarget during all three procedures, per manufacturer guidelines.
Of the six-vial blood culture set initially collected, only one aerobic vial (BACTEC™ Plus; BD Diagnostics) tested positive. Gram staining showed gram-positive cocci consistent with staphylococci. This vial was analyzed using the loop-mediated isothermal amplification-based eazyplex® MRSA kit (Amplex Diagnostics), which detects S. aureus complex species (S. aureus, S. argenteus, S. schweitzeri, S. roterodami), S. epidermidis, and mecA/mecC resistance genes. No target genes were detected in this case. The positive blood culture vial was subcultured on Columbia blood agar (BD Diagnostics, Heidelberg, Germany), yielding white, hemolytic colonies similar to those from tissue samples, both negative for clumping factor (Pastorex Staph-Plus, Bio-Rad, Marnes-la-Coquette, France). After protein extraction procedure (PE), MALDI-TOF MS identified all isolates from tissue and blood cultures as S. pseudintermedius with median score of 2.22 (range = 2.11–2.28). To exclude contamination, two additional blood culture sets were collected earlier; one aerobic vial was positive, and its colonies were identified as S. pseudintermedius. Except for these two positive aerobic vials, all other blood cultures remained negative after 6 days of incubation. No other bacteria or fungi were detected in clinical or screening samples throughout treatment.
The antimicrobial susceptibility testing (AST) of the isolates was performed using the VITEK 2 system (bioMérieux, Marcy l’Etoile, France) and VITEK software (Version 9.04) following the manufacturer’s instructions with the AST-P-654 test cards. Phenotypic methicillin resistance was further assessed by disk diffusion testing using oxacillin (1 μg), as recommended by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) for screening in S. pseudintermedius, S. intermedius, S. schleiferi, and S. coagulans. AST results were interpreted according to the EUCAST Breakpoint Tables for MICs and zone diameters, version 14.0. All isolates were found to be susceptible to all tested antibiotics, except for erythromycin and clindamycin (Table 1).
DNA isolation, library preparation and whole genome sequencing
Bacteria were cultured on blood agar plates (Becton, Dickinson and Company [BD], USA) at 37 °C for 16–24 h. Cells were harvested using a 1 μL inoculation loop, and genomic DNA was extracted with the NucleoSpin® Microbial DNA Kit (Macherey-Nagel, Germany) according to the manufacturer’s instructions. DNA purity and quality were assessed using the NanoDrop™ One/OneC Spectrophotometer (Thermo Fisher Scientific, USA), while DNA concentrations were determined using the Qubit™ 1X dsDNA Assay Kit and Qubit™ 4 Fluorometer (Thermo Fisher Scientific, USA). DNA was either used immediately for library preparation or stored at −80 °C until further processing.
Due to the use of two different sequencing platforms, separate library preparation protocols were followed. For Illumina short-read sequencing, libraries were prepared using the Illumina DNA Prep Kit following the manufacturer’s instructions, and sequencing was performed on a MiSeqDx system (Illumina, USA) using a 2 × 300 bp paired-end configuration.
For Oxford Nanopore (ONT) long-read sequencing, libraries were prepared using the Rapid Barcoding Kit SQK-RBK114.24 (Oxford Nanopore Technologies, UK) as per the manufacturer’s protocol. Sequencing was conducted using a MinION device equipped with an R10.4.1 flow cell (FLO-MIN114).
Assembly and polishing of consensus sequences
Raw Illumina reads were processed using the platform’s default basecalling settings, and the resulting FASTQ files were subsequently used for hybrid assemblies. For Oxford Nanopore Technologies (ONT) sequencing, raw signal data (POD5 files) were basecalled using the super-accurate (SUP) model [email protected] with the Dorado v0.9.1 basecaller1. The resulting ONT FASTQ reads were first used for genome size estimation with Long Read-based Genome size Estimation (LRGE) (Hall et al., 2024), followed by downsampling to 100 × coverage using Rasusa (Randomly Subsample Sequencing Reads or Alignments) (Hall, 2022). Downsampled FASTQ files were then assembled using the Autocycler pipeline v0.2.1 in fully automated mode, utilizing the following long-read assemblers: Flye, Miniasm, NextDenovo, and Raven. Resulting consensus sequences were polished with Medaka v2.0.1, applying the bacterial methylation model r1041_e82_400bps_bacterial_methylation2. Polished assemblies were reoriented using Dnaapler (Bouras et al., 2024), aligning sequences to start at the dnaN gene for chromosomes or the rep gene for plasmids, thereby standardizing orientation across all sequenced isolates. Final assemblies were hybrid-polished using high-quality Illumina short reads. These reads were quality-trimmed with Trimmomatic v0.39, and final polishing was performed using Polypolish v0.6.0 (Bouras et al., 2024; Wick and Holt, 2022; Bolger et al., 2014). The resulting high-quality consensus sequences were used in all downstream analyses.
