Comparative analysis of the diversity within the B. bronchiseptica fimX locus
Tracy L. Nicholson, Sarah M. Shore

TL;DR
This study explores genetic diversity in the fimX locus of Bordetella bronchiseptica, a respiratory pathogen, revealing multiple subunit gene types and recombination as a source of variation.
Contribution
The study identifies six distinct fimbrial subunit gene types in the fimX locus and suggests homologous recombination as a driver of diversity.
Findings
Six different fimbrial subunit gene types were identified in the fimX locus of B. bronchiseptica.
Genomic variability in the fimX locus includes differences in gene number and subunit types.
Homologous recombination is likely responsible for the diversity observed in the fimX locus.
Abstract
Bordetella bronchiseptica is a highly contagious veterinary bacterial respiratory pathogen with a broad host range that can cause a variety of clinical disease outcomes ranging from asymptomatic carriage to severe pneumonia. B. bronchiseptica fimbriae mediate attachment to respiratory epithelium and are considered to serve as potential protective antigens. Several comparative genomic studies involving B. bronchiseptica strains have demonstrated an overall low level of genomic variability. The region located within the fimX locus is one of the few regions in which genomic variability has been reported. The goal of this study was to comprehensively evaluate the genomic variability harbored within the fimX locus among B. bronchiseptica strains. Our analysis revealed that the genetic variability identified within the fimX locus included both the number of genes harbored, as well as the type…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
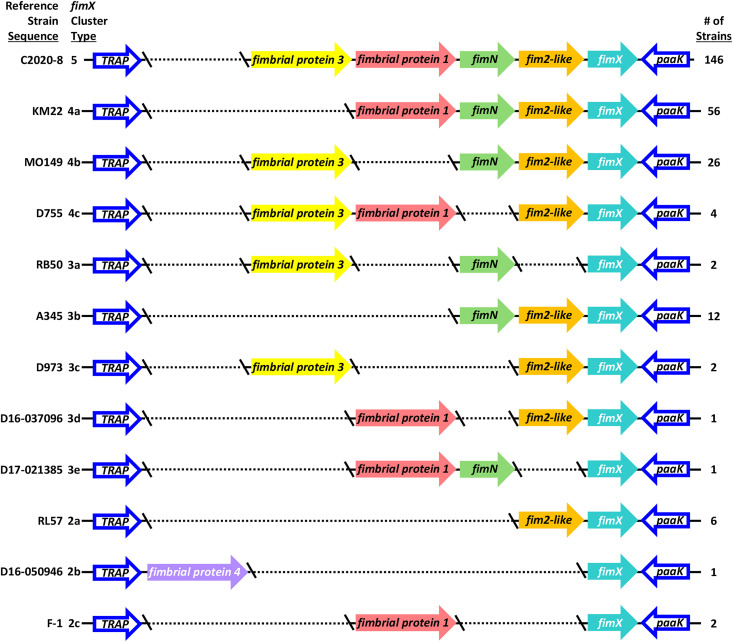 Fig 1
Fig 1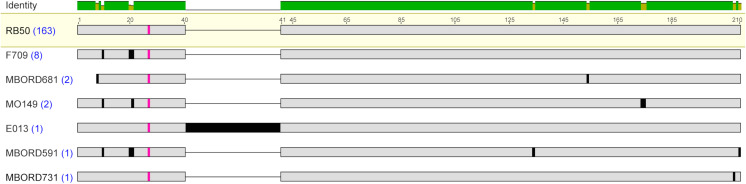 Fig 2
Fig 2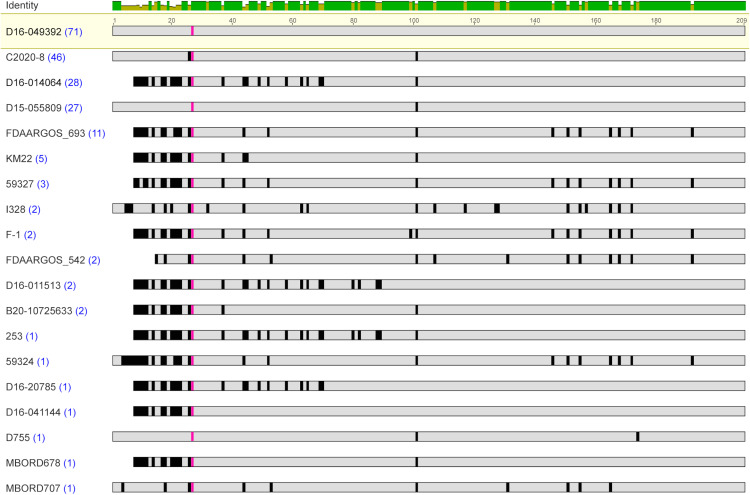 Fig 3
Fig 3 Fig 4
Fig 4 Fig 5
Fig 5 Fig 6
Fig 6 Fig 7
Fig 7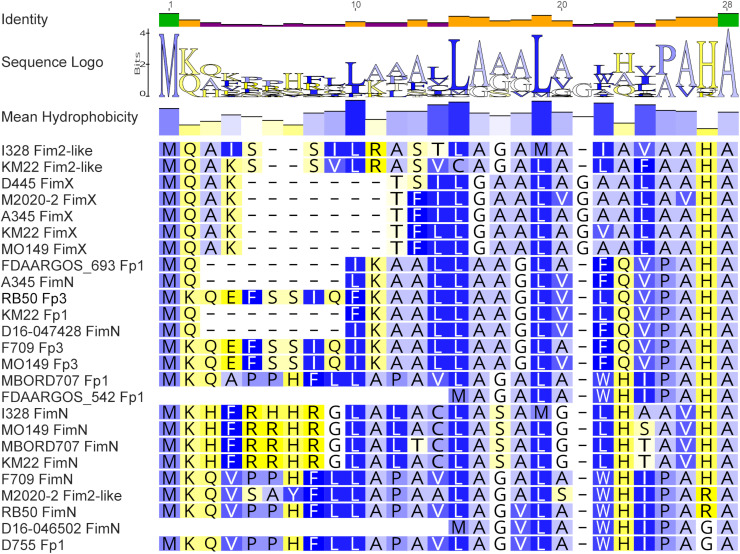 Fig 8
Fig 8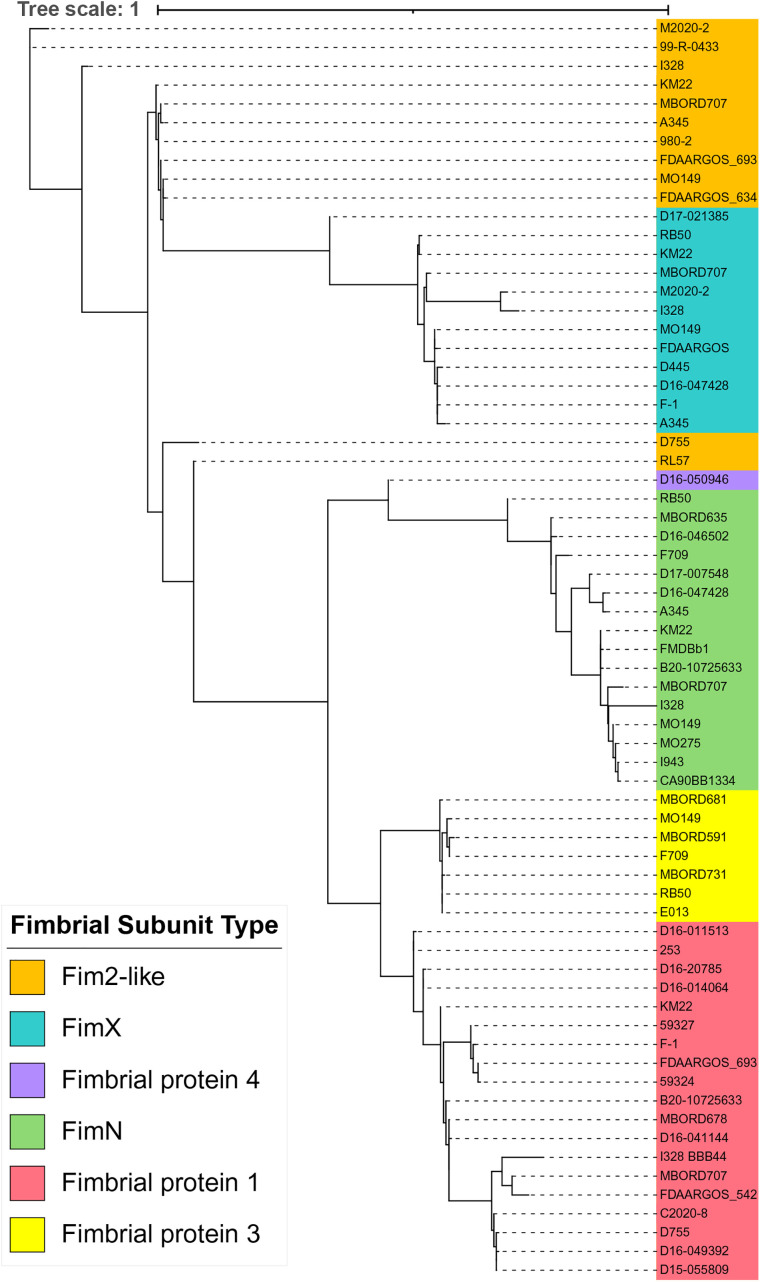 Fig 9
Fig 9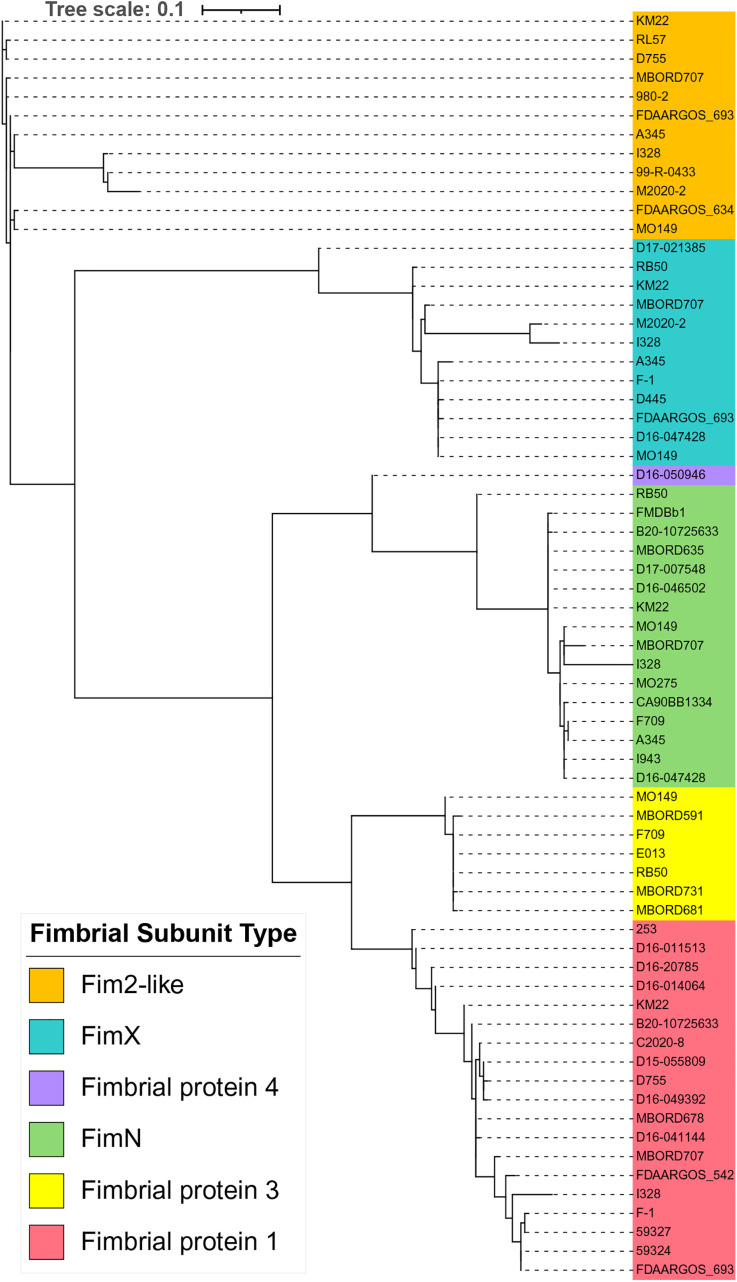 Fig 10
Fig 10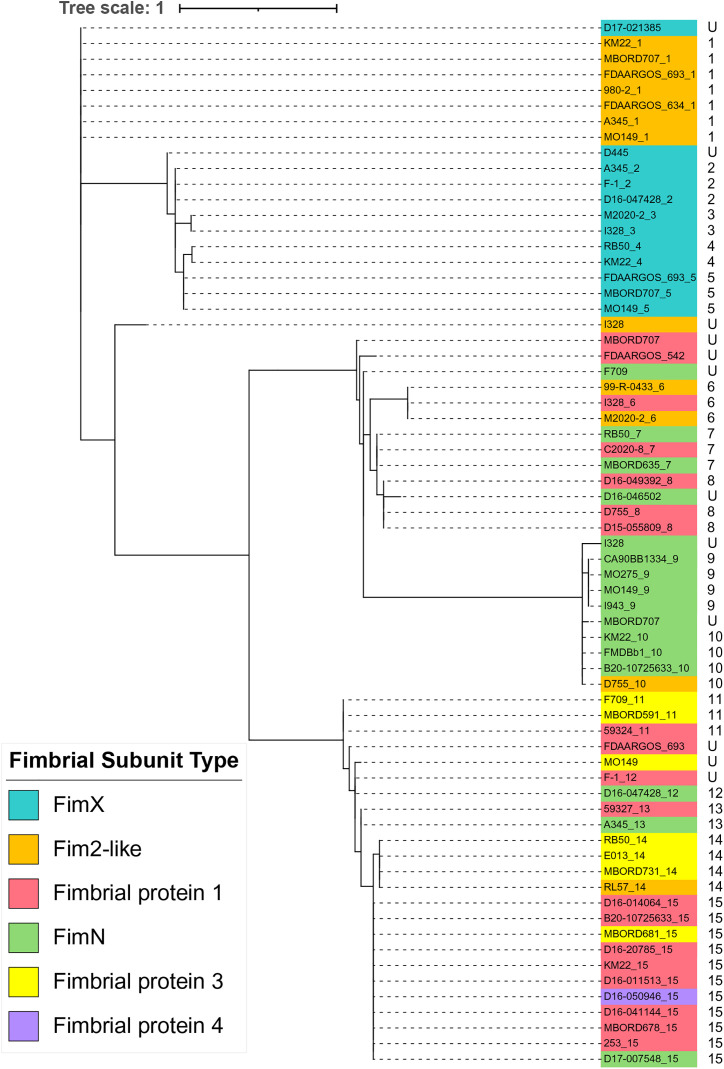 Fig 11
Fig 11- —http://dx.doi.org/10.13039/100007917Agricultural Research Service
- —http://dx.doi.org/10.13039/100007917Agricultural Research Service
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Infections and Vaccines · Pneumonia and Respiratory Infections · Herpesvirus Infections and Treatments
Introduction
Bordetella bronchiseptica is a highly contagious respiratory bacterial veterinary pathogen. It can serve as the primary cause or as a contributor to a spectrum of clinical disease outcomes ranging from asymptomatic carriage to severe bronchopneumonia [1–3]. B. bronchiseptica is closely related to Bordetella pertussis and Bordetella parapertussishu, the bacterial pathogens responsible for causing whooping cough in humans [4–7]. The human-restricted pathogens, B. pertussis and B. parapertussishu, are regarded to have evolved independently