Familial interstitial lung disease: emerging insights into screening and genetic risk
Ana Paola Hernández Cristancho, Lurdes Planas-Cerezales, Maria Molina-Molina, Raphael Borie, Wim A. Wuyts, Meritxell Jodar, Jacobo Sellares, Fernanda Hernandez-Gonzalez

TL;DR
This review explores how genetics and imaging help understand and screen for familial pulmonary fibrosis, a type of lung disease that runs in families.
Contribution
The paper integrates recent advances in genetics, radiology, and clinical strategies for early detection and prevention of familial pulmonary fibrosis.
Findings
Genetic variants in telomere-related genes, surfactant proteins, and MUC5B influence familial pulmonary fibrosis risk and progression.
High-resolution CT scans detect early lung abnormalities in at-risk relatives, especially those with known genetic mutations.
Combining genetic, radiologic, and biomarker data may improve risk assessment and enable early intervention for familial pulmonary fibrosis.
Abstract
Familial pulmonary fibrosis (FPF) is increasingly recognized as a distinct entity within the spectrum of interstitial lung diseases (ILDs), characterized by a significant genetic contribution involving genetic variation telomere-related genes, surfactant protein genes, and the MUC5B promoter polymorphism. These variants influence disease susceptibility, clinical course, and prognosis. Moreover, high-resolution computed tomography (HRCT) has revealed interstitial lung abnormalities (ILAs) as early manifestations in at-risk relatives, particularly amongst individuals with pathogenic variants, highlighting its central role in early detection. Despite substantial progress, significant challenges persist, particularly regarding the unidentified genetic variants in a considerable proportion of cases and the psychosocial impact associated with familial screening. Some studies suggest that…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
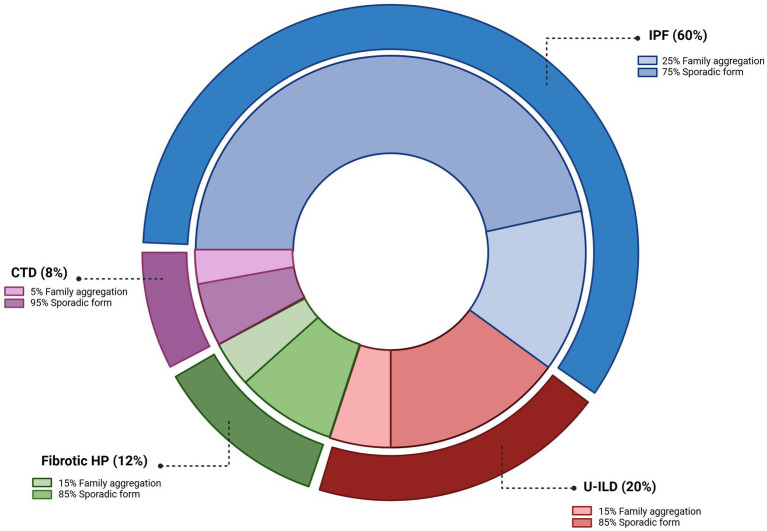 Figure 1
Figure 1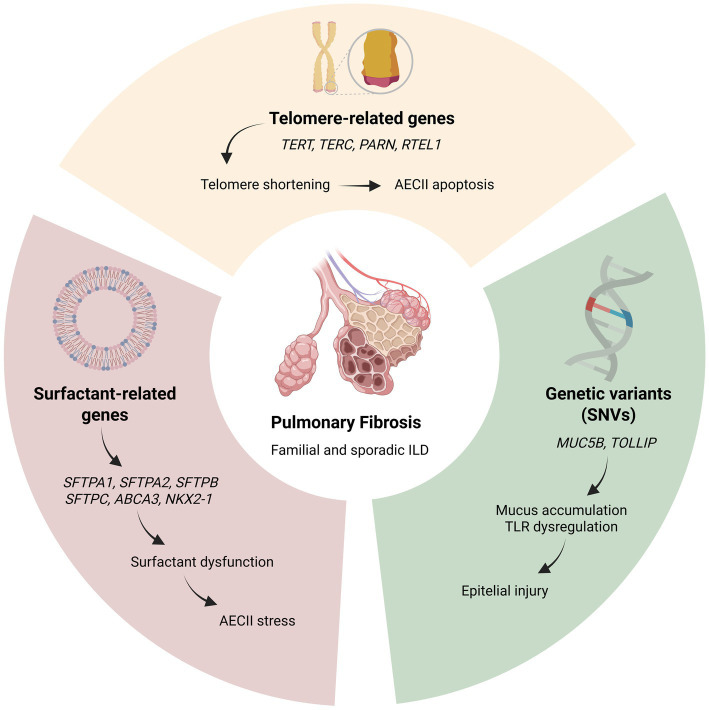 Figure 2
Figure 2| Domain | Criteria |
|---|---|
| Symptoms | Any amount of dyspnoea and/or cough that a clinician attributes to ILD |
| Physiology | Any abnormality in FVC, TLC, or DLCO attributed to ILD (value or |
| Meets physiologic criteria for progressive pulmonary fibrosis attributed to ILD | |
| Imaging (HRCT scan) | Fibrotic abnormalities (honeycombing and/or reticulation with traction bronchiectasis) involving ≥5% of total lung volume by visual estimate |
| Progressive fibrotic abnormality on serial HRCT | |
| Presence of a major fibrotic ILD pattern (UIP/probable UIP, fibrotic HP, fibrotic NSIP) | |
| Pathology | Presence of a major fibrotic ILD pattern (UIP/probable UIP, fibrotic HP, fibrotic NSIP) |
| Domain, Criteria | Recommendations |
|---|---|
| Chest HRCT scan in relatives of FPF patients | For adults over 50 years with a |
| Chest HRCT scan in relatives of IPF patients | For adults over 50 years with a |
| Timing of screening | Screening is generally recommended to begin |
| When the initial HRCT scan is | |