Genome annotation, comparative genomics and phylogenetic analyses
Initially, the consensus sequences were analyzed using the TORMES pipeline v1.3.03 (Quijada et al., 2019) for genome annotation via Prokka (Seemann, 2014) and identification of antimicrobial resistance genes using the CARD, ResFinder, and ARG-ANNOT databases (McArthur et al., 2013; Zankari et al., 2012; Gupta et al., 2014) (Supplementary Table S1). In addition, the sequences of S. pseudintermedius HGW2412 and pHGW2412 were annotated using the Bakta pipeline4 (Schwengers et al., 2021) (Supplementary Table S1).
Subsequently, Prokka-annotated sequences were employed for virulence gene identification. To this end, the complete protein dataset of the Virulence Factor Database (VFDB5) was downloaded, and Staphylococcus-specific virulence factors were extracted. These were used in a BLAST+ (v2.16.0+) search6 against the HGW2412 proteome. Hits were filtered using the following thresholds (Supplementary Table S1): alignment coverage (= align_l/s_end) ≥ 50%, sequence identity ≥ 30%, and bit score ≥ 100.
To assess the phylogenetic context of HGW2412, 5,500 S. pseudintermedius genomes were retrieved from the NCBI Reference Sequence Database (RefSeq)7. Pairwise genomic similarity was computed using the Fast Average Nucleotide Identity algorithm (FastANI)8 (Jain et al., 2018), and the 100 most closely related genomes were selected for downstream analysis.
These 100 genomes, along with HGW2412, were first subjected to MLST profiling in SeqSphere+ v10.5.0 using the Solyman et al. (2013) scheme, and a minimum spanning tree was constructed.
In parallel, core SNP-based phylogenetic analysis was conducted. An optimal k-mer size of 17 was determined using Kchooser4, and SNP calling was performed with kSNP4.1 (Hall and Nisbet, 2023). A maximum likelihood (ML) phylogenetic tree was then inferred from the core SNP matrix using RAxML v8.2.12 (Stamatakis, 2014), applying the GTRGAMMA substitution model and 1,000 bootstrap replicates. The annotated ML tree, including information on isolation source, country, and bootstrap values, was visualized using Interactive Tree of Life (iTOL v7.1.1) (Letunic and Bork, 2024).
AMR gene profiling
To investigate AMR determinants, a total of 5.500 S. pseudintermedius genomes and their associated metadata were downloaded from the RefSeq database (NCBI9) as described above. AMR gene detection was performed using ABRicate10 with four reference databases: NCBI AMRFinderPlus, CARD, ResFinder, and ARG-ANNOT (Zankari et al., 2012; Gupta et al., 2014; Feldgarden et al., 2019; Jia et al., 2017). Because the different databases can report partially distinct AMR genes, multiple hits corresponding to the same gene within a genome were collapsed into a single entry, ensuring that each AMR gene was counted only once per isolate. The results from all databases were then merged into a unified presence/absence matrix reflecting the similarity of each detected gene to its respective reference sequence (Supplementary Table S2). A minimum sequence identity threshold of 80% was applied, and gene names were standardized by replacing outdated nomenclature with the currently accepted designations. To calculate AMR gene prevalence across the isolates, all hits were binarized (presence = 1 or 100%). This matrix was subsequently used to identify AMR gene clusters employing the Self-Organizing Tree Algorithm (SOTA) implemented in MeV v4.9.0 (TM4 Software Suite, USA) using default settings exceptional the number of cycles which was adjusted to in total 10 cycles. Fewer cycles resulted in overly coarse clustering with high within-cluster variability, whereas more cycles produced overly fine clusters, some of which contained only very few isolates. The SOTA dendrogram depicts the hierarchical relationships among clusters; however, the accompanying heatmap represents relative internal node values rather than true gene prevalence. Therefore, AMR gene prevalence was recalculated for each cluster and visualized as a heatmap adjacent to the dendrogram in Figure 2. The whole ABRicate results table and all matrices (complete heatmap table of 5,501 sequences and individual SOTA clusters 1–10) are provided in Supplementary Table S2.
