The Yin–Yang of Stress and Senescence: Integrated Stress Response and SASP Crosstalk in Stem Cell Fate, Regeneration, and Disease
Douglas M. Ruden

TL;DR
Stem cell fate is shaped by a balance between protective and harmful stress responses, which influence regeneration and disease.
Contribution
The paper introduces a Yin–Yang framework to explain how ISR and SASP interact to regulate stem cell behavior and tissue outcomes.
Findings
ISR and SASP pathways act as a Yin–Yang system, balancing stem cell adaptation and maladaptation.
Chronic ISR and unresolved SASP contribute to stem cell exhaustion and disease.
ISR–SASP crosstalk influences tissue niches and long-term stem cell potential.
Abstract
Stem cell fate decisions are increasingly understood through the dynamic interplay of two fundamental stress-adaptive programs: the integrated stress response (ISR) and the senescence-associated secretory phenotype (SASP). These pathways act as a Yin–Yang system, balancing beneficial and detrimental outcomes across development, tissue homeostasis, and disease. On the yin (protective) side, transient ISR activation and acute SASP signaling foster adaptation, embryonic patterning, wound healing, and regeneration. On the yang (maladaptive) side, chronic ISR signaling and unresolved SASP output drive stem cell exhaustion, fibrosis, inflammation, and tumorigenesis. This duality highlights their roles as both guardians and disruptors of stem cell integrity. Mechanistically, ISR regulates translational control via eukaryotic initiation factor 2 alpha (eIF2α) phosphorylation and activating…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
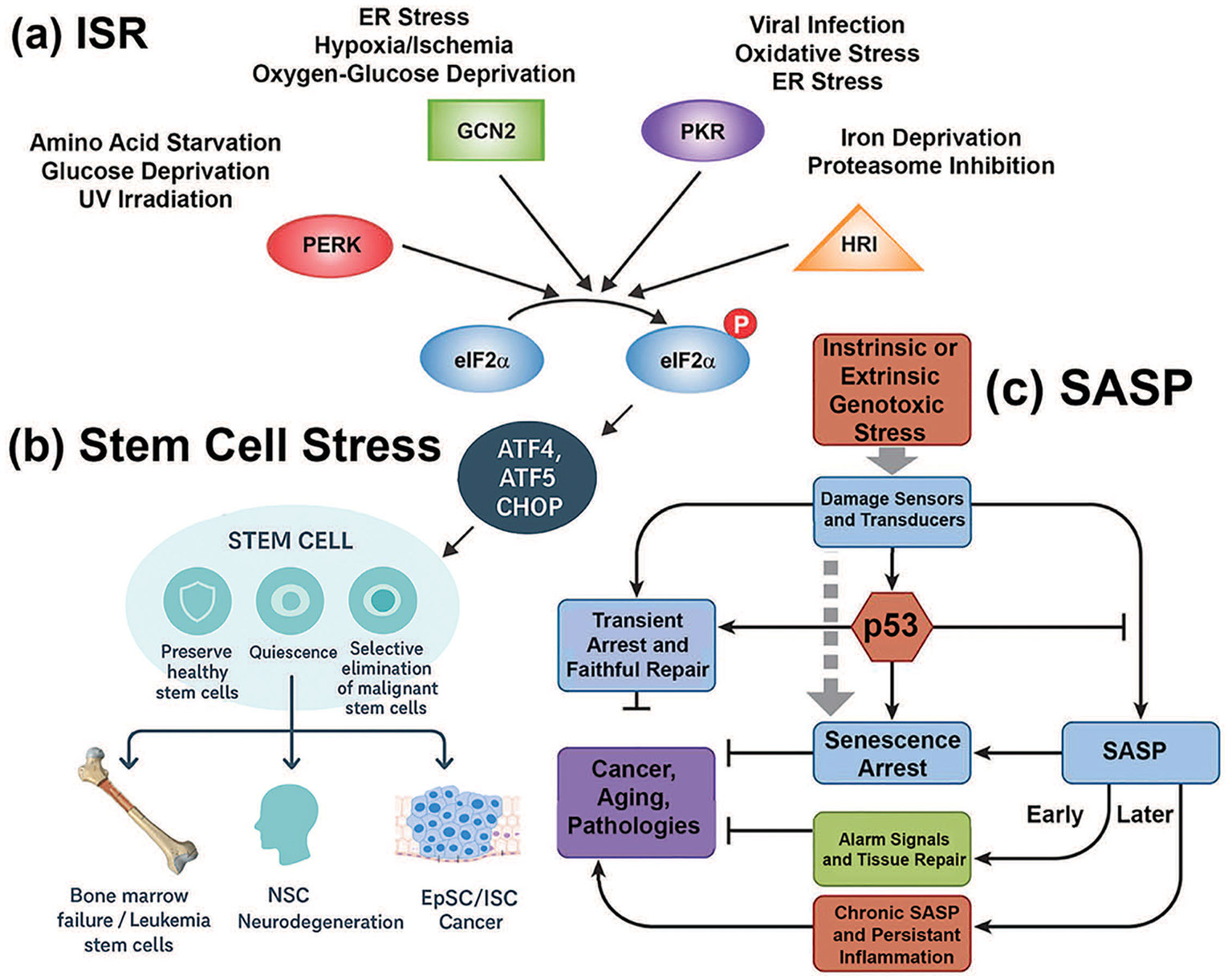 Figure 1
Figure 1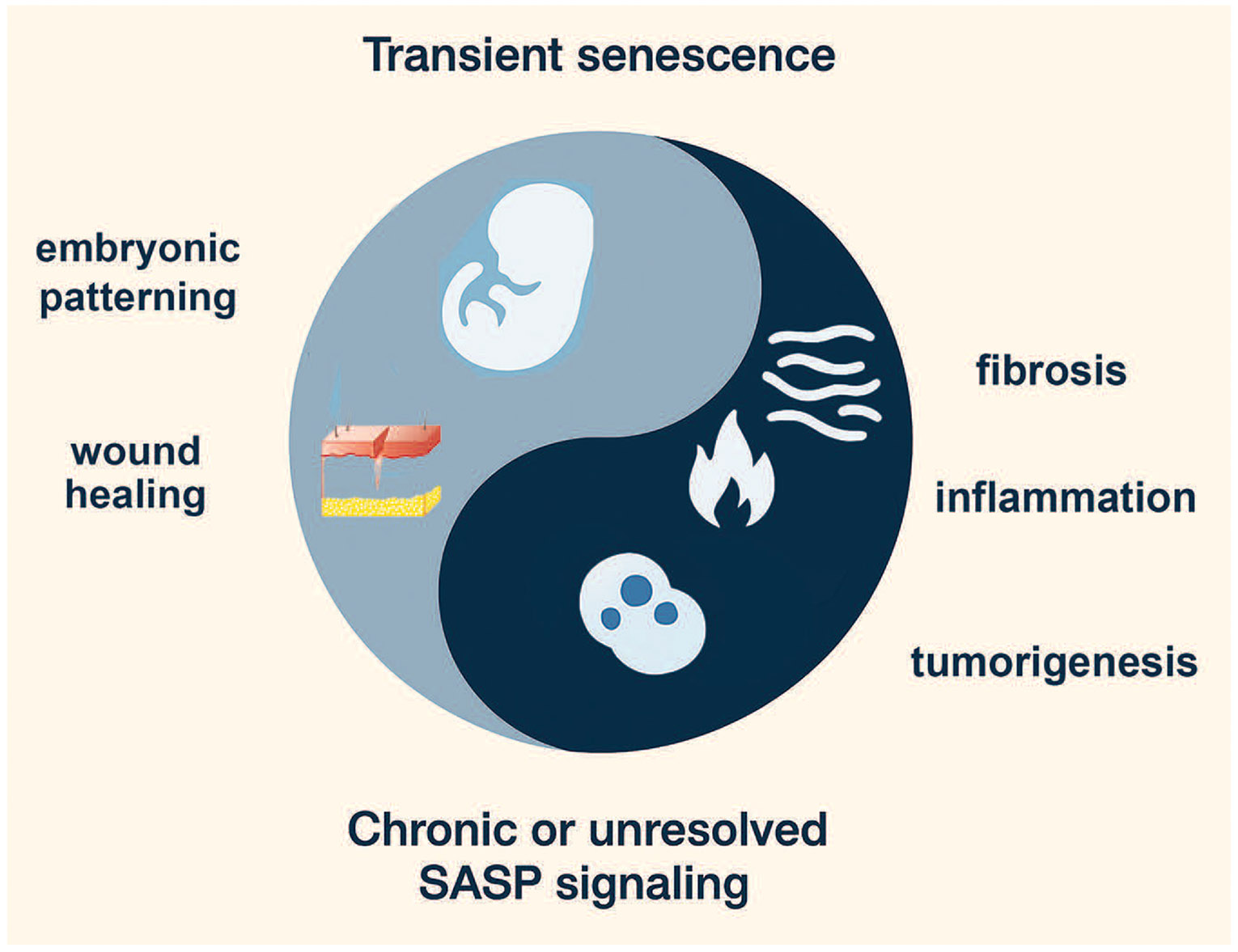 Figure 2
Figure 2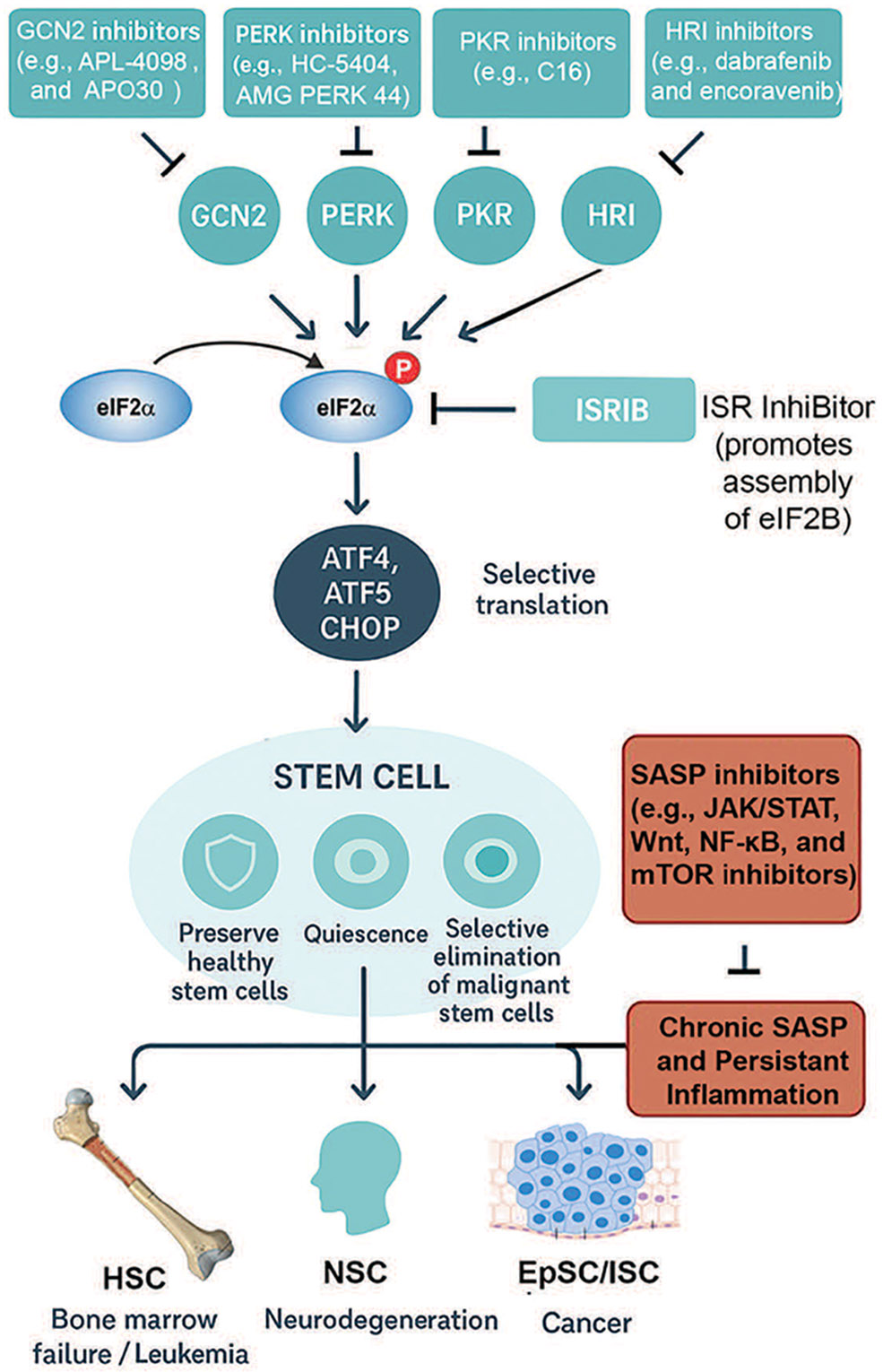 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTelomeres, Telomerase, and Senescence · Pluripotent Stem Cells Research · RNA regulation and disease
Introduction
1
Cellular stress responses have evolved to maintain tissue homeostasis by enabling cells to sense, adapt, or undergo programmed elimination in the face of environmental and metabolic perturbations. Two central processes that exemplify this adaptive capacity are the integrated stress response (ISR) (Fig. 1a) [1,2] and the senescence-associated secretory phenotype (SASP) (Fig. 1c) [3]. Although traditionally studied in distinct contexts, these pathways are increasingly recognized as intersecting networks that shape the biology of stem cells in health and disease (Fig. 1b) [4].
The ISR is a highly conserved signaling axis that modulates protein synthesis in response to diverse stressors such as nutrient deprivation, hypoxia, oxidative stress, and endoplasmic reticulum (ER) stress (Fig. 1a) [1,2]. The pathway converges on phosphorylation of the eukaryotic initiation factor eukaryotic initiation factor 2 alpha (eIF2α), resulting in global translational attenuation coupled with selective upregulation of stress-responsive genes, particularly the activating transcription factor 4 (ATF4) [5]. Through this mechanism, the ISR functions as a rheostat that balances adaptive survival with apoptotic elimination [5]. In stem cells, where proteostasis and energy balance are tightly linked to fate decisions, ISR activation can preserve quiescence, promote lineage bias, or trigger differentiation depending on context [4].