| MUC5B promoter variant testing | It has been strongly advised |
| Genetic testing for | |
| Telomere length measurement as initial screening | Telomere length assessment is |
| Telomere length testing may be valuable | |
| Baseline telomere length in ILAs | Routine baseline telomere length testing in all patients with ILAs is |
| Testing may be appropriate in cases with a |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInterstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Neonatal Respiratory Health Research · Pulmonary Hypertension Research and Treatments
Introduction
1
Interstitial lung diseases (ILDs) encompass a heterogeneous group of disorders characterised by variable degrees of inflammation and fibrosis of the pulmonary interstitium (1). Despite their clinical, radiological, and physiological overlap (2), their aetiology is diverse. Environmental exposures and autoimmune conditions are well-recognised risk factors, but genetic predisposition has emerged as a key determinant of disease susceptibility and progression (3). Within this evolving landscape, interstitial lung abnormalities (ILAs) have been identified as an important radiological entity, defined as non-dependent, bilateral parenchymal abnormalities detected on high-resolution computed tomography (HRCT), including ground-glass or reticular opacities, lung distortion, traction bronchiectasis, and/or honeycombing involving more than 5% of a lung zone by visual assessment, but not meeting the diagnostic criteria for ILD (Table 1) (4, 5).
Idiopathic pulmonary fibrosis (IPF) was long regarded as a sporadic condition. Yet, over the past two decades, accumulating evidence of familial clustering has reshaped this paradigm. Approximately 20–25% of patients with IPF report a family history of ILD, while 5–10% of those with fibrotic non-IPF ILDs—such as hypersensitivity pneumonitis (HP) or connective tissue disease-associated ILD (CTD-ILD)—also have affected relatives (6). The recognition of familial pulmonary fibrosis (FPF) has been reinforced by landmark genetic discoveries, including the MUC5B promoter polymorphism, strongly associated with ILD susceptibility with usual interstitial pneumonia (UIP) pattern, and telomere-related gene (TRG) variants recognised as the most frequent monogenic cause (7, 8). These insights have deepened our understanding of pathogenesis and raised expectations for targeted therapeutic strategies (6).
Nevertheless, major uncertainties remain. Up to three-quarters of patients with FPF lack an identifiable variant, and no universally accepted genetic panel is standardized for clinical use. Furthermore, the predictive value of genetic findings for therapeutic response is largely unexplored, and strategies for screening at-risk relatives remain fragmented and controversial. Beyond the well-established associations, new susceptibility variants continue to be identified, underlining the complexity of gene–environment interactions and the urgent need for integrative models that combine radiological, functional, genetic, and biomarker data.