AMR gene prevalence and cluster analysis of S. pseudintermedius genomes. (A) A total of 5,500 S. pseudintermedius genomes from the NCBI RefSeq database, together with the HGW2412 genome, were screened for AMR determinants using ABRicate v1.0.1 (https://github.com/tseemann/abricate). AMR gene prevalences were calculated across all genomes. Colors and letter codes denote antibiotic classes to facilitate interpretation. (B) Presence–absence profiles of all detected AMR genes were used as input for clustering with the Self-Organizing Tree Algorithm (SOTA). The resulting hierarchical structure is shown alongside AMR gene prevalence patterns for each cluster, enabling comparison of resistance gene compositions across clusters. Cluster identifiers and the number of genomes per cluster are indicated. The HGW2412 isolate is assigned to Cluster 7 and highlighted accordingly. Figures were generated using GraphPad Prism v9.5.1 (GraphPad Software, USA) and MeV v4.9.0 (TM4 Software Suite, USA). aac6-aph2, Aminoglycoside N-acetyltransferase; ant2, Aminoglycosid 2′-phosphotransferase; ant4, Aminoglycosid 4′-phosphotransferase; ant6, Aminoglycosid 6′-phosphotransferase; ant9, Aminoglycosid 9′-phosphotransferase; aph2, Aminoglycoside 2″-phosphotransferase; aph(3″)-III, Aminoglycoside 3″-phosphotransferase III; aph-Stph, Aminoglycoside phosphotransferase from Staphylococcus species; sat, Spectinomycin acetyltransferase; spc, Spectinomycin resistance gene; spd, Spectinomycin dehydratase; spw, Spectinomycin resistance gene; str, Streptomycin resistance gene; blaI, Beta-lactamase repressor; blaR, Beta-lactamase regulatory protein; blaZ, Beta-lactamase; mecI, Methicillin resistance repressor; mecR, Methicillin resistance regulatory protein; mecA, Methicillin resistance protein; blaTEM, Beta-lactamase TEM; erm, Erythromycin methyltransferase; lsaE, lincosamide and streptogramin A resistance protein E; lnu, Lincosamide nucleotidyltransferase; mefE, Macrolide efflux pump; tetK/L, Tetracycline efflux pump; tetM/O, Tetracycline resistance ribosomal protection protein; tetR, Tetracycline resistance repressor; cat, Chloramphenicol acetyltransferase; fexA, phenicol efflux pump; fosD, Fosfomycin resistance gene; fusC, Fusidic acid resistance gene; dfr, Dihydrofolate reductase; mupA, Mupirocin resistance protein; optrA, oxazolidinone resistance protein A.
Results
Patient history and microbiological analyses
A 23-year-old man presented with a closed, unstable compression fracture of the fourth lumbar vertebra following a fall from a second-story balcony. He initially underwent transpedicular stabilization from L2 to L5, and his early postoperative recovery was uneventful. Despite elevated infection markers, he left the hospital against medical advice without receiving antibiotics. Three days later, he returned with worsening back pain, fever, and markedly increased inflammatory markers; imaging revealed spondylodiscitis at L4. During revision surgery, purulent material was observed around the osteosynthesis, and five tissue samples were collected for microbiological analysis. Culture of these samples produced white, hemolytic colonies that were catalase-positive but negative for clumping factor. After protein extraction, MALDI-TOF MS confirmed all isolates as S. pseudintermedius. Blood cultures obtained during readmission also grew S. pseudintermedius in two aerobic vials, while other cultures remained negative. Notably, the patient had no contact with pets either prior to the fall or after discharge, a detail specifically inquired about by the treating physicians given that S. pseudintermedius is typically associated with animals. No mecA or mecC resistance genes were detected, and antimicrobial susceptibility testing indicated that the isolates were susceptible to all tested agents except erythromycin and clindamycin. Collectively, these findings identify S. pseudintermedius as the pathogen responsible for the patient’s postoperative spondylodiscitis. Detailed patient history and microbiological analyses information’s are provided in the Materials and Methods section.
Treatment and outcomes
The S. pseudintermedius infection posed a therapeutic challenge due to the patient’s documented penicillin allergy. Although the strain was methicillin-susceptible, beta-lactam antibiotics were contraindicated, necessitating alternative treatment. Empirical therapy with intravenous clindamycin (600 mg TID) was started for 3 days. Following species identification and antimicrobial susceptibility testing, therapy was changed to intravenous daptomycin (8 mg/kg body weight, corresponding to 1,250 mg once daily) for 3 weeks, in accordance with the AST results and the patient’s clinical condition. Five days after the first revision surgery, a second extensive debridement and lavage were performed. The patient responded well, with significant decreases in CRP and leukocyte counts within 2 weeks, no new neurological deficits, and stable follow-up imaging showing no progression of spondylodiscitis. Upon completion of intravenous therapy, the patient was discharged on oral levofloxacin and rifampicin to ensure eradication of residual bacteria, considering their pharmacokinetics and broad-spectrum activity.
