Isolation, RP‐UHPLC‐ESI‐LIT‐Orbitrap‐MS/MS‐Based Metabolic Profiling and Anticandidal Activity of the Root and Leaf Secondary Metabolites of Monotes Kerstingii Gilg (Dipterocarpaceae)
Sorelle Kache Fotsing, Dominique Ngnintedo, Yanick Kevin Dongmo Melogmo, Alena Soboleva, Kevine Dongmo Jumeta, Bruno Ndjakou Lenta, Fabrice Fekam Boyom, Andrej Frolov, Norbert Arnold, Ludger A. Wessjohann, Norbert Sewald, Bonaventure Tchaleu Ngadjui, Ghislain Wabo Fotso

TL;DR
This study identifies new antifungal compounds in the leaves and roots of Monotes kerstingii and evaluates their effectiveness against Candida species.
Contribution
The first isolation of (cis)-stilbene-coumarins from nature and characterization of six new metabolites with antifungal activity.
Findings
Leaf hydroethanolic extract showed the strongest anticandidal activity against C. krusei, C. parapsilosis, and C. albicans.
Six new metabolites, including a glycosylated stilbene and three cis stilbene-coumarins, were identified.
Stilbene 7 was the most active compound against multiple Candida species.
Abstract
The anticandidal activity of Monotes kerstingii leaf and root crude extracts was evaluated against five clinical Candida isolates: C. albicans, C. parapsilosis, C. krusei, C. glabrata and C. tropicalis. Extracts from both organs displayed Minimal Inhibitory Concentrations (MICs) ranging from 3.9 to 2000 µg/mL. Out of the five Candida species, the leaf hydroethanolic extract (EMKL) was the most active with MIC values of 3.9, 15.6 and 31.5 µg/mL on C. krusei, C. parapsilosis and C. albicans, respectively. The chemical investigation of these extracts led to the characterization of six previously undescribed metabolites, including a glycosylated stilbene: kerstingioside (1), three cis stilbene‐coumarins: cis‐kerstilbcoumarin A–C (2–4) among which two as inseparable cis/trans‐mixtures, one flavanone: kerstingiiflavanone (5) and one fatty acid glycoside, monestoside B (6), alongside with 22…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
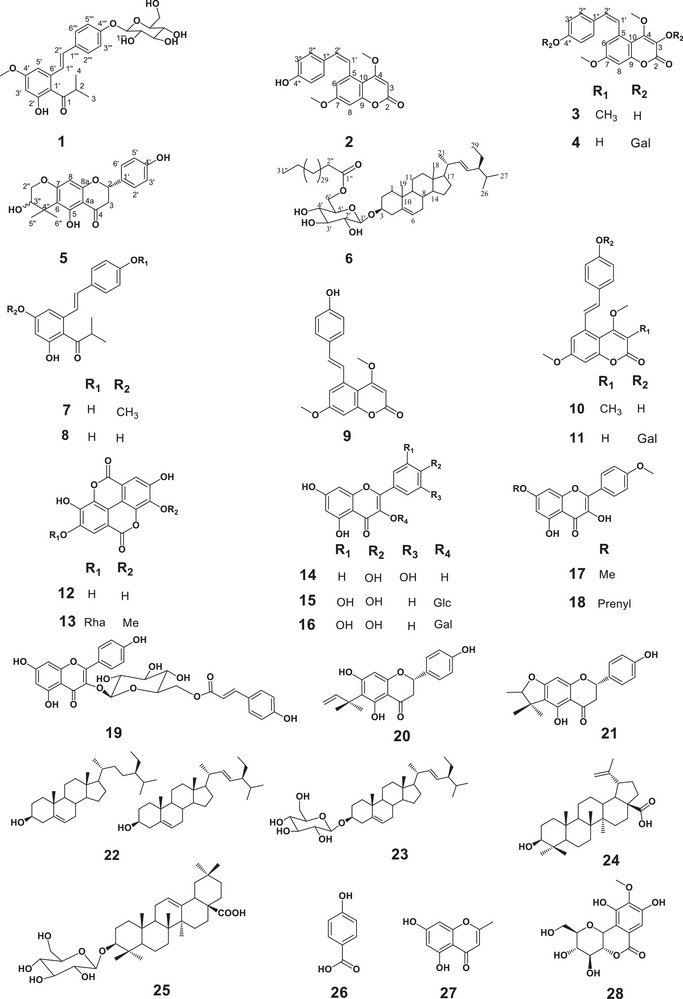 FIGURE 1
FIGURE 1| Pos. | 1 | ||
|---|---|---|---|
|
|
| HMBC | |
| 1 | 214.1 (C) | ||
| 2 | 3.36 (m) | 43.4 (CH) | C‐1 and C‐1′ |
| 3 | 1.16 (d, 7.3) | 18.3 (CH3) | C‐2, C‐3, C‐4, and C‐1 |
| 4 | 1.14 (d, 7.3) | 18.7 (CH3) | |
| 1′ | 115.8 (C) | ||
| 2′ | 159.0 (C) | ||
| 3′ | 6.97 (d, 2.0) | 105.4 (CH) | C‐4′, C‐5′, and C‐1′ |
| 4′ | 162.9 (C) | ||
| 5′ | 6.76 (d, 2.0) | 101.9 (CH) | C‐4′, C‐1′, and C‐1′′ |
| 6′ | 138.9 (C) | ||
| 1′′ | 7.03 (d, 16.8) | 132.9 (CH) | C‐5′ and C‐2′′ |
| 2′′ | 6.79 (d,16.8) | 123.2 (CH) | C‐1′′ and C‐1′′′ |
| 1′′′ | 130.0 (C) | ||
| 2′′′ | 7.31 (d, 8.4) | 129.1 (CH) | C‐3′′′ and C‐4′′′ |
| 3′′′ | 6.78 (d, 8.4) | 116.6 (CH) | C‐2′′′, C‐4′′′, and C‐1′′′ |
| 4′′′ | 157.1 (C) | ||
| 1′′′′ | 4.99 (d, 7.5) | 102.4 (CH) | C‐4′′′ and C‐3′′′′ |
| 2′′′′ | 3.42 (m) | 75.0 (CH) | |
| 3′′′′ | 3.46 (m) | 78.4 (CH) | |
| 4′′′′ | 3.35 (m) | 71.4 (CH) | |
| 5′′′′ | 3.46 (m) | 78.4 (CH) | C‐2′′′′ and C‐3′′′′ |
| 6′′′′ | 3.91 (dd, 12.1, 2.0); 3.68 (dd, 12.1, 6.2) | 62.7 (CH2) | C‐4′′′ and C‐5′′′ |
| 4′‐OMe | 3.88 (s) | 56.0 (CH3) | C‐4′ |
| Pos. | 2 | 3 | 4 | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
| HMBC |
|
| HMBC |
|
| HMBC | |
| 2 | 168.4 (C) | 163.1 (C) | 161.7 (C) | ||||||
| 3 | 5.71 (s) | 87.5 (CH) | C10, C4, and C2 | 110.7 (C) | 5.71 (s) | 87.8 (CH) | C10, C4, and C2 | ||
| 4 | 168.9 (C) | 165.9 (C) | 168.3 (C) | ||||||
| 5 | 137.5 (C) | 126.9 (C) | 137.1 (C) | ||||||
| 6 | 6.91 (d, 2.6) | 99.4 (CH) | C8 and C5 | 6.09 (d, 2.7) | 114.2 (CH) | C8 | 6.49 (brs) | 114.1 (CH) | C8, C10, C1′, and C7 |
| 7 | 161.5(C) | 161.4 (C) | 161.7 (C) | ||||||
| 8 | 6.53 (d, 2.6) | 114.1 (CH) | C10, C7, and C2′′ | 6.46(d, 2.7) | 100.2 (CH) | C10, C6 | 6.89 (brs) | 99.8 (CH) | C10 and C6 |
| 9 | 156.5 (C) | 155.0 (C) | 155.9 (C) | ||||||
| 10 | 106.8 (C) | 109.3 (C) | 106.9 (C) | ||||||
| 1′ | 6.49 (d, 12.1) | 128.4 (CH) | C1′′ | 6.09 (d, 12.0) | 128.7 (CH) | C2′′, C1′′ | 6.54 (d, 12.2) | 126.2 (CH) | , C1′′, C2′, and C10 |
| 2′ | 6.77 (d, 12.1) | 127.4 (CH) | C2′′ and C5 | 6.34 (d, 12.0) | 130.4 (CH) | C2′′ | 6.84 (d, 12.3) | 131.3 (CH) | C2′′ and C1′ |
| 1′′ | 130.4 (C) | 133.0 (C) | 130.8 (C) | ||||||
| 2′′ | 6.87 (d, 8.5) | 130.1 (CH) | C1′ and C2′′ | 6.35 (d, 8.1) | 127.3 (CH) | C2′′, C2′ | 6.98 (d, 8.5) | 130.0 (CH) | C2′′, C3′′, and C4′′ |
| 3′′ | 6.57 (d, 8.5) | 114.9 (CH) | C2′′ | 6.47 (d, 8.1) | 115.8(CH) | C1′′, C2′′ | 6.84 (d, 8.5) | 115.8 (CH) | C2′′ and C1′′ |
| 4′′ | 156.1 (C) | 156.8 (C) | 156.3 (C | ||||||
| 3‐Me | 1.60 (sl) | 10.1 (CH3) | C3, C4, C2 | ||||||
| 4‐OMe | 3.91 (s) | 56.5 (CH3) | C4 | 3.25 (s) | 60.5 (CH3) | C4 | 3.91 (s) | 56.7 (CH3) | C4 |
| 7‐OMe | 3.71 (s) | 55.3 (CH3) | C7 | 3.28 (s) | 55.9 (CH3) | C7 | 3.70 (s) | 55.7 (CH3) | C7 |
| 1′′′ | 4.81 (d, 7.7) | 100.2 (CH) | C4′′ | ||||||
| 2′′′ | 3.17 (m)* | 73.1 (CH) | C1′′′ | ||||||
| 3′′′ | 3.30 (m)* | 76.9 (CH) | C2′′′ | ||||||
| 4′′′ | 3.18 (m)* | 69.6 (CH) | |||||||
| 5′′′ | 3.35 (m)* | 76.5 (CH) | C1′′′, C3′′′, C4′′′, and C6′′′ | ||||||
| 6′′′ | 3.72 (dd, 5.7, 15.4); 3.48 (dd, 5.2, 15.4) | 60.6 (CH2) | C4′′′ and C5′′′ | ||||||
| Pos. | 5 | ||
|---|---|---|---|
|
|
| HMBC | |
| 2 | 5.45 (m) | 78.6 (CH) | |
| 3 |
3.26 (m) 2.68 (m) | 41.9 (CH2) | C4, C1′, and C2 |
| 4 | 197.2 (C) | ||
| 5 | 158.3 (C) | ||
| 6 | 114.1 (C) | ||
| 7 | 163.2 (C) | ||
| 8 | 5.98 (d, 2.2) | 90.3 (CH) | C8a, C7, C6, and C4a |
| 8a | 166.7 (C) | ||
| 4a | 102.9 (C) | ||
| 1′ | 128.7 (C) | ||
| 2′/6′ | 7.31 (d, 8.4) | 128.4 (CH) | C4′, C6′, C1′, and C2 |
| 3′/5′ | 6.79 (d, 8.4) | 115.1 (CH) | C4′, C5′, and C1′ |
| 4′ | 157.8 (C) | ||
| 2′′ | 3.70 (m) | 59.6 (CH2) | C3" |
| 3′′ | 4.31 (m) | 94.3 (CH) | C2′′ and C4′′ |
| 4′′ | 26.3 (C) | ||
| 5′′ | 1.44 (d,5.1) | 26.0 (CH3) | C6, C3", and C6′′ |
| 6′′ | 1.20 (d,7.6) | 20.3 (CH3) | C6, C3", and C5′′ |
| 5‐OH | 12.40 (s) | C5, C6, and C10 | |
| 4′‐OH | 9.58 (s) | C3′ and C4′ | |
| 3′′‐OH | 4.96 (s) | C2′′ and C3′′ | |
| Pos. | 6 | ||
|---|---|---|---|
|
|
| HMBC | |
| 1 | 37.3 (CH2) | ||
| 2 | 29.3 (CH2) | ||
| 3 | 3.53 (m) | 79.7 (CH) | C1′ |
| 4 | 38.9 (CH2) | ||
| 5 | 140.3 (C) | ||
| 6 | 5.35 (brs) | 122.1 (CH) | C4, C8, and C10 |
| 7 | 31.9 (CH2) | ||
| 8 | 32.0 (CH) | ||
| 9 | 50.2 (CH) | ||
| 10 | 36.7 (C) | ||
| 11 | 21.1 (CH2) | ||
| 12 | 39.8 (CH2) | ||
| 13 | 42.3 (C) | ||
| 14 | 56.8 (CH) | ||
| 15 | 24.3 (CH2) | ||
| 16 | 28.3 (CH2) | ||
| 17 | 56.1 (CH) | ||
| 18 | 0.67 (s) | 11.9 (CH3) | |
| 19 | 0.99 (s) | 19.0(CH3) | |
| 20 | 36.2(CH) | ||
| 21 | 0.81 (d, 6.8) | 18.8 (CH3) | |
| 22 | 5.35 (dd, 8.6, 15.1) | 130.2 (CH) | |
| 23 | 5.04 (dd, 8.6, 15.1) | 130.0 (CH) | |
| 24 | 45.8 (CH) | ||
| 25 | 29.2(CH) | ||
| 26 | 0.84 (d, 2.4) | 19.4 (CH3) | |
| 27 | 0.92 (d, 6.3) | 19.8 (CH3) | |
| 28 | 23.1 (CH2) | ||
| 29 | 0.85 (s) | 12.0 (CH3) | |
| 1′ | 4.37 (d, 7.7) | 101.2 (CH) | C3 |
| 2′ | 3.36 (m) | 73.5 (CH) | C1′, C3′, and C5′ |
| 3′ | 3.53 (m) | 76.1 (CH) | C4′, C2′, and C1′ |
| 4′ | 3.36 (m) | 70.3 (CH) | C3′ and C5′ |
| 5′ | 3.46 (Sl) | 73.8 (CH) | |
| 6′ | 4.37 (d, 7.7) and 4.30 (d, 12.1) | 63.4 (CH2) | C1′′ |