Moreover, radiological concepts have also evolved. ILAs are now recognized as early manifestations in at-risk relatives, with 15–30% of asymptomatic first-degree relatives of IPF patients demonstrating ILAs (4, 9, 10). Subpleural fibrotic ILAs, particularly in individuals with TRG variants or the minor T-allele of the MUC5B promoter polymorphism rs35705950, are associated with a higher risk of progression to overt ILD (11). By contrast, pulmonary function tests such as spirometry and DLCO, though valuable for longitudinal monitoring, lack sensitivity for detecting early disease, underscoring the central role of HRCT in screening strategies of patients with FPF (5).
In this review, we summarise current knowledge of the genetic basis of FPF, highlight emerging concepts in risk stratification, and critically discuss the implications for screening, early diagnosis, and clinical management of at-risk relatives, with an emphasis on current challenges and future opportunities.
Familial pulmonary fibrosis and ILA: a conceptual framework
2
FPF is broadly defined as the presence of fibrotic ILD in an individual together with at least one first- or second-degree relative affected by a fibrotic phenotype (12). Although HRCT is essential for ILD phenotyping, familial cases may display a broad spectrum of classic radiographic patterns (13, 14).
IPF is the most frequent phenotype within FPF (14), but other forms—including fibrotic HP, CTD-ILD, and unclassifiable ILD (U-ILD)—are also represented (Figure 1). Familial ILDs are typically associated with an earlier age of onset and a more aggressive clinical course compared with sporadic cases (15). While a positive family history alone is insufficient to establish a diagnosis, it carries significant prognostic implications (6). In another paper, it is suggested that the disease trajectory is similar with a certain family, pointing even more towards FPF as a strong prognostic factor (16).
Clinical spectrum of diagnoses amongst patients with familial pulmonary fibrosis (FPF). FPF most frequently presents with an idiopathic pulmonary fibrosis (IPF) phenotype, though other ILD subtypes are also observed. HP, hypersensitivity pneumonitis; CTD, connective tissue disease; U-ILD, unclassifiable interstitial lung disease. Adapted from Zhang and Newton (73) created in https://BioRender.com.
Beyond well-characterised familial clusters, first-degree relatives of patients with sporadic IPF also have an increased risk of ILAs. Although bilateral findings are required by definition, unilateral abnormalities may also be clinically relevant in selected high-risk populations. In particular, individuals with a strong family history of ILD or individuals with known pathogenic variants may be at increased risk of progression to ILD even when HRCT changes are unilateral (5, 17).
Three major ILA subcategories are recognised: (1) nonsubpleural ILAs: abnormalities without predominant subpleural localization; (2) subpleural non-fibrotic ILAs: abnormalities with predominant subpleural localisation but no evidence of fibrosis; and (3) subpleural fibrotic ILAs: abnormalities with predominant subpleural localization and imaging evidence of pulmonary fibrosis (18, 19). In both familial and sporadic IPF cohorts, 15–30% of asymptomatic first-degree relatives demonstrate ILAs on HRCT (4, 9, 10). This finding is strongly associated with older age, a history of smoking, the gain-of-function MUC5B T minor allele rs35705950 and shortened peripheral blood leukocyte telomere length (4, 5, 10, 20).
Genetic basis of familial ILD
3
The strongest evidence for a genetic predisposition to ILD comes from familial clustering, particularly in homozygous twins raised apart (21), multigenerational families (22, 23), and genetically related relatives (12). In FPF, disease susceptibility arises from the combined influence of common and rare genetic variants, estimated to account for approximately 12.4% and 25–30% of overall risk, respectively (24). There is a wide range between rare genetic variants with large effect sizes, typically associated with monogenic (Mendelian) disorders, and frequent genetic variants with smaller effect sizes that underlie polygenic or complex diseases (25). Rare gene variants identified through candidate gene and family-based studies predominantly affect pathways involved in telomere maintenance, surfactant metabolism, and mitotic spindle assembly. Importantly, a single gene may harbour multiple disease-associated variants that can be categorized as either common or rare.