At six-week follow-up, the patient was symptom-free with no signs of infection. The transpedicular dorsal spondylodesis was safely removed 6 months later after CT scans confirmed fracture healing.
Genetic characterization of S. pseudintermedius isolates
To complement the microbiological diagnostics, comprehensive WGS was performed on three S. pseudintermedius isolates of the patient. Two isolates were obtained from infected tissue and blood during the initial surgery, and a third isolate was collected 5 days later during wound revision. Hybrid assemblies were generated using both short-read (Illumina) and long-read (Oxford Nanopore Technologies) platforms. All three isolates contained an identical chromosome of 2,626,791 bp and a 3,100 bp plasmid (pHGW2412) (Figure 3). No SNPs or indels were detected between the isolates (data not shown), indicating infection by a single clonal strain. Therefore, all subsequent analyses were conducted using the genome of the initial isolate recovered from tissue.
Circular map of the S. pseudintermedius HGW2412 chromosome and its plasmid pHGW2412. The innermost circle displays the GC skew (blue and orange), followed by the GC content (black). The next two rings represent the reverse and forward strands, showing the coding DNA sequences (CDS, gray). Also annotated are tRNAs, rRNAs, ncRNAs, tmRNAs, replication origins (rep_origins), and other regulatory sequences. The figure was generated using Proksee (https://proksee.ca/).
Initial sequence analysis was performed using the TORMES pipeline (Quijada et al., 2019), including gene annotation with Prokka (Supplementary Table S1 – annotations tabs) and resistance gene screening via ABRicate (Zankari et al., 2012; Gupta et al., 2014; McArthur and Tsang, 2017) (Supplementary Table S1 – antibiotic resistance). The isolate harbored chromosomal genes encoding proteins responsible for resistance to various antibiotics, including β-lactams (blaZ), chloramphenicol (cat), macrolides (ermB), and aminoglycosides [aadE = ant(6)], aph-Stph, aph(3″)-III, and sat4A. Virulence factor screening with the Virulence Factor Database VFDB, see footnote^5^ revealed high-identity protein homologs of S. aureus toxins including beta-hemolysin (Hlb, 74.09%), staphylococcal enterotoxin C (Sec, 57.58%), superantigen-like protein SSL11 (Set26, 44.68%), and leukocidins LukS and LukF from S. intermedius (98.71 and 99.39% respectively) (Supplementary Table S1). Additional gene homologs were detected for iron acquisition systems (sfaABCD, sbnABCDEFGHI, and sirABC), capsule biosynthesis (cap8OP), adhesion (icaABC, vWb, and fnbB), tissue invasion (lip, geh, nuc, and aur), and immune evasion (sbi) (Supplementary Table S1). Furthermore, an autolysin gene homolog (aae) has been detected. Besides other functions, autolysins play a pivotal role in adherence and biofilm formation and have been described for several species such as S. aureus and various coagulase-negative staphylococci (Hussain et al., 2015; Heilmann et al., 1997; Biswas et al., 2006). The mere presence of homologs to these virulence factors suggests a considerable virulence potential of this isolate, which was supported by the course and severity of the infection.
The plasmid pHGW2412 encodes six distinct proteins, none of which resemble known antibiotic resistance determinants. Two proteins were associated with plasmid replication (Rep_1 and a mobilization protein) (Supplementary Table S1 – annotation pHGW2412). BLAST analyses revealed that pHGW2412-like sequences occur in other S. pseudintermedius genomes, often fragmented and unannotated, likely due to older, less accurate assembly methods. However, the exact biological function of the plasmid remains unknown.
For phylogenetic classification, our isolate was compared with 5,500 S. pseudintermedius genomes from the NCBI RefSeq database, identifying GCA_905169555 as the closest related genome using FastANI (Supplementary Table S1) (Jain et al., 2018). The 100 most similar genomes and our isolate were assigned to Multilocus Sequence Typing (MLST) (Solyman et al., 2013), which were visualized in a minimum spanning tree (Figure 4). Notably, none of the 100 closest genomes shared the sequence type (ST2051) of HGW2412, indicating high diversity. The most similar isolate GCA_905169555 differed by two housekeeping alleles, placing it in close proximity to our isolate (Figure 4). However, only one other ST2051 isolate was identified in the PubMLST database, derived from a cat in Wrocław (Poland) in 2017, but unfortunately, no associated whole-genome sequencing data are available.