In parallel, the SASP represents a hallmark feature of senescent cells, characterized by the secretion of pro-inflammatory cytokines, chemokines, growth factors, and matrix remodeling enzymes (Fig. 1c) [3]. Initially described as a driver of aging-related pathologies [5], the SASP is now recognized as a context-dependent mediator of tissue remodeling, regeneration, and developmental morphogenesis [6]. Transient, developmentally programmed senescence contributes to embryonic patterning [7], while injury-induced senescence accelerates wound healing and tissue repair [8]. Conversely, chronic or unresolved SASP signaling promotes fibrosis, inflammation, and tumorigenesis.
Stem cells are uniquely poised at the intersection of ISR and SASP biology (Fig. 1b) [9]. They require stress response pathways to sustain self-renewal and differentiation capacity, yet are vulnerable to maladaptive senescence signaling that can compromise their function [10]. Emerging evidence suggests that ISR activity in senescent cells directly shapes SASP composition, particularly by amplifying pro-inflammatory and proteolytic factors [11]. Conversely, SASP-mediated paracrine signaling can feed back onto stem cells, inducing plasticity or exhaustion depending on exposure dynamics [12].
The convergence of ISR and SASP thus represents a critical axis in developmental biology, tissue homeostasis, and age-associated disease [13]. This review synthesizes recent advances in understanding ISR–SASP crosstalk in stem cells, highlighting mechanisms, developmental roles, pathological consequences, and therapeutic opportunities. We also discuss methodological approaches that have enabled the dissection of these pathways, and we outline future directions aimed at leveraging ISR–SASP biology for regenerative and anti-aging interventions.
Methods
2
As this is a review article, Section 2 describes the systematic approach to literature selection, integration, and synthesis. We performed a structured literature search across PubMed, Web of Science, and Scopus databases from 2000–2025, using combinations of keywords including “integrated stress response,” “ISR,” “eIF2α phosphorylation,” “ATF4,” “senescence-associated secretory phenotype,” “SASP,” “stem cells,” “developmental senescence,” “inflammation,” and “regeneration.” We also screened preclinical trial databases and citation networks of key ISR–SASP papers to identify additional mechanistic or therapeutic studies.
Studies in mammalian models were prioritized, with supplemental evidence drawn from invertebrate systems where mechanistic insights were uniquely informative. Inclusion criteria required primary data on ISR or SASP signaling, stem cell outcomes, or disease relevance; exclusion criteria were studies lacking mechanistic detail, non-English publications, or conference abstracts without peer-reviewed follow-up.
Primary research articles, reviews, and preprints were included, with emphasis on mechanistic studies that directly interrogated ISR or SASP function in stem cells, development, or tissue repair. For ISR-related findings, we extracted details on eIF2α kinases (PERK, GCN2, PKR, HRI), downstream transcriptional programs, and functional consequences for stem cell fate. For SASP-related findings, we categorized secretome composition, temporal dynamics, and paracrine effects across developmental, regenerative, and pathological contexts. When available, we noted whether outcomes were acute/reversible or chronic/irreversible, to align with the proposed Yin–Yang model of ISR–SASP crosstalk.
The integration strategy was thematic, with findings organized into three domains: (1) ISR and stem cell biology, (2) SASP and development/regeneration, and (3) ISR–SASP crosstalk. Within each domain, evidence was synthesized into conceptual frameworks that highlight shared mechanisms, unique contexts, and therapeutic implications. All extracted studies were cataloged in an internal reference database, and key mechanistic examples are summarized in Table 1 to enhance transparency.
We also considered high-throughput multi-omics datasets, including transcriptomic, proteomic, and secretomic analyses, that provided system-level insights into ISR–SASP interactions. In cases where ISR and SASP were studied independently, we analyzed overlapping pathways, such as regulation of translation, NF-κB signaling, and cytokine production, to infer points of convergence. Therapeutic studies targeting ISR or SASP nodes were abstracted separately and synthesized into Table 2, with categorization by intervention point, disease context, and reported outcomes.
To ensure rigor, conflicting findings were examined considering experimental models, cell types, and stressors. For example, discrepancies in ISR’s effect on stem cell differentiation often reflected variations in nutrient stress versus ER stress. Similarly, SASP’s regenerative versus deleterious effects were parsed by distinguishing transient versus chronic senescence models. Where disagreements remained unresolved, we explicitly note these as knowledge gaps in the Discussion.
This methodology allows for a balanced synthesis of mechanistic insights, developmental roles, and translational opportunities, while acknowledging limitations such as model-specific effects, incomplete temporal mapping of ISR–SASP interactions, and a lack of longitudinal studies in human systems. Future reviews may benefit from meta-analytical approaches once sufficient quantitative datasets accumulate across ISR and SASP research.