Pathogenic variants in TRG and surfactant protein genes represent the most consistent genetic determinants of FPF. In parallel, common variants, particularly in MUC5B (T minor allele rs35705950) and TOLLIP (including rs111521887 and rs3750920T), have been shown to confer susceptibility across several ILD phenotypes with UIP pattern, including IPF and rheumatoid arthritis-associated ILD (RA-ILD) (26), and importantly, both variants are not only associated with IPF susceptibility but also with better survival in IPF patients (27).
In addition, genome-wide association studies (GWAS) suggest that common single-nucleotide polymorphisms (SNPs) contribute roughly 5–15% of the genetic liability for IPF in the general population (28), implicating genes related to telomere biology, alveolar epithelial barrier integrity, and host defense mechanisms, as shown in Figure 2. Beyond inherited variation, epigenetic processes—including DNA methylation, histone modifications, and non-coding RNAs such as miR-21 and miR-29—modulate fibrotic pathways, thereby influencing disease initiation, progression, and phenotypic heterogeneity (29). Despite these advances, a substantial portion of the genetic risk underlying IIPs remains unexplained.
Genetic basis of familial ILD (74). AECII, alveolar type II cell; TLR, Toll-like receptor. Created in https://BioRender.com.
Telomere dysfunction
3.1
Telomeres are six-nucleotide repeats located at the ends of chromosomes that prevent DNA degradation and fusion. The enzyme telomerase preserves this structure by maintaining and elongating telomeres. Increasing evidence indicates that telomere shortening and the consequent genomic instability are central drivers of fibrotic processes (30, 31). Although telomere attrition occurs as part of normal ageing, pathogenic variants in TRG accelerate this process and often result in premature shortening. Furthermore, different environmental factors may induce telomere attrition such as smoking and radiation. Once telomere length falls below a critical threshold—defined as under the 10th percentile for age (32)—DNA damage responses are triggered, leading to apoptosis (33), particularly in alveolar epithelial type II cells (AECII) (34).
The clinical spectrum associated with TRG variants is heterogeneous. Approximately 50% of individuals with TRG variants develop IPF, while others present with fibrotic HP (7–12%), CTD-ILD (2–3%), U-ILD (8–20%), or other idiopathic interstitial pneumonias (14–18%) (35, 36). Overall, TRG variants are identified in roughly one-quarter of FPF kindreds, with TERT variants accounting for 8–15% of cases (8, 36, 37), PARN and RTEL1 for 5–10% each (38), and TERC for 1–2% (36).
Importantly, short age-adjusted telomere length is not restricted to familial disease. It is found in around 50% of patients with FPF (38, 39), but also in sporadic ILD, including 20–60% of IPF (40, 41), 20–35% of fibrotic HP (42), and approximately 26% of RA-ILD (43). The mechanism leading to short telomere in those patients might include frequent variant in non-TRG and environmental risks factors (44). These findings underscore telomere dysfunction as a convergent pathogenic mechanism that extends across both familial and sporadic forms of ILD. Current evidence suggests that environmental exposures, particularly fine particulate air pollution (PM2.5), adversely affect patients with telomere dysfunction by promoting oxidative stress, genomic instability, and accelerated telomere shortening, thereby contributing to cellular senescence and pulmonary fibrosis progression. Recent studies further demonstrate associations between PM2.5 exposure, epigenetic alterations, and increased mortality in patients with fibrosing ILD (45).
Beyond the lung, telomere-related disorders exhibit incomplete penetrance and variable expressivity, which likely reflect the combined influence of environmental stressors, genetic background, and stochastic factors. A genetic anticipation phenomenon—characterized by earlier onset and more severe manifestations in successive generations—has also been observed in telomere biology disorder (TBD) due to progressive telomere shortening. Clinically, these entities are now recognized as part of a TBD, encompassing pulmonary fibrosis that may coexist with extrapulmonary involvement of variable severity, ranging from subtle laboratory abnormalities (such as premature greying, cytopenias or elevated liver enzymes) to overt organ failure (e.g., bone marrow failure, myelodysplastic syndrome/acute myeloid leukaemia or hepatic cirrhosis) (46).