Minimum spanning tree based on MLST analysis of S. pseudintermedius HGW2412 and its 100 closest sequences. Sequence types (STs) were assigned using the MLST scheme for S. pseudintermedius implemented in the SeqSphere+ software (Ridom, Germany), applying the “pairwise ignoring missing values” option for allele comparisons. “No group” refers to new or unknown STs that could not be classified and are placed in a separate group. For clarity, the Greifswald isolate HGW2412 (HGW2412_chr) is highlighted with a dashed red circle.
As no core-genome MLST scheme is available for S. pseudintermedius, phylogeny was reconstructed using a core-SNP approach, and the resulting tree was annotated with host and geographic metadata (Figure 5). Results revealed extensive genomic diversity and global distribution, hindering precise phylogenetic placement. HGW2412 clustered with isolates from North America, the Netherlands, Canada, Slovenia, and Kenya; German isolates appeared more distantly related. These findings were consistent with the MLST-based Minimum Spanning Tree (MST) (Figure 4), highlighting the substantial genetic heterogeneity even among the 100 most similar genomes.
Maximum likelihood (ML) tree illustrating the phylogenetic placement of S. pseudintermedius HGW2412 relative to its 100 closest genomes based on core-SNP analysis. The tree is unrooted, and available metadata, including isolation source and country of origin, are annotated. Metadata were obtained from NCBI RefSeq (isolation source, country of isolation). The Greifswald isolate was included in two forms, complete genome (HGW2412_full) and chromosome only (HGW2412_chr), and is highlighted in orange. The figure was generated using Interactive Tree Of Life (iTOL v7.1.1; https://itol.embl.de/).
Genomic AMR gene profiling in S. pseudintermedius
To contextualize the AMR gene repertoire of our isolate HGW2412 within the genetically diverse species S. pseudintermedius, we compared it with all RefSeq genomes available in June 2025 (n = 5,500). In total, 34 AMR genes were identified (Figure 2A and Supplementary Table S2 – results ABRicates/heatmap_5,501 sequences) and assigned to functional gene families. Thirteen genes were associated with aminoglycoside resistance; seven, including regulatory genes, with β-lactam inactivation; four with macrolide resistance; three with tetracycline resistance; two with phenicol resistance; and one gene each with resistance to fosfomycin, fusidic acid, trimethoprim, mupirocin, and oxazolidinones. Aminoglycoside resistance was dominated by aph-Stph (99.7%), while ant6, aph(3″)-III, and sat were detected in approximately 40% of genomes, and aac6-aph2 and aph2 in about 30%. Among β-lactam resistance genes, blaZ (78.4%), mecA (39.5%), and blaTEM-116 (0.14%, n = 8) were identified. MLS resistance genes included erm (42.3%) and mefA (27.0%), whereas lnu and lsaE were rare (2.4 and 0.8%). Tetracycline resistance was mainly associated with tetM or tetO (45.5%), while tetK or tetL occurred in 6.8%. Phenicol resistance genes included cat (14.4%) and fexA (0.4%). Dihydrofolate reductase genes (dfr) (trimethoprim resistance) were present in 37% of genomes. All remaining AMR genes occurred in fewer than 1% of isolates (Figure 2A and Supplementary Table S2 – heatmap_5,501 sequences).
Cluster (cl) analysis of AMR gene prevalence revealed three major groups (Figure 2B and Supplementary Table S2 – SOTA_cl:1 – SOTA _cl:10). Group 1 (clusters 1–2) exhibited a multidrug-resistant gene profile, with six aminoglycoside resistance genes present in >98% of genomes, high frequencies of blaZ (>98%) and mecA (100% in cluster 1, 83.2% in cluster 2), and erm genes in 98.4%. dfr genes were also common (>97% in cluster 1; 82.3% in cluster 2). Tetracycline resistance was dominated by tetM/tetO in cluster 2 (>91%), whereas tetK/tetL were more frequent in cluster 1 (33.8%). The mecA regulators mecI and mecR were detected exclusively in cluster 1. Group 2 (clusters 3–7) was characterized by the generally low prevalence of aac6-aph2 and the mecA regulatory genes mecI and mecR. Clusters 3–5 also showed low levels of blaI/blaR, and blaZ was absent in clusters 4 and 5. Our isolate HGW2412 belonged to cluster 7, which displayed high prevalences of ant6, aph(3″)-III, aph-Stph, and sat (99.7, 99.0, 100.0, and 99.0%), as well as blaI, blaR, blaZ, and erm (96.2, 85.3, 94.9, and 91.1%, respectively). Approximately 30% of cluster 7 genomes carried tetM/tetO, and nearly 60% carried the cat gene, which was also found in HGW2412. Group 3 (clusters 8–10) contained isolates with few AMR genes. Cluster 8, the largest cluster (n = 2,059; 37.4%), harbored aph-Stph (99.7%), the blaIRZ locus, and ~30% mefA, tetM, or tetO. Cluster 9 exhibited the lowest AMR gene content, containing only aph-Stph (100%) despite representing 13.3% of all genomes (n = 732). Cluster 10 resembled cluster 9 but showed higher prevalences of blaI (26.8%), blaZ (25.7%), and mefA (100%). In sum, a total of 42.5% (n = 2,337) of isolates (clusters 1–7) had gene combinations that predicted resistance to aminoglycosides, β-lactams, macrolides, sulfonamides/trimethoprim, and/or tetracyclines. By contrast, most genomes (57.5%, n = 3,164) harbored only a limited number of AMR genes, primarily predicting resistance to aminoglycosides, β-lactams, and/or macrolides, and were represented largely by clusters 8–10. Notably, the mecA gene was rarely detected in clusters 8–10. Finally, the most frequent AMR gene combinations were analyzed and computed for each cluster and are summarized in Table 2 and Supplementary Table S2. As expected, these patterns mirrored the overall AMR gene distribution; however, they also revealed substantial heterogeneity and a broad range of possible AMR gene constellations, particularly within clusters 1–7.