| 1′′ | 174.5 (C) | ||
| 2′′ | 34.3 (CH2) | ||
| 3′′‐29′′ | 29.5–29.9 (CH2) | ||
| 30′′ | 29.4 (CH2) | ||
| 31′′ | 0.88 (t, 6.9) | 14.1(CH3) | C30′′ |
|
| Molecular adduct ion |
|
| Error (ppm) | Elemental composition/ | Fragmentation patterns: | Assignment |
|---|---|---|---|---|---|---|---|
| 1.8 | [M+H] + | 329.0866 | 329.0867 | 0.3 | C14H17O9 + | 191.0 (10%), 209.0 (32%), 251.0 (72%), 253.0 (24%), 263.0 (24%), 275.0 (46%), 281.1 (24%), 293.1 (100%), and 311.1 (90%) | Bergenin |
| 5.6 | [M‐H]− | 461.0721 | 461.0725 | 0.9 | C21H17O12 − | 300.0 (1.9%), 315.0 (100%), 316.0 (6%), and 340.1 (0.11%) | 3‐ |
| 6.5 | [M+H] + | 475.1959 | 475.1963 | 0.8 | C25H31O9 + | 191.1 (0.2%), 219.1 (0.2%), 295.1 (0.3%), 311.1 (0.4%), 313.1 (100%), 355.1 (0.2%), and 457.2 (1.3%) | {5‐[(1 |
| 6.6 | [M+H] + | 487.1599 | 487.1591 | 1.6 | C25H27O10 + | 283.1 (0.9%), 297.1 (0.7%), 311.1 (40%), 312.1 (2%), 325.1 (100%), 326.1 (8%), and 353.1 (1.3%) | 5‐[(1 |
| 7.4 | [M‐H]− | 297.1130 | 297.1132 | 0.7 | C18H17O4 − | 189.1 (4%), 191.1 (10%), 203.1 (3.5%), 227.1 (100%), 253.1 (19%), 254.1 (4%), 267.1 (3.5%), and 282.1 (3%) | 1‐{2,4‐Dihydroxy‐6‐[(1 |
| 7.8 | [M+H] + | 325.1066 | 325.1071 | 1.5 | C19H17O5 + | 179.0 (20%), 217.0 (20%), 237.1 (24%), 265.1 (80%), 266.1 (40%), 282.1 (75%), 293.1 (66%), 294.1 (30%) 297.1 (100%), 309.1 (30%), and 310.1 (34%) | 5‐[(1 |
| 8.4 | [M‐H]− | 311.1289 | 311.1288 | 0.3 | C19H19O4 − | 189.1 (30%), 190.1 (60%), 205.1 (100%), 241.1 (26%), 253.0 (20%), 278.1 (14%), 281.1 (26%), 293.1 (20%), and 296.1 (52%) | 5‐[(1 |
| 8.5 | [M+H] + | 339.1228 | 339.1227 | −0.3 | C20H19O5 + | 193.1 (30%), 219.1 (20%), 231.1 (50%), 233.1 (28%), 245.1 (30%), 251.1 (15%), 264.1 (12%), 279.1 (100%), 280.1 (32%), 296.1 (80%), 307.1 (70%), 308.1 (15%), 311.1 (40%), 321.1 (28%), 323.1 (66%), and 324.1 (85%) | 5‐[(1Z/ |
| 11.3 | [MH‐H2O] + | 439.3569 | 439.3570 | 0.2 | C30H47O2 + | 241.2 (10%), 243.2 (6%), 259.2 (17%), 269.2 (10%), 287.2 (15%), 289.2 (5%), 301.2 (5%), 315.2 (8%), 393.4 (100%), and 421.3 (7%) | Betulinic acid |
| 13.2 | [M+H] + | 413.3773 | 413.3778 | 1.2 | C29H49O+ | 159.1 (2.5%), 173.1 (1.6%), 187.1 (1.8%), 201.2 (2.5%), 219.2 (2.2%), 241.2 (2.2%), 255.2 (4.5%), 259.2 (1.5%), 273.2 (2.5%), 283.2 (1.7%), 297.3 (3%), 311.3 (1.5%), and 395.4 (100%) | Stigmasterol |
|
| Molecular adduct ion |
|
| ∆ ppm | Elemental composition/ | Fragmentation patterns ( | Metabolites |
|---|---|---|---|---|---|---|---|
| 5.0 | [M‐H]− | 300.9988 | 300.9990 | 0.7 | C14H5O8 − | 179.0 (3%), 185.0 (21%), 201.0 (6.5%), 213.0 (3.5%) 229.0 (48%), 230.0 (5%), 245.0 (4%), 257.0 (100%), 258.0 (14%), 271.0 (3%), 273.0 (4%), and 284.0 (16%) | Ellagic acid |
| 5.2 | [M+H] + | 465.1027 | 465.1028 | 0.2 | C21H22O12 + | 303.0 (100%), 319.0 (68%), 329.0 (1.4%), 429.1 (2.2%), and 447.1 (5%) | Isoquercitrin |
| 5.3 | [M+H] + | 303.0500 | 303.0499 | −0.3 | C15H12O7 + | 137.0 (24%), 153.0 (26%), 165.0 (68%), 201.1 (10%), 229.0 (80%), 247.1 (36%), 257.0 (100%), 258.0 (18%), 274.0 (18%), 275.1 (16%), and 285.0 (66%) | Quercetin |
| 6.3 | [M+H] + | 595.1442 | 595.1446 | 0.7 | C30H27O13 + | 287.1 (95%), 291.1 (3.5%), 302.0 (7.5%), 309.1 (100%), 329.1 (25%), 353.1 (3%), 395.1 (6.5%), 431.1 (4%), 449.1 (3%), and 577.1 (18%) | Tiliroside |
| 7.7 | [M+H] + | 357.1332 | 357.1333 | 0.3 | C20H21O6 + | 147.0 (14%), 163.0 (24%), 219.1 (46%), 221.1 (26%), 237.1 (60%), 272.1 (18%), 273.1 (60%), 285.1 (100%), 301.1 (16%), and 339.1 (46%) | (2 |
| 8.3 | [M‐H]− | 313.0717 | 313.0718 | 0.3 | C17H13O6 − | 193.0 (0.2%), 255.0 (1%), 270.1 (0.4%), 283.0 (2.4%), 298.0 (100%), and 299.1 (5%) | 4′, 7‐dimethoxy kaempferol |
| 9.2 | [M+H] + | 341.1380 | 341.1384 | 1.2 | C20H21O5 + | 147.0 (0.6%), 165.0 (1.3%), 219.1 (2.3%), 220.1 (3.5%), 221.1 (100%), 285.1 (2.6%), and 299.1 (2.7%) | 6‐(1, 1‐dimethylallyl) naringenin |
| 10.9 | [M+H] + | 369.1331 | 369.1333 | 0.5 | C21H21O6 + | 221.1 (0.1%), 286.0 (0.2%), 297.1 (0.1%), 301.1 (100%), 313.1 (0.5%), and 327.1 (0.2%) | 7‐ |
| Extracts, fractions, and compounds | MICs (µg/mL) | ||||
|---|---|---|---|---|---|
| Clinical isolates | |||||
|
|
|
|
|
| |
|
| |||||
|
| 62.5 | 62.5 | 31.5 | 500 | 2000 |
|
| 250 | 250 | 250 | 1000 | / |
|
| 31.5 | 3.9 | 15.6 | / | / |
|
| 250 | 250 | 250 | / | / |
|
| |||||
|
| 500 | 2000 | / | / | / |
|
| 125 | 125 | 500 | 125 | 250 |
|
| 500 | 1000 | 125 | 500 | 250 |
|
| 62.5 | 1000 | / | 500 | / |
|
| 1000 | 2000 | / | / | / |
|
| 500 | 500 | / | 1000 | / |
|
| 1000 | 1000 | / | / | / |
|
| 2000 | / | / | / | / |
|
| / | / | / | / | 1000 |
|
| 500 | 1000 | 125 | 500 | 500 |
|
| 1000 | 1000 | 125 | 500 | 500 |
|
| 1000 | 1000 | 125 | 500 | 500 |
|
| |||||
|
|
|
|
| / |
|
|
| / | / |
| / | / |
|
|
| / |
| / |
|
|
| 0.0765 | 0.153 | 0.153 | 0.306 | 0.306 |
- —YaBiNaPA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNatural Compound Pharmacology Studies · Phytochemistry and Biological Activities · Plant-Derived Bioactive Compounds
Introduction
1
Candida species are commensal organisms present in some regions of the human microbial flora usually found on the skin and in the gastrointestinal and genital tracts. Recent global systematic analyses have highlighted the shifting epidemiology of invasive Candida infections, with an increasing incidence of uncommon and non‐albicans species that pose distinct clinical challenges in terms of antifungal susceptibility patterns and outcomes. The most common species responsible for invasive infections are C. albicans, C. glabrata, C. parapsilosis, C. tropicalis, and C. krusei. They can cause various infections in susceptible patients, including the elderly, hospitalized patients (nosocomial infections), and immunosuppressed individuals. Invasive Candida infections are among the most common fungal infections worldwide [1]. Amongst these species, Candida albicans accounted for approximately 37.1% of candidemia cases, with C. glabrata representing 30.4% and C. parapsilosis representing 13.5% in culture‐confirmed bloodstream infections in the United States from 2017 to 2021 [2, 3]. The strategy for controlling candidiasis depends on the patient's immune status, location, and infection severity [4]. Several drugs are recommended for the management of candidiasis, including polyenes, azoles, echinocandins, and 5‐flucytosine (5FC) [5]. The emergence of antifungal resistance to the most prescribed antifungal drugs is a serious threat to public health worldwide. More than 34 000 cases and 1700 deaths were recorded due to drug‐resistant Candida spp [2]. The significant prevalence of candidiasis and its correlation with both inadequate access to healthcare and drug resistance highlight the urgent need for new treatment alternatives. To combat this, medicinal plants can play an important role, as they serve as a critical foundation for the development of novel pharmaceuticals generally. Therefore, the chemical and pharmacological investigation of plants traditionally used to treat skin diseases is an appropriate approach for discovering Candida growth inhibitors. African tropical forests have a rich biodiversity, with approximately 8620 plant