from a B. bronchiseptica common ancestor [5–7]. In addition to their close genetic relatedness, B. bronchiseptica, B. pertussis, and B. parapertussishu, commonly referred to as the ‘classical bordetellae’, all harbor many of the same virulence factors that are similarly regulated [5,8–10]. Despite these similarities, these Bordetella species differ in traits such as host specificity, disease severity, and duration of infection. The host-specific B. pertussis infects humans, has no animal reservoir, and lacks the ability to survive in the environment. In contrast, B. bronchiseptica, infects a variety of animals, often establishing chronic infections that range from lethal pneumonia to asymptomatic carriage and is capable of surviving in the environment [2,4,11,12]. Although rare and predominately occurring in immunocompromised individuals, human infections of B. bronchiseptica have been reported [13–16].
Fimbriae are some of the virulence factors produced by classical bordetellae [1,2,5,8,9]. Vaccines and therapies targeting the fimbrial antigens have been successful in both human and veterinary applications [17–23]. Fimbriae produced by Bordetella are considered members of the type I pili family and are assembled and exported by the chaperone–usher pathway, which has been extensively investigated in uropathogenic Escherichia coli (UPEC) [24–30]. Studies have demonstrated that B. bronchiseptica genomes contain several fimbrial subunit genes, most of which are unlinked