Discussion
3
The Yin–Yang Model of ISR–SASP Crosstalk
3.1
We propose a “Yin–Yang” model to conceptualize how the integrated stress response (ISR) and the senescence-associated secretory phenotype (SASP) jointly regulate stem cell fate decisions (Fig. 2; Table 1). In this framework, protective (“Yin”) and maladaptive (“Yang”) outcomes are not fixed categories but dynamic states along a continuum. Three criteria define this balance. First, temporal dynamics: acute and transient activation of ISR or SASP can be adaptive, allowing cells to restore proteostasis, repair damage, or signal for regeneration, whereas chronic and sustained activation often tips toward dysfunction, senescence, or degeneration [32]. Second, intensity of signaling: mild or moderate stress can bias stem cells toward survival, quiescence, or lineage choice, while overwhelming stress activates irreversible checkpoints such as apoptosis or senescence [32,33]. Third, functional outcomes: ISR–SASP interactions produce context-dependent effects, ranging from tissue renewal and developmental patterning [6] to chronic inflammation, stem cell exhaustion, or oncogenic transformation [34,35].
Importantly, the Yin–Yang model emphasizes a continuum rather than a binary switch, where protective and pathological outcomes arise from the same signaling modules depending on timing, strength, and cellular context. For example, ISR-mediated translational control can either preserve hematopoietic stem cell quiescence under nutrient stress or, if prolonged, drive maladaptive senescence programs [36,37]. Similarly, the SASP can promote tissue repair in acute settings but, when chronic, foster inflammatory microenvironments that impair stem cell function and promote cancer [6,31]. Recognizing this spectrum is essential for interpreting ISR–SASP crosstalk in development, aging, and disease, and for designing interventions that tilt the balance toward adaptive states [31].
ISR as a Guardian of Stem Cell Integrity
3.2
The integrated stress response (ISR) acts as a central quality-control mechanism for stem cells, balancing adaptation with the preservation of regenerative potential (Fig. 1a and Table 1) [4,15]. At the molecular level, ISR signaling converges on eukaryotic initiation factor 2 alpha (eIF2α) phosphorylation, which transiently suppresses global protein synthesis while permitting selective translation of stress-adaptive transcripts, particularly those governed by ATF4 [5,38]. In hematopoietic stem cells (HSCs), this pathway preserves quiescence during nutrient limitation and oxidative stress, ensuring a reserve of stem cells is maintained for future regenerative needs [4,14,39].
Similarly, epidermal stem cells rely on ISR activity during amino acid deprivation, biasing differentiation programs toward epidermal fates while restraining hair follicle entry [15,16]. In neural stem cells, ISR signaling mitigates ER stress and maintains proteostasis, safeguarding against premature differentiation [17,18]. By dynamically adjusting protein synthesis, metabolism, and survival, the ISR provides a stress-buffering shield: cells capable of adapting resume renewal and tissue contribution, while cells with insurmountable damage undergo apoptosis or senescence [4,40]. This selective mechanism maintains the long-term integrity of stem cell compartments, ensuring tissue resilience throughout life.
SASP as a Double-Edged Regulator of Stem Cell Fate
3.3
The senescence-associated secretory phenotype (SASP) represents the paracrine counterpart to the cell-intrinsic ISR, together forming a Yin–Yang system of stem cell regulation (Fig. 1c) [12]. Whereas ISR primarily dictates the survival-versus-death balance within stressed stem cells [4], the SASP reshapes the microenvironment, influencing both neighboring stem cells and tissue repair dynamics [12].
SASP initiation begins with intrinsic or extrinsic genotoxic stress that activates canonical DNA damage sensors, leading to stabilization of p53 and p21-mediated cell cycle arrest [41,42]. Early SASP signaling is often transient and context-dependent, functioning as an acute “alarm” to mobilize immune clearance and stimulate regenerative pathways (Table 1) [43,44]. For example, short-lived SASP activity in epithelial stem cell niches facilitates wound healing and tissue remodeling, echoing a yin state of controlled stress adaptation [45].
Over time, unresolved damage drives a chronic SASP, orchestrated largely through NF-κB and C/EBPβ transcriptional programs [46,47]. This prolonged pro-inflammatory secretome shifts the balance toward a yang state, fostering fibrosis, chronic inflammation, and tumor promotion (Fig. 1b,c) [48]. In hematopoietic stem cells, chronic SASP signaling contributes to bone marrow failure and the expansion of pre-leukemic clones [49]; in neural stem cells, it exacerbates neurodegenerative processes by impairing neurogenesis and promoting inflammatory gliosis [50,51]; in epithelial compartments, it reinforces malignant transformation through persistent cytokine and growth factor release [3,31].