species known to have medicinal properties [6]. Among these plants, the Monotes genus, found across African countries, comprises 36 species. They are exploited in traditional medicine for their effect against diarrhoea, typhoid, cough, and skin diseases [7, 8]. It is worth mentioning that Monotes kerstingii is the only species found in Cameroon (Northern Region) to the best of our knowledge. However, few chemical studies have been conducted to date in this genus, including Monotes engleri Gilg and Monotes africanus Welw. & Kirk ex A. DC, C‐, and O‐prenylated flavonoids, coumarins and stilbene‐coumarins were isolated [9, 10]. The chemical study carried out on the stem bark and leaves of Monotes kerstingii Gilg revealed that stilbene‐coumarins and ellagic acid derivatives were the main constituents of the bark, whilst flavonoids were present only in the leaves [7]. In our continuous search for secondary metabolites with antimicrobial potential [7, 11], our current work was designed to investigate the bioactive constituents of the root extract of M. kerstingii against five clinical isolates of Candida, along with a deeper investigation of the leaf extract. This report includes the anticandidal activity of Monotes kerstingii root and leaf extracts and the isolation of their secondary metabolites, among which the first cis‐stilbene‐coumarin reported from nature, together with the rapid targeted UHPLC‐ESI‐LIT‐Orbitrap‐MS/MS‐based metabolic profile.
Results and Discussion
2
Structure Elucidation of the Isolated Compounds
2.1
The chemical investigation of the root and leaf extracts of M. kerstingii using Diaion HP‐20, silica gel, and Sephadex LH‐20 in combination with semi‐preparative HPLC led to the isolation and characterization of 28 secondary metabolites, including six previously undescribed metabolites (1–6).
Structure Elucidation of Compound 1
2.1.1
Compound 1 has been obtained from the n‐hexane/ethyl acetate (80:20, v/v) mixture as a pink oil, soluble in methanol. It gave positive ferric chloride and Molisch tests, suggesting its phenolic nature and the presence of a sugar moiety. Its molecular formula, C_25_H_30_O_9,_ was deduced from its HR‐ESIMS spectrum (Figure S1), which showed the pseudo molecular ion peak [M + H] ^+^ at m/z 475.1969 (calcd. 475.1963 for C_25_H_31_O_9_ ^+^). The combined analysis of its ^1^H and ^13^C‐NMR spectra in conjunction with HSQC and DEPT spectra (Figures S2–S8) revealed signals of two *trans‐*ethylenic protons at δ H/C 6.79 (1H, d, *J *= 16.8 Hz)/123.2 and 7.03 (1H,d, *J *= 16.8 Hz)/132.9, an AA′BB′ system for a *para‐*disubstituted aromatic ring at δ H/C 6.78 (2H, d, *J *= 8.4 Hz)/116.6 and δ H/C 7.31 (2H, d, *J *= 8.4 Hz)/129.1 together with an AB system of two meta coupling aromatic protons at δ H/C 6.76 (1H, d, *J *= 2.0 Hz)/101.9 and 6.97 (1H, d, *J *= 2.0 Hz)/105.4. This information suggested that 1 is likely to be a stilbene derivative [7, 12]. Additional coupled protons at δ H/C 3.36 (1H, m)/43.4, 1.16 (3H, d, 7.3)/18.3 and 1.14 (3H, d, 7.3)/18.7 were observed via COSY analysis (Figure S9), which clearly exhibited two geminal methyl moieties of an isopropyl group. Moreover, the HMBC spectrum of 1 (Figures S10–S12) showed important correlations between proton H‐2 (δ H 3.36) with a carbonyl C‐1 (δδ C 214.1), C‐3 (δ C 18.3), C‐4 (δ C 18.7), and C‐1′ (δ C 115.8) together with correlations of meta‐coupled protons H‐3′ (δ H 6.76) and H‐5′ (δ H 6.97) with C‐1′ (δ C 115.8). The presence of an isobutyryl moiety connected to the aromatic ring at C‐1′ was then confirmed. Additionally, the correlation between the methoxyl protons (δ H 3.88) and the meta‐coupled protons H‐3′ (δ H 6.76) and H‐5′ (δ H 6.97) with the same oxygenated carbon C‐4′ (δ C 162.9) allowed elucidation of the methoxyl group at C‐4′. The ^1^H and ^13^C‐NMR data of compound 1 (Table 1) in combination with the ^1^H‐^1^H COSY spectrum also revealed the presence of a sugar moiety. The signal of the anomeric proton was observed at δ H/C 4.99 (1H, d, *J *= 7.5 Hz)/102.4 together with one oxygenated methylene group with two diastereotopic protons at δ H/C 3.91(1H, dd, 12.1, 2.0)/62.7 and δ H/C 3.68 (1H, dd, 12.1, 6.2)/62.7, along with four oxygenated methine protons between 3.35 and 3.46 ppm. The chemical shift value of C‐4′′′′ at δ C 71.4 suggests that the sugar moiety is a glucose. These data, together with the high value of the coupling constant (J = 7.5 Hz) of H‐1′′′′ compared with those published in the literature [13] allowed us to identify the sugar as β–D‐glucopyranosyl. Based on the HMBC correlations between the anomeric proton H‐1′′′′ at 4.99 ppm with C‐4′′′ (δ C 157.1) and C‐3′′′′ (δ C 78.4), the sugar was located at position 4′′′ of the stilbene moiety. This was further supported by the NOESY correlations between the aromatic protons H‐1′′′′ (δ H 4.99) with H‐3′′′′ (δ H 3.46) and H‐5′′′′ (δ H 3.46). Thus, compound 1 was characterized as {5‐[(1E)‐2‐(4‐β‐D‐glucopyranosylphenyloxy)ethenyl]‐4′‐methoxyphenyl}‐2‐methyl‐1‐propanone to which the trivial name kerstingioside was given.
Structure Elucidation of Compound 2
2.1.2
Compound 2 has been obtained as a yellow powder soluble in DMSO. Its molecular formula was determined to be C_19_H_16_O_5_ by HR‐ESI‐MS (Figure S15), which showed the pseudo molecular ion peak [M+H]^+^ at m/z 325.1083 (calcd. 325.1071 for C_19_H_17_O_5_ ^+^). Its ^1^H‐NMR spectrum (Figure S16) displayed two olefinic protons at δ H 6.77 (1H, d, *J *= 12.1 Hz) and δ H 6.49 (1H, d, *J *= 12.1 Hz), which were attributed to the protons H‐2′ and H‐1′ of the stilbene moiety. The coupling constant value around 12 Hz suggests a cis configuration of the double bond [14]. (Jo et al., 2011). Analysis of its ^1^H and ^13^C‐NMR associated with COSY, HSQC, HMBC and NOESY (Figures S16–S21) exhibited signals of an AA'BB′ system for the *para‐*substituted aromatic ring at [δ H/C 6.57 (2H, d, *J *= 8.5 Hz)/114.9; 6.87 (2H, d, *J *= 8.5 Hz)/130.1]. In addition, an AB system of two aromatic protons at [δ H/C 6.91 (1H, d, J = 2.6 Hz)/99.4; 6.53 (1H, d, J = 2.6 Hz)/114.1] was distinguished. The spectra also display one signal of olefinic proton H‐3 of the stilbene‐coumarin at δ H 5.71 (1H, s)/87.5 [7]. Two signals are attributable to two methoxy groups at [δ H/C 3.91 (3H, s)/56.5; 3.71 (3H, s)/55.3] (Table 2). When comparing the ^1^H and ^13^C chemical shifts of the cis derivative with those of the known trans, no significant differences were observed in the coumarin moiety. However, the protons of the cis‐double bond appeared shielded with ∆δ (H‐1′) −0.99 ppm and ∆δ (H‐2′) value −0.46 ppm compared to the trans geometric isomer. The significant variation of the chemical shift of H‐2′′ ∆δ (H‐2′′) (−0.55 ppm) was observed. This can be explained by steric hindrance in the cis stereoisomer compared to the trans form [7, 12, 15]. Thus, compound 2 was clearly established as 5‐[(1Z)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one. To the best of our knowledge, it is the first cis stilbene‐coumarin reported from nature to which the trivial name cis‐kerstilbcoumarin A was given.