Discussion
This case illustrates the infectious potential of S. pseudintermedius, a species traditionally associated with veterinary medicine but increasingly reported as a human pathogen, particularly in immunocompromised individuals (Pomba et al., 2017). According to the recent case report/overview by Jones et al. (2025), S. pseudintermedius has been implicated in a wide range of clinical manifestations, including skin and soft tissue infections, wound and postoperative wound infections, dog-bite associated infections, pneumonia, rhinosinusitis and otitis, bacteremia, and even infections of internal organs, such as the gastrointestinal tract, joints, bones, and prosthetic implants, underscoring its growing clinical importance. Although human cases were long considered sporadic, both recent and earlier case reports indicate that S. pseudintermedius infections may occur more regularly and perhaps more often go unrecognized than previously assumed, particularly among individuals in close contact with companion animals such as dog owners and veterinarians (Somayaji et al., 2016; Jones et al., 2025). Interestingly, in this case, it is not known how the patient encountered S. pseudintermedius. Although the patient had no documented animal contact, indirect transmission via environmental surfaces, healthcare settings, or unrecognized community reservoirs cannot be ruled out, highlighting the need for broader surveillance of S. pseudintermedius in non-animal settings. We suspect that transmission of the pathogen occurred in the hospital, as early signs of infection were already detectable during hospitalization. Very recently, S. pseudintermedius was isolated from the nasal cavity of a healthcare workers and hospital environmental surfaces, a finding that lends further support to our hypothesis (Besharati et al., 2025).
Before the taxonomic separation of S. pseudintermedius from S. intermedius, the enterotoxigenic potential of S. intermedius, posing public health risks such as outbreaks, had already been documented (Becker et al., 2001). Its virulence factors include cytotoxins, exfoliative toxins, superantigens, and cell wall–associated proteins, which play key roles in the initiation and spread of infections, particularly skin and soft tissue infections and in evading host immune responses (Bannoehr et al., 2011; Pitchenin et al., 2018). Additionally, S. pseudintermedius is capable of forming biofilms, significantly enhancing its resistance and persistence in clinical settings (Teixeira et al., 2024). Enzymatic virulence mechanisms, such as proteases and thermonucleases, further contribute to pathogenicity, with plasma coagulation mediated by von Willebrand factor binding protein emerging as a particularly important factor (Pickering et al., 2021). The comprehensive WGS analysis of HGW2414 revealed a wide array of these diverse virulence factors (Supplementary Table S1). Interestingly, many of these factors share orthologs not only with S. aureus, but also with S. intermedius, S. haemolyticus, and S. epidermidis, suggesting evolutionary parallels in their pathogenic strategies (Bannoehr et al., 2011; Myrenas et al., 2024; Glajzner et al., 2023). These findings indicate that the virulence of S. pseudintermedius may not be exclusively linked to zoonotic infections, emphasizing its potential significance in human disease.