Recent evidence highlights mechanistic crosstalk between ISR and SASP. For instance, ATF4-driven metabolic rewiring in stressed stem cells modulates SASP factor production, while chronic SASP cytokines (e.g., IL-6, IL-8) can feed back to activate ISR via PERK and PKR signaling in recipient cells [4,6,52]. This bidirectional loop links intrinsic stress-buffering with extrinsic niche remodeling, tightly integrating cellular and tissue-level outcomes [4,6,52]. Importantly, this interaction occurs along a continuum: acute SASP provides transient regenerative cues, whereas chronic SASP drives degeneration, with mixed or partial SASP states occupying the intermediate spectrum [6,41,53].
Thus, the SASP operates as a double-edged regulator of stem cell fate—capable of mobilizing repair when tightly controlled, but also fueling pathology when persistent. Explicitly positioning the SASP within the Yin–Yang framework clarifies how temporal dynamics, stress intensity, and tissue context determine whether SASP signaling promotes resilience or degeneration.
Wnt Signaling as a Central Axis of the Differentiation-Associated Stress Response
3.4
The Wnt signaling network functions as a developmental axis that couples environmental stress with lineage specification and tissue repair (Table 1) [22,23]. Canonical Wnt/β-catenin signaling broadly maintains stemness and self-renewal in multiple stem cell populations [54], including embryonic stem cells [55] and adult intestinal crypt progenitors [54,56]. Under homeostatic conditions, canonical Wnt activity stabilizes β-catenin, enabling the transcription of renewal-associated targets that preserve tissue growth and regenerative capacity [57].
Stress, however, destabilizes this equilibrium and can bias Wnt signaling toward non-canonical branches. Non-canonical Wnt pathways, including planar cell polarity and calcium-dependent cascades, are activated under redox, metabolic, or ischemic stress [58]. This switch functions as a developmental checkpoint within the development associated stress response (DASR): it restrains indefinite self-renewal and promotes context-dependent differentiation or tissue remodeling. For example, ischemic injury in muscle and heart progenitors activates Wnt5a-mediated non-canonical signaling, which promotes repair but also depletes the long-term stem cell reservoir [59].
In embryogenesis, the canonical-to-non-canonical Wnt transition safeguards patterning fidelity under fluctuating nutrient and oxygen conditions [60,61]. By constraining unbalanced renewal, Wnt-mediated DASR ensures that differentiation trajectories proceed despite environmental instability. In adult tissues, however, maladaptive persistence of non-canonical Wnt activity has been linked to pathology: chronic Wnt5a-mediated non-canonical signaling contributes to fibrotic remodeling, impaired stem cell pool maintenance, and even tumor progression [62,63].
Thus, Wnt signaling is not a static determinant of stemness, but rather a stress-responsive rheostat within the DASR framework [64,65]. By toggling between canonical and non-canonical outputs, it integrates environmental inputs, developmental timing, and repair demands—determining whether stem cells preserve self-renewal, commit to differentiation, or exhaust their regenerative capacity [64,65].
The Wnt pathway indeed occupies a central position in our proposed Yin–Yang framework for integrating the integrated stress response (ISR) and the developmental adaptive stress response (DASR). However, Wnt signaling is not acting in isolation; rather, it operates within a network of feedback regulators and transcriptional hubs that modulate cell fate under stress. Among these, FoxO1 represents a particularly compelling candidate as an additional nodal regulator [66,67]. FoxO1 not only mediates stress-induced transcriptional programs downstream of AKT and AMPK but also interfaces directly with Wnt/β-catenin signaling [66,67]. Under stress, FoxO1 can sequester β-catenin away from TCF/LEF transcriptional complexes, shifting the balance from proliferation and differentiation (DASR-like) toward cytoprotective and quiescent states (ISR-like) [66,67]. Thus, FoxO1 may serve as a molecular switch that dynamically tunes Wnt output depending on energetic and redox context [66,67]. This crosstalk supports the notion that the Yin–Yang relationship between ISR and DASR is governed by a small number of multifunctional signaling hubs, of which Wnt and FoxO1 are central exemplars.
Developmental Stress as Yin: Patterning and Wound Healing
3.5
The “yin” (protective) side of the developmental stress response reflects its constructive role in patterning, morphogenesis, and wound repair (Fig. 2). During embryogenesis, controlled stress signaling sculpts lineage allocation and tissue organization [60,61]. For example, hypoxia-inducible factors (HIFs) activated by physiologic oxygen gradients orchestrate vascular and neural development, guiding pattern formation through spatially restricted stress cues [17,18]. Similarly, oxidative stress at low levels serves as a morphogen-like signal, regulating cardiac and skeletal muscle development [68,69].
Beyond embryogenesis, these stress-responsive pathways are redeployed in adult wound healing. Acute stress responses at injury sites activate ISR, Wnt, and TGF-β networks, which together coordinate cell migration, fibroblast activation, angiogenesis, and tissue closure [70]. In this context, stress is neither purely damaging nor protective but operates as a developmental toolkit that can be re-engaged in regenerative settings [70]. The yin perspective thus emphasizes stress as an instructive, organizing force that promotes adaptation and renewal [60,61].