Structure Elucidation of Compound 3
2.1.3
Compound 3 was obtained from the n‐hexane/acetone (80:20, v/v) solvent system as a greenish powder, soluble in acetone. Its molecular formula was determined to be C_20_H_18_O_5_ based on its HRESIMS spectrum (Figure S22), which showed a pseudo molecular ion peak [M‐H]^−^ at m/z 337.1084 (calcd for C_20_H_17_O_5_ ^−^, m/z 337.1081). The NMR data of 3 (Figures S23–S35) were obtained from a mixture that showed pairs of peaks at nearly identical chemical shifts, differing only in integration and intensity, suggesting a mixture of two stereoisomers. These data were almost similar to those of the stilbene‐coumarin, 5‐[(1E)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐3‐methyl‐2H‐1‐benzopyran‐2‐one 10 previously described in the literature and isolated from the stem barks of the plant [11, 12]. However, in addition to the signals of 10 present in the mixture, we observed two additional ethylenic protons, more shielded at δ H 6.09 (1H, d) and δ_H_ 6.34 (1H, d), with a coupling constant of 12 Hz, indicating a cis configuration [15]. The protons of the trans counterpart were observed downfield at δ H 7.32 (1H, d, J = 16.1 Hz) and δ H 6.56 (1H, d, J = 16.1 Hz). Therefore, compound 3 was elucidated as 5‐[(1Z)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐3‐methyl‐2H‐1‐benzopyran‐2‐one, a previously undescribed stilbene‐coumarin to which the trivial name cis‐kerstilbcoumarin B was given. The occurrence of this stereoisomer in nature, together with its spectroscopic data (Table 2), is in this study reported for the first time.
Structure Elucidation of Compound 4
2.1.4
Compound 4 has been obtained in the system n‐hexane/acetone 25:75 (v/v) as a white powder soluble in DMSO. Its molecular formula was determined to be C_25_H_26_O_10_ based on its HRESIMS spectrum (Figure S36), which showed a pseudo‐molecular ion peak [M + Na] ^+^ at m/z 509.1411 (calcd. for C_25_H_26_NaO_10_ ^+^ m/z 509.1419). The NMR data of 4 (Figures S37–S49), like those of 2 were also obtained from those of an inseparable mixture showing pairs of peaks at near identical chemical shifts, suggesting a mixture of cis and trans stilbene‐coumarin derivatives. In addition of the signal observed in 2, those of a sugar moiety were observed at δ H/C 4.81 (m)/100.2 (C‐1′′′), δ H/C 3.17 (m)/73.1 (C‐2′′′), δ_H/C_ 3.30 (m)/76.9 (C‐3′′′), δ_H/C_ 3.18 (m)/69.6 (C‐4′′′), δ H/C 3.35 (m)/76.5 (C‐5′′′), and two diastereotopic protons of the carbinol group δ_H/C_ 3.72 (dd, 5.7, 15.4) and 3.48 (dd, 5.2, 15.4)/60.6 (C‐6′′′), This sugar was identified to a β‐galactopyranosyl unit in comparison with published [7, 15]. The sugar was located at C‐4′′ based on HMBC analysis (Table 2). All these findings, combined with the literature data, allowed us to establish the structure of 4 as 5‐[(1Z)‐2‐(4‐β‐D‐galactopyranosyloxyphenyl)ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one, a cis‐stilbene‐coumarin also reported from nature for the first time, to which the trivial name cis‐kerstilbcoumarin C was given.
It is worth noting that in our previous study on the stem bark of this plant collected in the same area, no *cis‐*stilbene‐coumarin was found. The occurrence of the less stable cis stereoisomers in the roots can be explained by the fact that roots are not exposed to sunlight, unlike the stem bark. Therefore, as the far North region of Cameroon is very sunny, the cis stereoisomer may quickly isomerize to the most stable trans stereoisomers in the stem bark while exposed to UV light.
Structure Elucidation of Compound 5
2.1.5
Compound 5 has been obtained from the mixture n‐hexane/acetate 80:20 (v/v) as a white powder soluble in DMSO. It gave a positive ferric chloride and Shinoda test, suggesting its flavonoid nature. Its molecular formula was assigned as C_20_H_20_O_6_ based on its positive‐mode HRESI‐MS spectra (Figures S50 and S51), with a pseudo molecular ion peak [M+H]^+^ at m/z 357.1343 (calcd for C_20_H_21_O_6_ ^+^ m/z 357.1333) corresponding to 11 degrees of unsaturation. The ^1^H‐NMR spectrum (Figures S52 and S53) showed the signal of a chelated proton at δ H 12.42 together with the characteristic signals of the C‐ring of flavanones at δ H 5.45 (1H, m, H‐2) and two diastereotopic protons at δ H 3.27 (1H, m, H‐3a) and 2.67 (1H, m, H‐3b). This information was supported by the ^13^C‐NMR and DEPT spectra of 5 (Figures S54 and S55), which showed characteristic signals of C‐2, C‐3, and C‐4 at δ C 78.6, 41.9 and 197.2, respectively [10]. Interpretation of its COSY and HSQC spectra (Figures S56 and S57) revealed signals at δ H/C 6.79 (2H, d, *J *= 8.4 Hz)/114.1 and δ H/C 7.31 (2H, d, *J *= 8.4 Hz)/128.4, which agrees with the presence of an AA'BB′ system for the *para‐*disubstituted aromatic ring characteristic for the B‐ring [10]. Besides these signals, a singlet was observed at δ H/C 5.98 (1H, s)/90.3 in the aromatic region, revealing a penta‐substituted ring A. This singlet was attributed to H‐8 based on its HMBC correlations (Figure S58) with C‐4a, C‐6, C‐7, and C‐8a. The presence of an eleventh degree of unsaturation in 5 (ten for the flavanone skeleton) suggested that 5 possesses another ring, linearly fused to the A‐ring at C‐6/7. This information was confirmed by cross‐analysis of the ^1^H and ^13^C‐NMR spectra of compound 5 (Table 3) showing signals of two geminal methyl groups at δ H/C 1.44 (3H, d, *J *= 5.1 Hz, H‐5′′)/26.0 and 1.20 (3H, d, *J *= 7.6 Hz, H‐6′′)/20.3, linked to carbon C‐4′′(26.3) together with an oxymethine protons δ H/C 4.31 (1H, m, H‐3′′)/94.3 and an oxymethylene group at δ H/C 3.70 (2H, m, H‐2′′)/59.6. This was corroborated by HMBC correlations between H‐5′′ and H‐6′′ with C‐6 (114.1), C‐3“(94.3), C‐6′′ (20.3), and C‐5′′ (26.0); between H‐2′′ and C‐3”(94.3) and finally between H‐3′′ with C‐4“(26.3) and C‐2”(59.6). The absolute configurations of the stereogenic centre at C‐2 were deduced from the ECD spectrum of 5 (Figure S59) and its optical rotation, by comparison with previously described compounds. Indeed, the absolute configuration S of C‐2 was deduced by the comparison of the UV and ECD spectrum of (2S)‐naringenin and those of 5 recorded in methanol/CDCl_3_ 8:2, showing a positive cotton effect at 292 nm (ε = −34295.3) based on the Snatzke rule, which led to an empirical assignment of the absolute configuration [16] (Figures S59 and S60). Thus, compound 5 was characterized as (2S)‐4',5‐dihydroxy‐[3‐hydroxy‐2,2‐dimethyl‐3,4‐dihydropyrano(5,6:6,7)]flavanone to which the trivial name kerstingiiflavanone was given.
Structure Elucidation of Compound 6
2.1.6
Compound 6 has been obtained from a mixture of n‐hexane and acetone (75:25, v/v) as a yellow, amorphous powder, soluble in chloroform. It gave a positive Liebermann–Burchard test characteristic of steroids and a positive Molisch test, suggesting that 6 is likely to be a steroid bearing a sugar moiety. Its molecular formula was deduced as C_68_H_122_O_7_ based on its HRESIMS spectrum (Figure S61), which showed a pseudo‐molecular ion peak [M+H] ^+^ at m/z 1051.9351 (calcd. for 1051.9263, C_68_H_123_O_7_ ^+^). Its ^1^H and ^13^C‐NMR spectra coupled with COSY, HSQC and DEPT spectra (Figures S62–S71) show signals of three olefinic protons divided into two groups including one broad singlet at δ H/C 5.35 (1H, brs)/122.1 ppm characteristic of the double bond δ ^5^ of a steroid and two doublets of a doublet at δ H/C 5.35 (1H, dd, *J *= 7.3 Hz, *J *= 12.5 Hz)/130.2 and 5.04 (1H, dd, *J *= 8.7 Hz, *J *= 15.2 Hz)/130.0 characteristic of δ ^22^. This suggested that 6 possesses a stigmata‐5,22‐diene skeleton [7]. A multiplet at δ H/C 3.53 (1H, m)/79.7 was attributed to the oxymethine at C‐3 [17]. Furthermore, the ^1^ H and ^13^C‐NMR data of compound 6 (Table 4) in combination with the DEPT and ^1^H‐^1^H COSY spectra reveal the presence of a sugar moiety. The signal of the anomeric proton appeared at δ H/C 4.37 (1H, d, *J *= 7.7 Hz)/101.2 along with four oxygenated methine protons between δ H 3.36 and 3.53 ppm and one oxymethylene group with two diastereotopic protons at δ H/C 4.37 (1H, d, *J *= 7.7 Hz)/63.4 and δ H/C 4.30 (1H, d, *J *= 12.1 Hz)/63.4. According to the high value of the coupling constant (*J *= 7.7 Hz), the sugar was identified as β–D‐glucopyranosyl [13]. The downfield part of the spectra shows the presence of seven methyl groups, with six of them at δ H/C 0.67 (3H, s)/12.0, δ H/C 0.99 (3H, s)/19.0, δ H/C 0.81 (3H, d, *J *= 6.8 Hz)/18.8, δ H/C 0.92 (3H, d, *J *= 6.3 Hz)/19.8, δ H/C 0.84 (3H, d, *J *= 2.4 Hz)/19.4 and δ H/C 0.85 (3H, s)/12.1 being characteristic of the steroidal skeleton [17]. Furthermore, a triplet at δ H/C 0.88 (3H, t, *J *= 6.9 Hz)/14.1 was attributed to a terminal methyl attached to an aliphatic chain. This was confirmed by the ^1^H and ^13^C‐NMR spectra of 6, in which a broad singlet at δ_H/C_ 5.35 (1H, brs)/122.1 was observed. Moreover, the ^13^C‐NMR displayed the signal of the carbonyl group of an ester at 174.5 ppm, suggesting the presence of a fatty ester [CH_3_ (CH_2_)n COO‐] in 6. The analysis of the molecular formula of 6 deduced the length of the chain (*n *= 31). Then, the aliphatic long chain was further located at C‐6′ of the sugar moiety based on the long‐range correlations of the HMBC spectrum (Figures S72–S75) between the two diastereotopic protons H‐6′ at δ H/C 4.37 (1H, d, 7.7) and 4.30 (1H, d, 12.1)/63.4 and the carbonyl of the acyl group C‐1′′ at δ C 174.5 ppm. Hence, the structure of compound 6 was elucidated as 3‐O‐[6’‐O‐psylloyl‐β‐D‐glucopyranosyl]‐β‐sitosterol, a previously undescribed fatty glucoside to which the trivial name monestoside B was given.