Whole-genome sequencing further enabled the identification of genetic determinants responsible for the observed antibiotic resistance, which partially correlated with phenotypic antimicrobial susceptibility testing (AST) results. Our isolates harbored blaZ resistance genes, which confer resistance to penicillin but not oxacillin. As expected, they were phenotypically methicillin-susceptible, which ultimately facilitated effective treatment and the patient’s recovery. Interestingly, genetic determinants associated with aminoglycoside and chloramphenicol resistance were detected; however, phenotypic resistance to gentamicin was not observed, highlighting the necessity of conventional AST. As mentioned above, our isolate HGW2414 was methicillin-susceptible, yet the increasing prevalence of MRSP markedly complicates the management of infections caused by this species in both veterinary and human medicine (Besharati et al., 2025; Monteiro et al., 2025). Unfortunately, research on antimicrobial-resistant S. pseudintermedius has predominantly focused on animal isolates, with limited reports on human cases involving MRSP (Paul et al., 2011). Our global genomic analysis of 5,501 S. pseudintermedius sequences confirms the frequent occurrence of multidrug-resistant strains (Figure 2 and Supplementary Table S2), theoretically resistant to aminoglycosides/streptothricin, β-lactams, macrolides/lincosamides/streptogramin, tetracyclines, sulfonamides/trimethoprim, and/or phenicols. Some of these putatively multidrug-resistant isolates also carried additional resistance determinants against mupirocin, fosfomycin, fusidic acid, or oxazolidinones, although these were rare. Overall, the analyzed genomes were grouped into three major groups comprising ten distinct AMR gene clusters, within which the resistance patterns were distributed. Overall, 42.5% (cluster 1–7) of all analyzed genomes displayed this multidrug-resistant profile, underscoring the clinical and epidemiological significance of such isolates within the species. In contrast, 57.5% (cluster 8–10) of isolates harbored only a limited number of AMR genes, suggesting that they could theoretically be treated with older, well-established antibiotics, potentially preventing further resistance acquisition. Our genomic AMR findings are broadly consistent with previous studies on antibiotic resistance in S. pseudintermedius (Tyson et al., 2021), although earlier reports often focused on phenotypic resistance rather than AMR gene prevalence (Besharati et al., 2025; Monteiro et al., 2025; Nocera and De Martino, 2024; Morais et al., 2023). Notably, Monteiro et al. (2025) reported a high proportion of fluoroquinolone-resistant isolates, whereas in our dataset we detected, e.g., no plasmid-mediated fluoroquinolone resistance genes, such as qnr or aac(6′)-Ib-cr. This likely reflects the well-described mechanism in staphylococci, in which point mutations in the target genes gyrA (DNA gyrase subunit A) and grlA (topoisomerase IV subunit A) confer fluoroquinolone resistance, a mechanism that was not assessed in our analysis (Tyson et al., 2021; Morais et al., 2023). Cluster analyses revealed patterns of AMR gene accumulation in specific isolates, potentially linked to other genetic determinants, such as the frequent occurrence of MRSP among particular sequence types (Besharati et al., 2025; Nocera and De Martino, 2024). Both, our data and previous reports highlight the high prevalence of mecA-positive S. pseudintermedius isolates, reinforcing the need for systematic AMR screening of S. pseudintermedius in animal and human medicine. Such surveillance is essential to optimize therapy, avoid unnecessary antibiotic use, and mitigate the spread of multidrug-resistant strains among zoonotic pathogens. Thus, the multidrug-resistant profile of MRSP and, to a lesser extent MSSP, especially regarding resistance to multiple key antibiotic classes, underscores the need for strong antimicrobial stewardship, informed by both phenotypic and genomic resistance testing, even in MSSP (Kadlec and Schwarz, 2012).
Since the source of infection could not be identified, phylogenetic analyses may aid in the spatial classification of the isolates. Advances in molecular typing have greatly enhanced our understanding of the SIG species (Bannoehr et al., 2007), but a specific MLST scheme for S. pseudintermedius was only introduced in 2013, accompanied by the establishment of a public database to improve surveillance of this species (Solyman et al., 2013). However, S. pseudintermedius exhibits substantial clonal diversity, which complicates precise epidemiological surveillance using MLST (Morais et al., 2023; Grist et al., 2025). While certain sequence types (e.g., ST45, ST71, ST258) are more prevalent, genetic diversity remains remarkably high even within a single geographic region or same isolation hosts (Solyman et al., 2013; Morais et al., 2023; Grist et al., 2025; Damborg et al., 2016; Videla et al., 2018). This was also evident in our analysis, where even those genomes identified as highly similar by FastANI differed by at least two alleles in the MLST scheme (Figure 4). We identified sequence type ST2051 for the HGW2412 isolate, a ST previously found in Poland. It is possible that both isolates are genetically closely related, a hypothesis supported by their geographic proximity; however, no WGS data of the polish isolate were available to confirm this. To address this epidemiological and diagnostic limitation, WGS combined with core genome MLST (cgMLST) enables high-resolution genotyping through genome-wide, gene-by-gene allele calling of conserved loci. This approach is recombination-robust, standardized, and scalable, making it ideally suited for routine laboratory use, including the surveillance of multidrug-resistant bacteria (Mellmann et al., 2016). To date, no cgMLST scheme exists for S. pseudintermedius, likely due to its high genetic diversity, which poses challenges for standardized scheme development. However, establishing such a scheme would be highly desirable, particularly in the context of the “One Health” frame work. Our core-SNP analysis confirmed the MLST results (Figure 4) and highlighted substantial genetic variability, which impeded precise phylogenetic resolution (Figure 5). The effectiveness of the core-SNP approach became particularly evident in the identification of closely related sequences, especially those from Spain, as well as from Slovenia and the USA (Figure 5); yet a clear geographic association could not be established based on these results. However, in addition to molecular-based methods, MALDI-TOF MS has made significant advances and greatly facilitated the species-level identification of SIG members (Canver et al., 2019; Decristophoris et al., 2011), although it is not suitable for resolving epidemiological questions.