Pathological Stress as Yang: Fibrosis, Inflammation, and Tumors
3.6
In contrast, the “yang” (maladaptive) dimension of developmental stress emerges when adaptive programs become chronic or dysregulated, fueling pathology (Fig. 2) [29-31]. Prolonged activation of ISR and Wnt pathways shifts their roles from protective to maladaptive, driving fibrotic scarring, chronic inflammation, and tumorigenesis [29-31]. In fibrotic disease, persistent TGF-β and stress-induced Wnt activation lead to sustained fibroblast activation and extracellular matrix deposition, replacing regenerative repair with rigid scarring [71]. Inflammation represents another maladaptive outcome, where chronic ISR and NF-κB activation perpetuate cytokine storms, exhausting tissue stem cell pools and disrupting regenerative balance [72].
Tumor biology can be seen as an extreme form of stress maladaptation: cancer stem cells exploit ISR to resist nutrient and oxidative stress, while hijacking Wnt and other developmental pathways to sustain unchecked proliferation [29-31]. The Yin–Yang model thus underscores the duality of stress responses—protective and patterning in acute, controlled contexts, but pathological and destructive when stress is excessive or unresolved. Understanding this continuum provides a framework for therapies that re-bias stress signaling toward adaptation and repair while preventing pathological drift.
Molecular Crosstalk between ISR and SASP
3.7
A growing body of evidence indicates that the integrated stress response (ISR) and the senescence-associated secretory phenotype (SASP) are linked not only conceptually but also through direct molecular cross-regulation [4,11]. At the core of the ISR is the phosphorylation of eIF2α by stress-sensing kinases (PERK, PKR, HRI, and GCN2), which reduces global protein synthesis while selectively enhancing translation of activating transcription factor 4 (ATF4) [2]. ATF4, in turn, activates transcriptional programs that overlap substantially with SASP regulators [2]. For instance, ATF4 can upregulate IL-6 and IL-8, two canonical SASP cytokines, through cooperation with NF-κB and AP-1 [3,41]. Conversely, chronic ISR signaling promotes expression of CHOP, which not only drives apoptosis but also contributes to the pro-inflammatory arm of SASP [2].
Reciprocally, SASP factors themselves can reinforce ISR signaling in recipient cells. Cytokines such as IFN-γ and TNF-α activate PKR, leading to renewed eIF2α phosphorylation [73,74], while amino acid–depleting enzymes secreted during senescence (e.g., indoleamine 2,3-dioxygenase) activate GCN2 [75,76], thereby amplifying ISR activity. This creates a feed-forward loop in which senescent cells impose stress on neighboring stem or progenitor cells, nudging them toward maladaptive fates [77,78]. In tissue contexts, this ISR–SASP crosstalk contributes to both regenerative responses—such as transient activation of ISR to facilitate wound healing [4,6]—and degenerative outcomes, including fibrosis and stem cell exhaustion under chronic stress [4,6].
Thus, the mechanistic interface between ISR and SASP can be conceptualized as a dynamic circuit: ATF4-driven ISR promotes SASP-like transcriptional outputs, while SASP-derived cytokines and metabolites reactivate ISR in surrounding cells [4,6]. Identifying the molecular nodes of this circuit, such as ATF4–NF-κB convergence or PKR-mediated ISR activation by SASP cytokines, will be critical for therapeutic targeting [6,79].
Therapeutic Modulation of ISR–SASP Crosstalk
3.8
The intersection between the integrated stress response (ISR) and the senescence-associated secretory phenotype (SASP) provides a promising but complex therapeutic axis [11,73]. Both pathways serve as adaptive mechanisms: the ISR preserves stem cell integrity by buffering proteotoxic and metabolic stress, whereas the SASP coordinates tissue-level responses by releasing pro-inflammatory and remodeling factors [11,73]. However, when chronically engaged, their crosstalk can amplify dysfunction—driving bone marrow failure, neurodegeneration, and tumorigenesis (Fig. 3; Table 2) [11,73]. Thus, interventions must be designed to selectively restore beneficial signaling while suppressing maladaptive persistence.
One approach involves pharmacologic tuning of the ISR. Small-molecule inhibitors of eIF2α kinases (e.g., ISRIB analogs) can restore global translation and attenuate excessive ATF4-driven pro-apoptotic programs [4,38], thereby protecting hematopoietic and neural stem cell pools under chronic stress [4]. Conversely, transient ISR activation may be desirable in cancer, where sustained protein synthesis in malignant stem cells supports growth [80,81]. Here, PERK activators or modulators of amino acid-sensing pathways (GCN2 agonists) could shift damaged cells toward apoptosis rather than repair [2,75].