Figure S76 summarizes the key COSY and HMBC correlations for compounds 1–6. Based on the interpretation of NMR analyses, combined with data published in the literature twenty‐two other secondary metabolites were fully characterized including: 5‐[(1E)‐2‐(4‐hydroxyphenyl)ethenyl]‐4‐methoxyphenyl}‐2‐methyl‐1‐propanone (7) [12], 1‐{2,4‐dihydroxy‐6‐[(1E)‐2‐(4‐hydroxyphenyl)ethenyl]‐phenyl}‐2‐methyl‐1‐propanone (8) [12], 5‐[(1E)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐3‐2H‐1‐benzopyran‐2‐one (9) [7], 5‐[(1E)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐3‐methyl‐2H‐1‐benzopyran‐2‐one (10) [12], 5‐[(1E)‐2‐(4‐β‐D‐galactopyranosyloxyphenyl)ethenyl]‐4,7‐dimethoxy‐3‐2H‐1‐benzopyran‐2‐one (11) [7], ellagic acid (12) [18], 3‐O‐methyl ellagic acid‐4′‐O‐αL‐rhamnopyranoside (13) [19], quercetin (14) [20], isoquercitrin (15) [21], hyperoside (16) [21], 4′,7‐dimethoxy kaempferol (17) [22], 7‐O‐(3‐methyl‐2‐butenyl) kaempferide (18) [23], tiliroside (19) [24], 6‐(1, 1‐dimethylallyl) naringenin (20) [10], 5, 4′‐dihydroxy‐4″, 4″‐dimethyl‐5″‐methyl‐5″‐H‐dihydrofurano [2″,3″:6,7] flavanone (21) [10], mixture of stigmasterol and β‐sitosterol (22) [17] stigmasterol glucoside (23) [17], betulinic acid (24) [25], 3‐O‐β‐D‐glucopyranosyl oleanolic acid (25) [26], 4‐hydroxybenzoic acid (26) [27], noreugenin (27) [28] and bergenin (28) [29] (Figure 1).
Structures of compounds 1–28 isolated from the roots and leaves of M. kerstingii.
RP‐UHPLC‐ESI‐LIT‐Orbitrap‐MS/MS
2.2
To confirm that all isolated compounds were initially present in the extracts, a rapid targeted UHPLC‐ESI‐LIT‐Orbitrap‐MS/MS profiling was performed. Liquid chromatography—high resolution Orbitrap mass spectrometry allowed annotation of ten and eight individual metabolites in the total ion current chromatograms (TICs) of M. kerstingii root and leaves extracts, respectively (Tables 5 and 6; Figures S77–S95). Even though mass spectrometry is one of the most widely used methods for characterization of metabolites, this method has some intrinsic limitations. Thus, it does not allow for distinguishing cis/trans isomers, for example, 5‐[(1E)‐2‐(4‐hydroxyphenyl) ethenyl]‐4,7‐dimethoxy‐3‐methyl‐2H‐1‐benzopyran‐2‐one and 5‐[(1Z)‐2‐(4‐hydroxyphenyl) ethenyl]‐4, 7‐dimethoxy‐3‐methyl‐2H‐1‐benzopyran‐2‐one (Table 5), or isoquercitrin and hyperoside (Table 6).
Anticandidal Activity
2.3
Using the dichloromethane–methanol 1/1 (v/v) and ethanol–water 7/3 (v/v) mixtures, six extracts were obtained from leaves, stem barks and roots. These crude extracts were further screened for their anticandidal activity at a single dose (2000 µg/mL) against five clinical isolates of Candida spp. from Laboratory SION (Cameroon), including C. albicans, C. krusei, C. parapsilosis, C. tropicalis, and C. glabrata. The results are summarized in Table S1. It appears from this table that, the crude extracts at the high entry assay concentration, MKL, can inhibit growth in all five clinical isolates. MKR, MKW, and EMKS were only inactive against C. glabrata, whilst EMKR was inactive against C. tropicalis and C. glabrata. Extracts that showed activity at 2000 µg/mL were selected to determine their MICs, and the most potent extracts were further fractionated and their MICs determined.
Minimum Inhibitory Concentrations (MICs) of Selected Crude Extracts, Fractions, and Isolated Compounds
2.3.1
The MICs (µg/mL) of selected extracts, fractions obtained from them, and isolated compounds were determined and are presented in Table 7 below. The negative control was made up with culture media and fungal suspension while fluconazole was used as positive control.
Elaborated cutoff points for antimicrobial activities by Tamokou et al. [30] classified the activity of extracts and fractions as follows: highly active: MIC below 100 µg/mL, significantly active: 100 ≤ MIC ≤ 512 µg/mL, moderately active: 512< MIC ≤ 2048 µg/mL, Low activity: MIC > 2048 µg/mL, considered not active: MIC > 10 mg/mL. It appears from Table 7 that the MICs range from 3.9 to 2000 µg/mL for the crude extracts, from 62.5 to 2000 µg/mL for the fractions and 7.81 µg/mL to 62.5 µg/mL for the isolated compounds. The results show the influence of the plant parts used, the extraction solvent, and the Candida spp. Targeted toward the antifungal acitivity. Overall, leaf extracts (MKL and EMKL) were the most active compared to the root (EMKR, MKR), which showed significant to moderate activity (MICs: 125–1000 µg/mL) against the same strains. The hydroethanolic extract from leaves (EMKL) was the most active, with the MICs of 3.9 µg/mL, 15.6 µg/mL and 31.5 µg/mL against C. krusei, C. parapsilosis and C. albicans, respectively. The dichloromethane/methanolic extract of the leaves (MKL) exhibited high activity even though lower than the hydroethanolic extract with MICs of 31.5 µg/mL, 62.5 µg/mL and 62.5 µg/mL against C. parapsilosis, C. krusei and C. albicans, respectively. C. krusei appeared to be the most susceptible strain, with MICs of 62.5 µg/mL, and 3.9 µg/mL recorded for MKL and EMKL, respectively. In the same line, the aqueous residue MKL (F5MKL) was the most active one amongst fractions with a MIC of 62.5 µg/mL against C. albicans, followed by the ethyl acetate fraction (F3MKL) that exhibited significant activity against all five strains with the MICs ranging from 125 to 500 µg/mL. The n‐butanolic fraction (F4MKL) of the same extract had MICs ranging from 125 to 1000 µg/mL, indicating significant activity against four strains (C. albicans, C. krusei, C. parapsilosis, and C. tropicalis). Out of the 28 isolated compounds, 17 were screened based on the amount isolated. Three of them, compounds .7, 14, and 15, exhibited strong to weak activity. Compound 15, with the MIC ranging from 62.5 to 500 µg/mL was the most active against C. albicans. More interestingly, compound 7, obtained from the dichloromethane/methanolic extract of M. kerstingii root, exhibited potent activity against C. albicans, C. krusei, and C. parapsilosis, with MICs of 7.81, 15.62, and 15.62 µg/mL, respectively. These results agree with those obtained by Fotso et al. in 2019 [7], in which compound 7 was intensely active against the fungus Septoria tritici. Previous phytochemical investigations highlighted the presence of flavonoids, stilbenes, coumarins, stilbene‐coumarins, and a steroid in extracts from the stem bark and leaves of M. kerstingii [7]. Phenolic compounds such as tannins, flavonoids, and simple phenols have shown their ability to destabilize the membrane of microorganisms [31, 32]. The antimicrobial effects of several flavonoids may be attributed to the inhibition of nucleic acid synthesis. Tannins can induce protein chelation, thereby inactivating microbial adhesins, membrane enzymes, and membrane transport proteins [33, 34]. Regarding antifungal activity, it has been shown that stilbene and some analogues are tyrosinase inhibitors [35]. These mechanisms of action might explain the observed anticandidal activity.
Insight Into Oral Acute Toxicity
2.4
For oral acute toxicity testing, EMKL and MKL were selected based on their MICs. The limit test dose of 2 g/kg in rats showed no mortality when distilled water was used as the vehicle. The rats were individually observed for the first 30 min and 2 h, and then periodically for 24 h, and daily for 14 days. After 24 h of observation, no death was recorded, suggesting that the LD_50_ is up to 2 g/kg of body weight. The body weight of the animals increased with the same trend as the control group (Figures S96 and S97). There was no significant difference (P ≥ 0.05) between the relative organ weights of the experimental group and the control group. These findings suggest that EMKL and MKL do not directly affect the body or the metabolism of any vital organ. Behavioral observations for each animal treated with MKL and EMKL did not show signs of toxicity. The extracts tested at 2 g/kg body weight can be classified as nontoxic substances.
Conclusions
3
The anticandidal‐guided isolation of Monotes kerstingii Gilg root and leaf extracts, followed by a rapid targeted UHPLC‐ESI‐LIT‐Orbitrap‐MS/MS‐based metabolic analysis, was performed. Thus, 28 compounds (1–28) were isolated and eighteen annotated from both extracts, among which six constituents are reported here for the first time. The roots were found to be rich in stilbenes and stilbene‐coumarins, while the leaves were found to contain mainly flavonoid derivatives. It is worth noting that, unlike the stem bark, in which only trans stilbene‐coumarins could be isolated, the roots of the plants were found to contain various cis‐stereoisomers, probably due to their protection from sunlight, which induces cis‐trans isomerization in stilbenes. This study accordingly reports the first isolation of a cis‐stilbene‐coumarin from nature. The anticandidal screenings revealed that the leaf crude extract was more active than that from the root. However, the most active antifungal compound in the plant was a stilbene isolated from the root extract, which exhibited potent activity against 3 of the five tested Candida strains. The toxicity study conducted with the most active leaf extract showed an acute LD50 greater than 2 g/kg body weight. These results justify the use of Monotes kerstingii in traditional folk medicine for the treatment of skin diseases, although long‐term and subtoxic‐dose effects still need to be studied. From the leaf extract of this plant, an effective standardized phytomedicine can be formulated and safely integrated into the health care system for sustainable development.