Surfactant related genes
3.2
Beyond their essential role in maintaining alveolar stability, surfactant proteins contribute to pulmonary host defense by regulating immune responses and inflammation. They constitute up to 10% of total surfactant weight and include five key proteins: SP-A1, SP-A2, SP-B, SP-C, and SP-D, encoded by SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD, respectively. Gene variants in SFTPA1, SFTPA2, SFTPB, and SFTPC are recognised monogenic causes of ILD and lung cancer (25, 47, 48).
Amongst these, SFTPB variants most frequently lead to severe neonatal respiratory failure, whereas SFTPA1, SFTPA2, and SFTPC genetic variation are implicated in both pediatric and adult ILD, particularly pulmonary fibrosis (49). In addition, gene variant involved in surfactant lipid transport, such as ABCA3 (ATP binding cassette subfamily A member 3), and regulatory genes including NKX2-1 (encoding thyroid transcription factor 1, TTF1), which governs transcription of both surfactant proteins and ABCA3, have also been identified (12). Collectively, these findings highlight disruption of surfactant homeostasis as a distinct group of monogenic causes of ILD (49).
From a clinical perspective, the identification of a rare surfactant gene variant in a proband has implications for family members. Genetic testing and radiological screening of unaffected individuals with pathogenic variants may enable early recognition of subclinical disease, providing opportunities for timely intervention and counselling (50).
MUC5B and disease susceptibility
3.3
Mucins (MUC) are gel-forming glycoproteins that constitute key components of airway mucus. The human genome encodes 17 mucins, of which MUC5AC and MUC5B are the most abundantly expressed in the airways (51). MUC5B, located on chromosome 11, encodes mucin 5 subtype B, a glycoprotein secreted by submucosal glands and airway secretory cells (51). It plays a central role in mucus production, mucociliary clearance, host defense, and the maintenance of airway and lung homeostasis (52).
Aberrant expression of MUC5B has been implicated in several respiratory diseases, including chronic obstructive pulmonary disease (COPD), asthma, cystic fibrosis (53), and severe community-acquired pneumonia (54). In the context of pulmonary fibrosis, excessive MUC5B production promotes mucus accumulation in the distal airways and impairs mucociliary clearance. This accumulation favours the retention of inhaled particles—such as cigarette smoke and air pollutants, both recognised risk factors for IPF—and contributes to epithelial injury. Moreover, excess MUC5B disrupts surfactant function and hampers AECII cell repair mechanisms (7). Collectively, these processes foster persistent epithelial damage, fibroproliferation, and honeycomb cyst formation, which are hallmark features of progressive pulmonary fibrosis (55).
Moreover, the MUC5B promoter variant rs35705950 is a strong genetic risk factor for rheumatoid arthritis-associated ILD (RA-ILD), particularly in patients with a UIP pattern and in familial forms of ILD. While this variant does not increase susceptibility to rheumatoid arthritis itself, it confers a markedly increased risk of ILD amongst RA patients, with effect sizes comparable to those observed in IPF (56).
Toll-interacting protein (TOLLIP)
3.4
TOLLIP encodes a ubiquitin-binding protein that regulates multiple steps of Toll-like receptor (TLR) signalling. It is expressed across diverse lung cell populations—including monocytes, macrophages, regulatory T cells, and alveolar epithelial type I cells—in both healthy individuals and patients with IPF (57). Four isoforms (A–D) have been identified; three are expressed in human mononuclear cells, where they modulate innate immune responses and influence epithelial cell apoptosis (57).
Through its roles in inflammation regulation, autophagy, and vacuolar trafficking, TOLLIP has been implicated in a broad range of pulmonary diseases, including IPF, COPD, asthma, and respiratory infections (57). In the context of IPF, SNPs within the TOLLIP locus on chromosome 11 have been associated not only with disease susceptibility but also with prognosis (58). These findings suggest that TOLLIP functions as both a regulator of innate immunity and a genetic determinant of interindividual variability in fibrotic lung disease.