A complementary strategy is direct SASP modulation. Senolytics (e.g., dasatinib, navitoclax) eliminate senescent cells, reducing the inflammatory burden that exacerbates stem cell exhaustion [82-84]. Senomorphics, such as JAK inhibitors or mTOR modulators, instead dampen SASP factor production without killing senescent cells, preserving their initial tumor-suppressive arrest [83,85]. Notably, ISR–SASP crosstalk creates opportunities for synergy: ISR attenuation may reduce the persistence of DNA damage signaling that fuels SASP [4], while SASP suppression alleviates the pro-inflammatory milieu that chronically re-engages ISR pathways [6,86].
Context-specific modulation is critical. In regenerative medicine, transient ISR enhancement coupled with senomorphic therapy may preserve stem cell quiescence and prevent fibrosis during tissue repair [87]. In cancer therapy, the inverse may apply: ISR activators can drive tumor stem cells toward death, while senolytics remove SASP-amplified niches that support relapse [88]. Similarly, in neurodegenerative disease, balancing ISR inhibitors with SASP dampening could mitigate proteostasis collapse and chronic inflammation [89].
Ultimately, the therapeutic value of targeting ISR–SASP crosstalk lies in its dual-level control: intracellular stress adaptation and extracellular niche regulation. Precision in timing, dosage, and tissue context will determine whether interventions reinforce resilience or inadvertently accelerate decline.
Conclusion
4
The ISR and SASP are deeply integrated pathways that together orchestrate stem cell responses across development, regeneration, aging, and disease. The ISR acts as a master regulator of translation and survival, balancing adaptation and apoptosis to safeguard stem cell pools [1,2]. The SASP, long viewed as a pathological driver of aging, is now recognized as a dynamic program with critical roles in embryogenesis and wound healing [3]. Their convergence is most evident in senescent cells, where ISR signaling modulates SASP composition, amplifying inflammatory outputs with far-reaching consequences for tissue microenvironments and stem cell function [26,27].
Therapeutically, the ISR–SASP axis offers an attractive target for modulating stem cell fate in regenerative medicine, mitigating age-related decline, and controlling cancer progression [6,97,98]. Yet, interventions must be carefully tuned to distinguish between transient, beneficial roles and chronic, deleterious effects [6,97,98]. This review highlights the need for integrated frameworks that connect developmental biology with aging research, emphasizing that the ISR–SASP interplay represents both a biological constraint and an opportunity for therapeutic innovation.
Future Studies and Caveats
5
Future studies must dissect the temporal dynamics of ISR–SASP interactions at single-cell and spatial resolution, particularly within stem cell niches across development, adulthood, and aging [99]. Multi-omics approaches integrating translatomics, secretomics, and epigenomics will be crucial to map how ISR rewires SASP outputs [100]. Caveats include model-specific limitations: murine studies may not fully recapitulate human senescence programs, and in vitro stem cell assays often lack the complexity of in vivo microenvironments. Furthermore, interventions that blunt SASP or ISR could inadvertently disrupt essential developmental or regenerative functions. Addressing these challenges will be key to translating ISR–SASP biology into safe and effective therapies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science. 2020;368(6489):eaat 5314. doi:10.1126/science.aat 5314.32327570 PMC 8997189 · doi ↗ · pubmed ↗
- 2Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–95. doi:10.15252/embr.201642195.27629041 PMC 5048378 · doi ↗ · pubmed ↗
- 3CoppéJP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5(1):99–118. doi:10.1146/annurev-pathol-121808-102144.20078217 PMC 4166495 · doi ↗ · pubmed ↗
- 4Kalinin A, Zubkova E, Menshikov M. Integrated stress response (ISR) pathway: unraveling its role in cellular senescence. Int J Mol Sci. 2023;24(24):17423. doi:10.3390/ijms 242417423.38139251 PMC 10743681 · doi ↗ · pubmed ↗
- 5Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr. 2012;3(3):307–21. doi:10.3945/an.112.002113.22585904 PMC 3649462 · doi ↗ · pubmed ↗
- 6Alqahtani S, Alqahtani T, Venkatesan K, Sivadasan D, Ahmed R, Sirag N, SASP modulation for cellular rejuvenation and tissue homeostasis: therapeutic strategies and molecular insights. Cells. 2025;14(8):608. doi:10.3390/cells 14080608.40277933 PMC 12025513 · doi ↗ · pubmed ↗
- 7Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155(5):1119–30. doi:10.1016/j.cell.2013.10.041.24238961 · doi ↗ · pubmed ↗
- 8Wilkinson HN, Hardman MJ. Cellular senescence in acute and chronic wound repair. Cold Spring Harb Perspect Biol. 2022;14(11):a 041221. doi:10.1101/cshperspect.a 041221.35817510 PMC 9620855 · doi ↗ · pubmed ↗