Experimental Section
4
General Experimental Procedures
4.1
Optical rotation indices were determined in methanol/CDCl_3_ 8:2 on a JASCO polarimeter P‐2000 digital (JASCO, Tokyo, Japan) using a 1 nm cell. Column chromatography was carried out on silica gel 230–400 mesh (Merck, Darmstadt, Germany), 70–230 mesh (Merck), or Sephadex LH‐20 (Sigma‐Aldrich, Munich, Germany). Thin‐layer chromatography (TLC) was performed on Merck pre‐coated silica gel (60 F254) aluminum foil, and compound spots were detected by spraying with diluted sulfuric acid before heating the plate to about 100°C, or by visual inspection under a UV lamp at 254 nm and 365 nm. High‐resolution mass spectra were obtained with an Orbitrap Compact Spectrometer (Bruker, Germany) equipped with an HR‐ESI source. The spectrometer was operated in positive and negative modes (mass range: 50–1500, scan rate: 1.00 Hz) with automatic gain control to provide high‐accuracy mass measurements with a deviation of 0.4 ppm using Na formate as a calibrant. The following parameters were used for experiments: spray voltage of 4.5 kV, and capillary temperature of 200°C. Nitrogen was used as sheath gas (4 L/min). The optical rotations were measured using a PerkinElmer polarimeter. The ^1^H and ^13^C‐NMR spectra were recorded on Bruker DRX 500 MHz and 600 MHz NMR spectrometers (Bruker Corporation, Brussels, Belgium) in deuterated solvents. Chemical shifts were reported in δ (ppm) using tetramethylsilane (TMS) (Sigma‐Aldrich) as an internal standard, while coupling constants (J) were measured in Hz. The analytical (Gradient A) and semi‐preparative RP HPLC (Gradient B) were performed on a Shimadzu Prominence system with a CBM‐20A communications bus module, an SPD‐M20A diode array detector, an FRC‐10A fraction collector, a DGU‐20A5R degassing unit, an LC‐20AT liquid chromatograph, and a SIL‐20A HT autosampler. For analytical HPLC, a YMC Pack Pro C18 column (5 µm, 250×4.5 mm I.D, YMC, USA) was used as a stationary phase with Gradient A: deionized water (Milli‐Q, Millipore, Schwalbach, Germany) with 0.025% TFA (trifluoroacetic acid; solvent A) and acetonitrile (solvent B) as mobile phase. The flow rate was set to 1 mL/min, and UV detection was performed at *λ *= 254 and 365 nm. Elution gradient: isocratic condition at 20% solvent B for 5 min, then increased to 80% in 20 min; isocratic condition at 80% for 5 min, then increased to 100% in 30 min. For semi‐preparative HPLC, a YMC Pack Pro C18 column (10 µm, 250×10 mm I.D., YMC, USA) was used as the stationary phase with the following gradient programs. Gradient B: Deionized water (Milli‐Q, Millipore, Schwalbach, Germany) with 0.025% TFA (Trifluoroacetic acid; solvent A) and acetonitrile (solvent B) as mobile phase. The flow rate was set at 3 mL/min, and UV detection was carried out at *λ *= 254 and 365 nm. Elution gradient: isocratic condition at 20% solvent B for 7 min, then increased to 80% in 29 min; isocratic condition at 80% for 7 min, then increased to 100% in 44 min. The HPLC methods used were able to separate the stereoisomers of the stilbene‐coumarins mixture (Figures S13 and S14).
Plant Material
4.2
The plant materials (leaves and roots) were harvested at Sakdjié in the North Region of Cameroon in March 2021. A fresh sample of this plant was identified at the National Herbarium of Cameroon by Mr Nana Victor and a specimen was registered under the voucher number N6661/SRFCAM. The samples were cut up, air‐dried for 2 weeks, and ground to obtain a fine powder.
Ethical Consideration
4.3
The experiments involving rats were conducted according to the ethical policies and procedures approved by the Cameroon National Ethical Committee (N° ref : FW‐IRB00001954).
Animal Material
4.4
For oral acute toxicity evaluation, adult female rats of the Wistar strain, weighing 90–110 g and aged 6–8 weeks, were randomly selected and marked to permit individual identification, and kept in their cages for 1 week for acclimation in the laboratory conditions (Lighting was artificial, the sequence being 12 h light and 12 h dark) before dosing. For feeding, conventional rodent laboratory diets were used, with unlimited access to drinking water.
Extracts Preparation
4.5
Small‐Scale Extractions
4.5.1
For the preliminary screenings, two extracts were prepared for each of the plant material (leaves and roots). The air‐dried and powdered leaves (52 g) was extracted twice by maceration for 48 h using (250 mL x2) of ethanol/water 7:3 (v/v) and methylene chloride/methanol 1:1 (v/v).The solvent was then filtered and evaporated under vacuum at 45°C to afford the crude extracts EMKL (8.0 g) and MKL (10.3 g), respectively. The same procedure was applied to the roots of the plant yielding the extracts EMKR (5.3 g) and MKR (8.6 g) for ethanol/water 7/3 (v/v) and methylene chloride/methanol 1/1 (v/v). respectively.
Large‐Scale Extractions
4.5.2
The air‐dried and powdered leaves (4.3 kg) was macerated twice in 15L of a mixture of dichloromethane/methanol (1:1) mixture for 48 h each. The solvent was each time then filtered and evaporated under vacuum at 45°C to afford 539.7 g of the crude extract MKL. Similarly, 3.2 Kg of the air‐dried and powdered roots was macerated twice in 12L of a mixture of dichloromethane/methanol (1:1) mixture for 48 h each. Filtration and evaporation of the solvents using a rotary evaporator under vacuum at 45°C led to the crude extracts labelled MKR (305.9 g).
Fractionation and Isolation of Compounds From MKR and MKL
4.6
The methylene chloride–methanol (1:1, v/v) crude extract of Monotes kerstingii root MKR (205.1 g) was dissolved in 250 mL of water and partitioned between n‐hexane (n‐hex: 500 mL × 3), ethyl acetate (EtOAc: 500 mL × 3), n‐butanol (n‐BuOH, 500 mL × 2) to give F1MKR (n‐hex, 35.19 g); F2MKR (EtOAc, 87.3 g); F3MKR (n‐BuOH, 38.92 g) and F4MKR (aqueous residue, 25.19 g). Then, 82 grams (82.3 g) of F2MKR was adsorbed and chromatographed over silica gel column chromatography (0.04 ‒ 0.063 mm, 22 × 122 cm) using a mixture of a gradient of acetone (Ace) and n‐hexane (n‐hex) from 100:0 to 0:100 (v/v). The column was monitored by a UV lamp (254 and 366 nm). More than six hundred fractions of 200 mL each were collected gradually as follows: n‐hex/Ace 97.5:2.5: [1–46], n‐hex/Ace 95:5: [47–111], n‐hex/Ace 92.5:7.5: [112–151], n‐hex/Ace 90:10: [152–193], n‐hex/Ace 87.5:12.5: [194–214], n‐hex/Ace 85:15: [215–223], n‐hex/Ace 82.5:17.5: [224–253], n‐hex/Ace 80:20: [254–288], n‐hex/Ace 77.5:22.5: [289–308], n‐hex/Ace 75:25: [309–359], n‐hex/Ace 70:30: [360–422], n‐hex/Ace 65:35: [423–500], n‐hex/Ace 60:40: [501–544], n‐hex/Ace 55:45 [545–563], n‐hex/Ace 50:50: [564–612], n‐hex/Ace 25:75: [613–685], and pure acetone [686–698]. From this column, nine pure compounds were directly obtained and characterized: the mixture of stigma sterol and β‐sitosterol (8–9, 22, 16.1 mg) [Rf 0.26: n‐hexane/ethyl acetate 85:15 (v/v)] as white needles, betulinic acid (99–109, 24, 8.1 mg) [Rf 0.66: n‐hexane/ethyl acetate 75:25 (v/v)] as white powder, 5‐[(1E)‐2‐(4‐hydroxyphenyl) ethenyl]‐4‐methoxyphenyl}‐2‐methyl‐1‐propanone (154–164, 7, 27.1 mg) [Rf 0.46: n‐hexane/ethyl acetate 80:20 (v/v)], as yellow crystal. Then, one white powder, three yellow powders and one red oil were obtained and identified as: 5‐[(1E)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐3‐methyl‐2H‐1‐benzopyran‐2‐one (213–226, 10, 7.2 mg) [Rf 0.30: n‐hexane/acetone 72.5:27.5 (v/v)], (1:1) mixture of 1‐{2‐hydroxy‐6‐[(1E)‐2‐(4‐hydroxyphenyl) ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one and 1‐{2‐hydroxy‐6‐[(1Z)‐2‐(4‐hydroxyphenyl) ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one (260–288**, 3**, 8.0 mg) [Rf 0.44: n‐hexane/ethyl acetate 75:25 (v/v)], 3‐O‐[6’‐O‐psylloyl‐β‐D‐glucopyranosyl]‐β‐sitosterol (295–307**, 6**, 14.0 mg) [Rf 0.57: n‐hexane/acetone 70:30 (v/v)], 5‐[(1E)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one (340–400**, 9**, 6000.2 mg) [Rf 0.54: n‐hexane/ethyl acetate 60:40 (v/v)] and 1‐{2, 4‐dihydroxy‐6‐[(1E)‐2‐(4‐hydroxyphenyl) ethenyl]‐phenyl}‐2‐methyl‐1‐propanone (300–312**, 8**, 6.4 mg) [Rf 0.43: DCM/MeOH 90:10 (v/v)]. Stigma sterol glucoside (577–582, 23, 20.9 mg) [Rf 0.45: DCM/MeOH 90:10 (v/v)] and 3‐O‐methylellagic acid‐4′‐O‐α‐L‐rhamnopyranoside (600–620, 13, 14.1 mg) [Rf 0.40: DCM/MeOH 90:10 (v/v)] as beige powders. The subfraction 650–665 was chromatographed on silica gel with an isocratic system of n‐hex/Ace 70:30 (v/v). Twenty‐five fractions of 100 mL were collected, and 14–19 were purified by Sephadex LH‐20 column chromatography using MeOH. Fractions of 1–2 mL were collected to afford a pink oil, namely {5‐[(1E)‐2‐(4‐β‐D‐glucopyranosylphenyloxy)ethenyl]‐4′‐methoxyphenyl}‐2‐methyl‐1‐propanone (23–31, 1, 15.0 mg) [Rf 0.39: n‐hexane/acetone 40:60 (v/v)] along with two white powders: bergenin (40–44, 28, 2.1 mg) [Rf 0.32: n‐hexane/ethyl acetate 40:60 (v/v)], and 4‐hydroxybenzoic acid (68–70, 26, 1.8 mg) [Rf 0.40: n‐hexane/ethyl acetate 40:60 (v/v)].
Thirty‐six grams (36.0 g) of F3MKR was adsorbed and chromatographed over silica gel column chromatography (0.04‒0.063 mm, 16 × 80 cm) using a gradient of methanol (MeOH) and ethyl acetate (EtOAc) from 100:0 to 0:100 (v/v). The column was monitored by a UV lamp (254 and 366 nm). One hundred and fifty fractions of 100 mL each were collected gradually as follows: EtOAc 100:0: [1–63], EtOAc/MeOH 95:5: [64–79], EtOAc/MeOH 85:15: [80–107], EtOAc/MeOH 70:30: [108–129], EtOAc/MeOH 50:50: [130–145] and pure MeOH [146–150] to afford stigma sterol glucoside (5–10, 23, 16.8 mg) [Rf 0.30: EtOAc/MeOH 90:10 (v/v)] and mixture of 5‐[(1Z)‐2‐(4‐β‐D‐galactopyranosyloxyphenyl)ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one and 5‐[(1Z)‐2‐(4‐β‐D‐galactopyranosyloxyphenyl)ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one (45–63, 4, 28.3 mg) [Rf0.33: ethyl acetate/methanol 90:10 (v/v)] in EtOAc–MeOH (100:0, v/v), Once reached EtOAc–MeOH 80:20 (v/v), 5‐[(1E)‐2‐(4‐β‐D‐galactopyranosyloxyphenyl) ethenyl]‐4, 7‐dimethoxy‐3‐2H‐1‐benzopyran‐2‐one (65–68, 11, 7.8 mg) [Rf0.33: ethyl acetate/methanol 90:10 (v/v)] and 3‐O‐methylellagic acid‐4′‐O‐α‐ L‐rhamnopyranoside (81–83, 13, 22.0 mg) [Rf 0.40: DCM/MeOH 90:10 (v/v)] were obtained. Compound 2 (1.4 mg) was obtained as a yellow powder described as 5‐[(1Z)‐2‐(4‐hydroxyphenyl)ethenyl]‐4,7‐dimethoxy‐2H‐1‐benzopyran‐2‐one through the purification of a 1:1 mixture of two stilbene‐coumarins derivatives (2+9) from the ethyl acetate fraction of MKR using analytical RP HPLC (Gradient A), two peaks were detected, respectively, at t R * *= 19.3 and 20.1 min. Therefore, 9.5 mg of this mixture was set for purification by semipreparative RP HPLC (Gradient B) to afford 9 (t R 28.4 min, 4.2 mg) and a 2.1 mg of a mixture of cis and trans (2+9) which was repurified (Gradient B) to afford 2 (t R 27.6 min, 1.4 mg) and 9 (t R 28.3 min, 0.3 mg). All reagents used were analytical grade. All the efforts to purify the mixtures 3 and 4 were unsuccessful.