Genetic variation in TOLLIP has been implicated in both susceptibility to pulmonary fibrosis and disease progression. While the minor C allele of TOLLIP rs5743890 appears protective against ILD development, it is paradoxically linked to worse survival and faster progression in established IPF. Other variants (rs111521887G, rs3750920T) increase ILD risk, though their prognostic impact is less consistent. Mechanistically, risk alleles are associated with reduced TOLLIP expression, enhanced epithelial apoptosis, and dysregulated innate immunity, promoting fibrotic remodelling (26).
Importantly, TOLLIP also has pharmacogenetic relevance, as IPF patients with the rs3750920 TT genotype benefit from N-acetylcysteine, whereas those with the CC genotype may experience harm, supporting a potential role for genotype-guided therapy; no such association has been observed with antifibrotic agents (59). In contrast, no association has been demonstrated between TOLLIP variants and response to antifibrotic agents such as nintedanib or pirfenidone (60).
Collectively, these findings indicate that TOLLIP genetic variants influence both disease susceptibility and progression in pulmonary fibrosis and may have therapeutic implications for NAC treatment, though not for antifibrotic therapies.
Clinical implications for at-risk relatives
4
One of the most pressing and unresolved challenges in FPF lies in defining appropriate clinical strategies for the relatives of affected individuals. Family members often express a strong interest in assessing their personal risk for developing ILD. However, despite increasing scientific attention, insufficient understanding prevents reliable predictions about individual susceptibility, timing of disease onset, or the mechanisms driving its pathogenesis—thereby limiting opportunities for proactive health management.
Although there is growing support for HRCT scan-based screening in asymptomatic relatives (12, 61), no consensus exists regarding fundamental aspects such as the optimal age to initiate screening, the frequency of follow-up, or the management of positive findings. Cohort studies suggest that 14–25% of asymptomatic relatives, at a mean age of approximately 50 years, already exhibit ILA (4, 5). Established risk factors—including older age, smoking, and shortened telomere length—contribute to risk stratification (9, 10, 62), but remain insufficient for precise prediction. Alarmingly, about 20% of these individuals progress to extensive ILA or clinical ILD within 5 years, despite having preserved pulmonary function at baseline (5). This underscores the limited sensitivity and specificity of pulmonary function testing as a screening tool, even though such tests remain valuable for longitudinal monitoring.
Genetic insights further highlight the complexity of FPF. Despite advances in sequencing technologies, up to 75% of patients lack an identifiable pathogenic variant. Moreover, even when gene variants are detected, the genetically complex and multifactorial nature of the disease makes the dichotomy of ‘with’ versus ‘without pathogenic variants’ misleading. Consequently, most families occupy an intermediate zone in which genetic information provides limited predictive value.
Together, these observations reveal a critical gap: current approaches are fragmented, and neither radiology nor genomics in isolation can provide the clarity that families seek. What is urgently needed is an integrated, evidence-based framework that bridges imaging, genetics, and clinical monitoring to truly inform decision-making in at-risk populations.
Screening challenges and preventive approaches in FPF
4.1
An important consideration in screening programmes for FPF is the potential impact on asymptomatic individuals. The identification of radiological, genetic, or biological abnormalities has been associated with feelings of regret and increased psychological burden amongst relatives who undergo screening (63). Consequently, any screening initiative should ideally be supported by appropriate multidisciplinary approach, including emotional, and decisions regarding testing must be accompanied by transparent and understandable about the implications of potential outcomes.
While prospective studies in FPF relatives are still lacking, practical recommendations can nonetheless be offered. These include minimising modifiable risk factors such as smoking, occupational exposures, and air pollution, as well as ensuring that vaccinations against respiratory pathogens are up to date. Indeed a recent prospective study showed that most relatives have at least one harmful respiratory exposure that may modified after genetic counselling (64). Although such measures do not replace future disease-modifying therapies, they may help reduce the risk of disease development or progression and remain relevant for improving overall health and resilience.
Genetic testing is most strongly indicated in patients with familial ILD, particularly those with two or more biologically related affected relatives, early-onset disease (<50 years), unusually severe or rapidly progressive disease, or syndromic features suggestive of a TBD. When testing is undertaken, the appropriate strategy is to begin with the index case or, in its absence, with an affected family member, as this increases the likelihood of identifying a segregating pathogenic variant. Testing unaffected relatives in the absence of this step may yield results that are challenging to interpret and of limited clinical value. It is important to recognise that only a positive genetic test result is unequivocally informative, but a negative result does not exclude genetic risk. To maximise diagnostic yield, testing should be reserved for patients with a high pre-test probability, as defined by clinical features and family history (65).