Conclusion
To support ongoing surveillance and research, the development of novel, accurate epidemiological tools, such as an internationally standardized cgMLST scheme for S. pseudintermedius, is essential. This scheme should be systematically validated across both animal and human S. pseudintermedius isolates to ensure its efficacy in One Health-based AMR gene surveillance. Equally important is the rapid and reliable identification of S. pseudintermedius in clinical settings (human and animal!), as timely recognition of the pathogen is critical not only for initiating targeted antimicrobial therapy and improving clinical outcomes, but also for preventing further spread and limiting the development of additional multidrug resistance.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bannoehr J. Ben Zakour N. L. Reglinski M. Inglis N. F. Prabhakaran S. Fossum E. . (2011). Genomic and surface proteomic analysis of the canine pathogen Staphylococcus pseudintermedius reveals proteins that mediate adherence to the extracellular matrix. Infect. Immun. 79, 3074–3086. doi: 10.1128/IAI.00137-11, 21576333 PMC 3147560 · doi ↗ · pubmed ↗
- 2Bannoehr J. Ben Zakour N. L. Waller A. S. Guardabassi L. Thoday K. L. van den Broek A. H. . (2007). Population genetic structure of the Staphylococcus intermedius group: insights into agr diversification and the emergence of methicillin-resistant strains. J. Bacteriol. 189, 8685–8692. doi: 10.1128/JB.01150-07, 17905991 PMC 2168937 · doi ↗ · pubmed ↗
- 3Bannoehr J. Guardabassi L. (2012). Staphylococcus pseudintermedius in the dog: taxonomy, diagnostics, ecology, epidemiology and pathogenicity. Vet. Dermatol. 23, 253–266. doi: 10.1111/j.1365-3164.2012.01046.x 22515504 · doi ↗ · pubmed ↗
- 4Barash N. R. Birkenheuer A. J. Vaden S. L. Jacob M. E. (2018). Agreement between parallel canine blood and urine cultures: is urine culture the poor man's blood culture? J. Clin. Microbiol. 56, 1–9. doi: 10.1128/JCM.00506-18, 29997202 PMC 6113489 · doi ↗ · pubmed ↗
- 5Becker K. Keller B. von Eiff C. Bruck M. Lubritz G. Etienne J. . (2001). Enterotoxigenic potential of Staphylococcus intermedius. Appl. Environ. Microbiol. 67, 5551–5557. doi: 10.1128/AEM.67.12.5551-5557.2001, 11722906 PMC 93343 · doi ↗ · pubmed ↗
- 6Becker K. von Eiff C. Keller B. Bruck M. Etienne J. Peters G. (2005). Thermonuclease gene as a target for specific identification of Staphylococcus intermedius isolates: use of a PCR-DNA enzyme immunoassay. Diagn. Microbiol. Infect. Dis. 51, 237–244. doi: 10.1016/j.diagmicrobio.2004.11.010, 15808314 · doi ↗ · pubmed ↗
- 7Besharati R. Haghbin A. Hashemi S. A. Vosoughi-Motlagh A. Azimian A. (2025). Molecular detection and characterization of methicillin-resistant Staphylococcus pseudintermedius (MRSP) ST 2361 in a healthcare setting. One Health 21:101265. doi: 10.1016/j.onehlt.2025.101265, 41283071 PMC 12637273 · doi ↗ · pubmed ↗
- 8Biswas R. Voggu L. Simon U. K. Hentschel P. Thumm G. Gotz F. (2006). Activity of the major staphylococcal autolysin Atl. FEMS Microbiol. Lett. 259, 260–268. doi: 10.1111/j.1574-6968.2006.00281.x, 16734789 · doi ↗ · pubmed ↗