The methylene chloride–methanol (50:50, v/v) crude extract of Monotes kerstingii leaves MKL (509.0 g) was treated with 500 mL of petroleum ether–ethyl acetate (99:1, v/v) to remove chlorophyll. The crude residue (379.8 g) was dissolved in 300 mL of water and partitioned between petroleum ether (PE: 500 mL × 1), n‐hexane (n‐hex: 500 mL × 3), ethyl acetate (EtOAc: 500 mL × 3), n‐butanol (n‐BuOH, 500 mL × 2) to give F1MKL (PE: 50.6 g), F2MKL (n‐hex: 10.4 g), F3MKL (EtOAc: 118.7 g), F4MKL (n‐BuOH, 72.4 g) and F5MKL (aqueous residue, 124.0 g). The EtOAc fraction F3MKL (115.9 g) was subjected to open silica gel column chromatography (0.04‒0.063 mm, 31 × 175 cm) using a mixture of n‐hex—EtOAc (75:25, v/v), n‐hex—EtOAc (50:50, v/v), n‐hex—EtOAc (25:75, v/v), EtOAc, and MeOH resulting in four major subfractions based on TLC profile: F_1_AE (10.1 g, n‐hex—EtOAc (75:25, v/v)), F_2_AE (21.8 g, n‐hex—EtOAc (50:50, v/v)), F_3_AE (8.3 g, n‐hex—EtOAc (25:75, v/v)), and F_4_AE (39.7 g, EtOAc, and MeOH).
The sub‐fraction F_2_AE (20.37 g) was chromatographed over silica gel column chromatography (0.04‒0.063 mm, 6 × 30 cm) eluting using a gradient of EtOAc in n‐hexane from 95:5 to 0:100 (v/v). The column was monitored by a UV lamp (254 and 366 nm). More than three hundred fractions of 100 mL each were collected gradually like pure n‐hex [1–6], n‐hex/EtOAc 95:5: [7–40], n‐hex/EtOAc 90:10: [41–73], n‐hex/EtOAc 85:15: [74–80], n‐hex/EtOAc 80:20: [81–90], n‐hex/EtOAc 75:25: [91–140], n‐hex/EtOAc 70:30: [141–177], n‐hex/EtOAc 65:35: [178–204], n‐hex/EtOAc 60:40: [205–215], n‐hex/EtOAc 55:45 [216–223], n‐hex/EtOAc 50:50: [224–245], n‐hex/EtOAc: [246–274], n‐hex/EtOAc 30:70: [275–298], n‐hex/EtOAc 20:80: [299–334], pure ethyl acetate [335–351] and EtOAc/MeOH 90:10: [352–363]. From this fraction, six compounds were directly filtered and washed with methanol, they were characterized as follows: noreugenin (55–58, 27, 8.07 mg) [Rf 0.61: n‐hexane/ethyl acetate 80:20 (v/v)] as a white powder, (2S)‐4',5‐dihydroxy‐[3‐hydroxy‐2,2‐dimethyl‐3,4‐dihydropyrano(5,6:6,7)]flavanone (78–88, 5, 20.05 mg) [Rf 0.76: n‐hexane/dichloromethane 20:80 (v/v)] as a white powder, quercetin (128–135, 14, 17.39 mg) [Rf 0.53: n‐hexane/DCM 20:80 (v/v)] as a yellow powder, tiliroside (279–282, 19, 16.42 mg) [Rf 0.81: EtOAc/MeOH 92.5:7.5 (v/v)]as a yellow powder, hyperoside (329–340, 16, 25.18 mg) [Rf 0.46: EtOAc/MeOH 92.5:7.5 (v/v)] as a yellow powder, isoquercitrin (350–355, 15, 9.31 mg) [Rf 0.46: EtOAc/MeOH 80:20 (v/v)] as a yellow powder. Purification of the sub‐fraction 41–80 obtained from F_2_AE over silica gel column chromatography (0.04‒0.063 mm, 2 × 30 cm) eluting using a gradient of EtOAc in n‐hexane from 100:0 to 80:20 (v/v). The column was monitored by a UV lamp (254 and 366 nm). Seventy‐five fractions of 10 mL each were collected like pure n‐hex [1–9], n‐hex/EtOAc 97.5:2.5: [10–27], n‐hex/EtOAc 95:5: [28–59], n‐hex/EtOAc 85:15: [60–68], n‐hex/EtOAc 80:20: [69–75] afforded four pure secondary metabolites labelled: 7‐O‐(3‐methyl‐2‐butenyl) kaempferide (12–19, 18, 2.97 mg) [Rf 0.39: n‐hexane/ethyl acetate 95:5 (v/v)] as yellowish needles, 5, 4′‐dihydroxy‐4″, 4″‐dimethyl‐5″‐methyl‐5″‐H‐dihydrofurano [2″, 3″:6, 7] flavanone (34–40, 21, 3.13 mg) [Rf 0.51: n‐hexane/ethyl acetate 90:10 (v/v)] as a beige powder, 4′, 7‐dimethoxy kaempferol (53–59, 17, 1.88 mg) [Rf 0.41: n‐hexane/ethyl acetate 90:10 (v/v)] as a beige powder and 6‐(1, 1‐dimethylallyl) naringenin (68–71, 20, 5.10 mg,) [Rf 0.47: n‐hexane/ethyl acetate 85:15 (v/v)] as a white powder. Purification of F_4_AE (36.7 g) was purified over silica gel column chromatography (0.04‒0.063 mm, 8 × 30 cm) eluting using a gradient of EtOAc in n‐hexane from 85:15 to 0:100 (v/v). The column was monitored by a UV lamp (254 and 366 nm). One hundred and eighty fractions of 100 mL each were collected gradually like n‐hex/EtOAc 85:15: [1–20], n‐hex/EtOAc 80:20: [21–40], n‐hex/EtOAc 75:25: [41–62], n‐hex/EtOAc 70:30: [63–89], n‐hex/EtOAc 60:40: [90–104], n‐hex/EtOAc 60:40: [105–115], n‐hex/EtOAc 50:50 [116–138], n‐hex/EtOAc 25:75 [139–174] and pure ethyl acetate [175–180], three yellow powders characterized as quercetin (43–50, 14, 4.02 mg) [Rf 0.53: n‐hexane/DCM 20:80 (v/v)], hyperoside (61–65, 16, 7.49 mg) [Rf 0.46: EtOAc/MeOH 92.5:7.5 (v/v)] and ellagic acid (136–147, 12, 9.14 mg) [Rf 0.34: EtOAc/MeOH 95:5 (v/v)] were obtained. F_3_AE (7.9 g) was chromatographed over silica gel column chromatography (0.04‒0.063 mm, 3 × 60 cm) eluting using a gradient of EtOAc in n‐hexane from 80:20 to 0:100 (v/v). The column was monitored by a UV lamp (254 and 366 nm), which allowed us to isolate hyperoside (16, 7.49 mg) and at n‐hex/EtOAc 25:75 3‐O‐β‐D‐glucopyranosyl oleanolic acid (25, 2.16 mg) [Rf 0.41: n‐hexane/ethyl acetate 20:80 (v/v)] as a white powder was obtained. Concerning the n‐butanolic fraction (F4MKL), only hyperoside was isolated (8.26 mg).
RP‐UHPLC‐ESI‐LIT‐Orbitrap‐MS/MS
4.7
2 µL of the leaf (MKL) and root (MKR) extracts at 2 mg/mL each was loaded on an ACQUITY UPLC reversed‐phase BEH column (C18‐phase, ID 1 mm, length 50 mm, particle size 1.7 µm, Waters GmbH, Eschborn, Germany) under isocratic conditions (95% A + 5% eluent B, 1 min), and separated using a linear gradient from 5% to 95% eluent B in 10 min using water and acetonitrile (both containing 0.1% v/v formic acid) as eluents A and B, respectively. The separations were performed with a Dionex Ultimate 3000 UHPLC System (Thermo Fisher Scientific, Bremen, Germany) at a flow rate of 400 µL/min and a column temperature of 40°C. The column effluents were transferred online into a hybrid LTQ‐Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), equipped with a heated electrospray ionization (HESI) source at 300°C and operated in both negative and positive ion mode. The analysis in negative mode was performed under an ion spray (IS) voltage of 3.5 kV, with a nebulizer and auxiliary gases set to 20 and 10 psig, respectively. The capillary temperature was set to 275°C. The analysis in positive mode was performed under an IS voltage of 4.0 kV, with a nebulizer and auxiliary gases set to 25 and 21 psig, respectively. The capillary temperature was set to 325°C.
Analytes were annotated in preliminary data‐dependent acquisition (DDA) experiments designed according to the double‐play algorithm. The DDA experiments comprised a survey FT‐scan with a mass resolution of 30000 (m/z 100–2000 in negative mode and m/z 220–2000 in positive mode) followed by dependent linear ion trap (LIT) scans for the three most abundant signals selected in each survey scan. Analytes were annotated in survey scans based on their tR, m/z, isotopic distributions, and tandem mass spectrometric (MS/MS) patterns. Thereby, collision‐induced dissociation (CID) was performed in LIT by resonance activation in the presence of He as a collision/cooling gas. The Normalized collision energy was 30%. The corresponding quasi‐molecular ions were isolated with a width of 2 m/z, and activation time and relative activation frequency were 10 ms and 0.250, respectively. Spectra were evaluated in Xcalibur 2.2 software (Thermo Fisher Scientific).