Importantly, screening strategies in FPF should not be limited to the detection of fibrosing lung disease. Given the multisystemic nature of telomere-related disorders, evaluation should also include potential extrapulmonary manifestations that may precede or accompany pulmonary fibrosis. Recognising these systemic features can improve early identification of at-risk individuals and refine risk stratification within affected families.
Moreover, about one-third of lung transplant recipients with PPF have telomere-dysfunction related to pathogenic TRG variants and/or short telomere length, and although carefully selected patients do not show consistently worse short- or mid-term outcomes compared with non-TRG variants/telomere shortening recipients, emerging evidence suggests that post-transplant care and treatment management may be more complex—given higher rates of complications such as CMV infection, cytopenias, and anastomotic problems—though definitive conclusions remain limited by heterogeneous data (66).
A positive result carries several potential implications. It may influence therapeutic decisions, such as transplant timing or the initiation of antifibrotic therapy; guide targeted screening of at-risk relatives; and support referral for specialised genetic counselling. These benefits must, however, be balanced against the limited variant interpretation, and the psychological impact of results. Together, these considerations highlight the need for multidisciplinary decision-making and careful integration of genetic testing into the broader clinical context of ILD.
In essence, successful FPF screening requires an integrated approach that combines clinical vigilance, preventive strategies, genetic counselling and psychological support, ensuring that early detection truly translates into meaningful patient benefit.
Evidence-based suggestions for screening
4.2
Given the growing recognition of FPF and the increasing availability of advanced imaging and genetic tools, the question of how and when to screen at-risk relatives has gained clinical relevance. Although high-quality evidence remains limited, recent studies and expert consensus provide a rationale for pragmatic, evidence-informed screening approaches.
A meticulous collection of the family history—ideally covering at least three generations—is a cornerstone of FPF evaluation. A detailed pedigree allows clinicians to identify inheritance patterns, anticipate variable disease expression, and guide genetic testing strategies. Importantly, family histories should be updated at each clinical visit, as new cases of interstitial lung disease or related extrapulmonary manifestations may emerge over time, refining the assessment of familial risk.
In this regard, the American Thoracic Society clinical statement presented evidence-based suggestions for evaluation and management of adults with ILA and a first-degree relative with pulmonary fibrosis (67). The evidence-based suggestions shown in Table 2 aim to guide clinicians in identifying ILA or ILD at early, potentially reversible stages (4, 10, 14, 62, 68–70). Together, these recommendations provide support to clinicians by standardizing the approach to ILAs. Although there are several important unanswered questions, it gives a direction for future initiatives, while emphasizing the need to identify treatment options for ILA.
Unresolved clinical challenges in asymptomatic relatives
4.3
The management of asymptomatic relatives of patients with FPF remains a major unresolved challenge. While many seek clarity regarding their individual risk, evidence is insufficient to define standardised protocols for screening or follow-up. Cohort studies indicate that a significant proportion of relatives may harbour ILA before symptoms emerge, yet predictors of progression are poorly understood (4, 14, 71). This uncertainty complicates decisions on the timing and intensity of surveillance and raises concerns about the psychological burden of monitoring individuals who may never develop clinically relevant disease.
Role of HRCT scan and lung function testing in early detection
4.4
HRCT scan is currently the most sensitive tool for detecting early ILD or ILA in at-risk relatives, though its optimal use remains debated. Critical questions—such as the appropriate age to begin screening, the interval between scans, and the management of indeterminate findings—remain unanswered. Moreover, HRCT entails radiation exposure and a risk of overdiagnosis or anxiety in otherwise healthy individuals (14). Emerging imaging technologies, including lung ultrasound and photon-counting computed tomography, may help address some of these limitations in the future. Pulmonary function tests, although insufficiently sensitive for early abnormalities detection, remain valuable for longitudinal monitoring once ILD is established (67).