Candida Growth Inhibition Assay
4.8
Preliminary Screening at a Single High Dose of Plant Crude Extracts
4.8.1
The inhibitory potential of the crude extracts at a single high concentration (2 mg/mL) was determined according to the protocol described by Clinical and Laboratory Standards Institute (CLSI), (2008) (M27‐A3 protocol) [36] with slight modifications. Thus, in a 96‐well microplate, 60 µL of Sabouraud Dextrose Broth (SDB) medium (Sigma‐Aldrich) was added to each well, followed by 40 µL of crude extracts (10 mg/mL in 10% DMSO). Subsequently, 100 µL of the fungal suspension standardized at 2 × 10^4^ CFU/mL using the 0.5 McFarland standard solution (2.5 × 10^6^ CFU/mL) was added to each well except for those of the sterility control. The plates were covered and incubated at 37°C for 48 h. Fluconazole (Sigma‐Aldrich) at 1.25 µg/mL was used as the positive control. The negative control consisted of culture medium and inoculum, and the sterility control consisted of culture medium alone. The tests were done in duplicate. At the end of the incubation period, 20 µL of a freshly prepared resazurin solution (0.15 mg/mL in sterile distilled water) was aseptically added, and the plates were re‐incubated under the same conditions for 30 min. The extracts in wells colored blue were considered active at 2000 µg/mL and selected for MIC determinations.
MIC Determinations of Selected Crude Extracts, Fractions, and Isolated Compounds
4.8.2
The MICs of the selected extracts/fractions and compounds were determined using the aforementioned protocol. The text was performed twice in duplicate measurements in sterile 96‐well microplates. The extracts/fractions, single compounds and fluconazole were prepared by microdilution in different concentrations ranging from 2000 to 1.95 µg/mL, from 500 to 0.48 µg/mL, and from 1.25 to 0.0383 µg/mL, respectively. The negative control was made up with culture media and fungal suspension, the positive control was constituted with fluconazole, fungal suspension, and culture media, while the sterility control was the culture media alone. The final inoculum concentration in each well was 10^4^ cells/mL, with a final volume of 200 µL. At the end of the incubation period, the MIC was determined using the resazurin‐based viable assay as previously described. The MIC was then defined as the lowest concentration of extract/fractions or compounds at which no change in resazurin color from blue to pink was observed.
Oral Acute Toxicity
4.9
Oral acute toxicity of the most active extracts (MKL and EMKL) was assessed according to OECD 423 guidelines using female Wistar albino rats [37]. Experimental animals were divided into three groups (three rats per group). They were fasted for 12 h before the assay with free access to water. The control group received distilled water, and the experimental groups received a single oral dose of 2 g extract/kg body weight. Animals were observed for any changes in general behavior and mortality for 30 min and periodically for 24 h after administration. The body weight of each animal was recorded every 2 days. The rats were observed for 14 days, and at the end of the treatment, the animals were humanely killed for relative organ weight evaluation.
Statistical Analysis
4.10
The raw data were statistically analyzed using OriginPro 2024 and GraphPad Prism software, version 8.0.1.244. The results were presented as mean ± standard deviation. Data comparison was performed using analysis of variance (ANOVA) followed by Fisher LSD's post‐test. P values ≤ 0.05 were considered significant
Spectroscopic Data
4.11
Kerstingiioside (1)
4.11.1
Pink oil (15.0 mg), silica gel 60 F254, n‐hexane/EtOAc 80:20 (v/v), [Rf 0.39: n‐hexane/acetone 40:60 (v/v)] ^1^ H‐NMR, ^13^C‐NMR (CD_3_OD, 500 and 125 MHz) and HMBC, see Table 1, HR‐ESI‐MS (+): [M+ H] ^+^ m/z 475.1963 (calcd. for C_25_H_31_O_9_ ^+^, m/z 475.1963), [M+ Na] ^+^ m/z 497.1792 (calcd. for C_25_H_30_NaO_9_ ^+^, m/z 497.1783), [2M+ Na] ^+^ m/z 971.3671 (calcd. for C_50_H_60_NaO_18_+, m/z 971.3672).
cis‐Kerstilbcoumarin A (2)
4.11.2
Yellow powder (1.4 mg), semipreparative RP HPLC, ^1^ H‐NMR, ^13^C‐NMR (DMSO, 500 and 125 MHz) and HMBC, see Table 2, HR‐ESI‐MS (+): [M+H] ^+^ m/z 325.1083 (calcd. for 325.1071 for C_19_H_17_O_5_ ^+^), [2M+H] ^+^ m/z 649.2072 (calcd. for 325.1071 for C_38_H_33_O_10_ ^+^),
cis‐Kerstilbcoumarin B (3)
4.11.3
Green powder (8.0 mg), silica gel 60 F254, n‐hexane/ethyl acetate 65:35 (v/v), [Rf 0.44: n‐hexane/ethyl acetate 75:25 (v/v)], ^1^ H‐NMR, ^13^C‐NMR (CD_3_COCD_3_, 500 and 125 MHz) and HMBC, see Table 2, HRESIMS (‐): [M‐H]^−^ m/z 337.1084 (calcd. for C_20_H_17_O_5_ ^−^ m/z 337.1076), [2 M + HCOO]^−^ m/z 721.2281 (calcd. for C_41_H_37_O_12_ ^−^ m/z 721.2290)
cis‐Kerstilbcoumarin C (4)
4.11.4
White powder (28.3 mg), silica gel 60 F254, n‐hexane/acetone 25:75 (v/v), [Rf0.33: ethyl acetate/methanol 90:10 (v/v)], ^1^ H‐NMR, ^13^C‐NMR (DMSO, 500 and 125 MHz) and HMBC, see Table 2, HR‐ESI‐MS (+): [M+H] ^+^ m/z 487.1592 (calcd. for 487.1599 for C_25_H_27_O_10_ ^+^), [M+Na] ^+^ m/z 509.1411 (calcd. for 509.1419 for C_25_H_26_NaO_10_ ^+^), [2M+Na] ^+^ m/z 995.2901 (calcd. for 995.2945 for C_50_H_52_NaO_20_ ^+^),
Kerstingiiflavanone (5)
4.11.5
White powder (20.0 mg), silica gel 60 F254, n‐hexane/acetate 80:20 (v/v), [Rf 0.76: n‐hexane/dichloromethane 20:80 (v/v)]^1^ H‐NMR, ^13^C‐NMR (DMSO, 500 and 125 MHz) and HMBC, see Table 3, HR‐ESI‐MS (+): [M+H]^+^ m/z 357.1343 (calcd. for 357.1333 for C_20_H_21_O_6_ ^+^), [M‐H]^−^ m/z 355.1172 (calcd. for 355.1187 for C_20_H_19_O_6_ ^−^), [M‐Cl]^−^ m/z 391.0938 (calcd. for 391.0953 for C_20_H_20_ClO_6_ ^−^).
Monestoside B (6)
4.11.6
Yellow amorphous powder (14.0 mg), silica gel 60 F254, n‐hexane/acetone 75: 25 (v/v), [Rf 0.57: n‐hexane/acetone 70:30 (v/v)] ^1^ H‐NMR, ^13^C‐NMR (CDCl_3_, 500 and 125 MHz) and HMBC, see Table 4, Positive HRESIMS m/z 1051.9351 [M+H] ^+^ (calcd. For C_68_H_123_O_7_ ^+^ m/z 1051.9269).
Author Contributions
Ghislain Wabo Fotso, Bruno Ndjakou Lenta, and Bonaventure Tchaleu Ngadjui: conceptualization. Sorelle Kache Fotsing, Alena Soboleva, Andrej Frolov, Dominique Ngnintedo, and Kevine Dongmo Jumeta: formal analysis and software. Bruno Ndjakou Lenta, Norbert Sewald, Ludger A. Wessjohann, and Norbert Arnold: funding acquisition and methodology. Sorelle Kache Fotsing, Yanick Kevin Dongmo Melogmo, and Alena Soboleva: investigation. Ghislain Wabo Fotso, Bruno Ndjakou Lenta, and Bonaventure Tchaleu Ngadjui: supervision, validation, and visualization. Bruno Ndjakou Lenta and Norbert Sewald: project administration. Ludger A. Wessjohann, Bonaventure Tchaleu Ngadjui, and Ghislain Wabo Fotso: data curation. Norbert Sewald, Bruno Ndjakou Lenta, Ludger A. Wessjohann, and Bonaventure Tchaleu Ngadjui: resources. Sorelle Kache Fotsing, Alena Soboleva, Dominique Ngnintedo, and Yanick Kevin Dongmo Melogmo: writing – original draft. Ghislain Wabo Fotso, Bruno Ndjakou Lenta, Bonaventure Tchaleu Ngadjui, Ludger A. Wessjohann, Fabrice Fekam Boyom, and Norbert Sewald: writing – review and editing.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting file 1: cbdv71026‐sup‐0001‐SuppMat.pdf.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. Pinho , I. M. Miranda , and S. Costa‐de‐Oliveira , “Global Epidemiology of Invasive Infections by Uncommon Candida Species: A Systematic Review,” Journal of Fungi 10, no. 8 (2024): 558, 10.3390/jof 10080558 CDC.39194884 PMC 11355942 · doi ↗ · pubmed ↗
- 2CDC Active Bacterial Core surveillance (AB Cs) . Population‐Based Active Surveillance for Culture‐Confirmed Candidemia — 10 Sites, United States, 2017–2021. MMWR Surveillance Summaries 74 no. SS‐4 (2023): 1–13.10.15585/mmwr.ss 7404 a 1PMC 1211550540424200 · doi ↗ · pubmed ↗
- 3S. Wolfgruber , S. Sedik , L. Klingspor , et al., “Insights From Three Pan‐European Multicentre Studies on Invasive Candida Infections and Outlook to ECMM Candida IV,” Mycopathologia 189 (2024): 70, 10.1007/s 11046-024-00871-0.39088098 PMC 11294264 · doi ↗ · pubmed ↗
- 4P. G. Pappas , C. A. Kauffman , D. R. Andes , et al., “Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America,” Clinical Infectious Diseases 62, no. 4 (2016): e 1–e 50, 10.1093/cid/civ 933.26679628 PMC 4725385 · doi ↗ · pubmed ↗
- 5S. Bhattacharya , S. Sae‐Tia , and B. C. Fries , “Candidiasis and Mechanisms of Antifungal Resistance,” Antibiotics 9 (2020): 312, 10.3390/antibiotics 9060312.32526921 PMC 7345657 · doi ↗ · pubmed ↗
- 6V. Kuete and T. Efferth , “Cameroonian Medicinal Plants: Pharmacology and Derived Natural Products,” Frontiers in Pharmacology 1 (2010), Oct 25: 123, 10.3389/fphar.2010.00123. e Collection 2010.21833168 PMC 3153003 · doi ↗ · pubmed ↗
- 7G. W. Fotso , L. Mogue Kamdem , M. Dube , et al., “Antimicrobial Secondary Metabolites From the Stem Barks and Leaves of Monotes Kerstingii Gilg (Dipterocarpaceae),” Fitoterapia 137 (2019): 104239, 10.1016/j.fitote.2019.104239.31201886 · doi ↗ · pubmed ↗
- 8B. C. Valentin , K. I. Pierre , M. M. Henry , F. M. Kasali , O. N. Philippe , and L. S. Jean Baptiste , “Ethnobotanical Study of Plants Used by Traditional Healers in Lubumbashi (Democratic Republic of Congo) in the Management of Typhoid Fever,” GSC Biological and Pharmaceutical Sciences 21, no. 01 (2022): 265–286, 10.30574/gscbps.2022.21.1.0403. · doi ↗