Towards multimodal risk prediction and personalised approach
4.5
A central limitation of the current landscape is that no single modality—imaging, lung function testing, genetic analysis, or biomarker measurement—can independently provide accurate prediction of disease risk. Future advances in FPF management will likely rely on integrated, personalized models that combine radiologic, physiologic, genetic, and molecular data. Such multidimensional approaches may enable early identification of high-risk individuals, refine surveillance strategies, and pave the way for preventive or early therapeutic interventions in FPF.
Ongoing research is increasingly focused on the discovery of biomarkers to refine risk stratification and disease monitoring. Shortened telomere length has been associated with a higher prevalence of ILAs and systemic manifestations (e.g., liver disease, bone marrow failure), although its limited sensitivity, specificity, and technical complexity currently preclude routine clinical use. Circulating biomarkers such as KL-6 have shown promise as indicators of alveolar injury and fibrotic activity, but their predictive value in asymptomatic relatives remains to be validated.
Notably, the PRECISIONS trial, which randomized patients with IPF carrying the TOLLIP rs3750920 TT genotype to receive oral N-acetylcysteine or placebo, demonstrated a pharmacogenetic interaction between N-acetylcysteine and the TOLLIP polymorphism, highlighting the critical need for molecularly guided, personalized therapeutic strategies and randomized clinical trials and representing an important step towards precision medicine in fibrotic ILD (72).
Conclusion
5
Genetic and epigenetic discoveries have deepened our understanding of ILD susceptibility, onset, and prognosis. However, no standardised genetic panel currently exists, and the therapeutic implications of these findings remain uncertain, highlighting the urgent need for further research and precision medicine approaches.
The screening of asymptomatic relatives continues to raise psychological and ethical concerns, underscoring the importance of counselling and transparent communication. Although antifibrotic agents are not yet suitable for preventive use, risk reduction strategies—such as smoking cessation, minimising environmental exposures, and maintaining appropriate vaccination—remain fundamental.
Looking ahead, the development of preventive therapies may transform ILD from a relentlessly progressive condition into one where early intervention and even prevention become possible, ultimately improving long-term outcomes for patients and their families.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Maher TM. Interstitial lung disease: a review. JAMA. (2024) 331:1655–65. doi: 10.1001/jama.2024.366938648021 · doi ↗ · pubmed ↗
- 2Travis WD Costabel U Hansell DM King TE Lynch DA Nicholson AG . An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. (2013) 188:733–48. doi: 10.1164/rccm.201308-1483 ST 24032382 PMC 5803655 · doi ↗ · pubmed ↗
- 3Lederer DJ Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. (2018) 378:1811–23. doi: 10.1056/NEJ Mra 170575129742380 · doi ↗ · pubmed ↗
- 4Hunninghake GM Quesada-Arias LD Carmichael NE Martinez Manzano JM de Frías SP Baumgartner MA . Interstitial lung disease in relatives of patients with pulmonary fibrosis. Am J Respir Crit Care Med. (2020) 201:1240–8. doi: 10.1164/rccm.201908-1571 OC 32011908 PMC 7233344 · doi ↗ · pubmed ↗
- 5Salisbury ML Hewlett JC Ding G Markin CR Douglas K Mason W . Development and progression of radiologic abnormalities in individuals at risk for familial interstitial lung disease. Am J Respir Crit Care Med. (2020) 201:1230–9. doi: 10.1164/rccm.201909-1834 OC 32011901 PMC 7233345 · doi ↗ · pubmed ↗
- 6Cutting CC Bowman WS Dao N Pugashetti JV Garcia CK Oldham JM . Family history of pulmonary fibrosis predicts worse survival in patients with interstitial lung disease. Chest. (2021) 159:1913–21. doi: 10.1016/j.chest.2021.01.02633484728 PMC 8173755 · doi ↗ · pubmed ↗
- 7Seibold MA Wise AL Speer MC Steele MP Brown KK Loyd JE . A common MUC 5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. (2011) 364:1503–12. doi: 10.1056/NEJ Moa 101366021506741 PMC 3379886 · doi ↗ · pubmed ↗
- 8Armanios MY Chen JJL Cogan JD Alder JK Ingersoll RG Markin C . Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. (2007) 356:1317–26. doi: 10.1056/NEJ Moa 06615717392301 · doi ↗ · pubmed ↗