and located throughout the chromosome [5,8,9,31–33]. While these major fimbrial subunit genes are unlinked on the chromosome, assembly and export of the fimbrial subunit products are mediated by the products of three genes, fimB, fimC, and fimD [30,34,35]. FimB shares similarity with the UPED chaperone protein, PapD, thought to prevent degradation of the fimbrial subunit proteins in the periplasmic space. FimC shares similarity with the UPEC usher protein, PapC, considered to aid in the transport of fimbrial subunit proteins across the outer membrane and secure the fimbrial structure [34]. FimD is considered to function as the adhesive tip of the fimbrial structure [36]. Current knowledge regarding the involvement of B. bronchiseptica fimbriae in adherence to the respiratory epithelium, as well as the involvement of B. bronchiseptica fimbriae as protective antigens, is derived from studies employing a B. bronchiseptica strain that is incapable of producing any fimbrial serotypes due to an in-frame deletion of the fimBCD genes [35,54]. These studies additionally demonstrate that the fimBCD genes function as the sole chaperone-usher system required for the assembly and export of B. bronchiseptica fimbrial subunit products [35,54].
Several comparative genomic studies involving B. bronchiseptica strains have demonstrated an overall low level of limited genomic variability [5,8,9,31–33]. One of the few regions in which genomic variability has been reported is the region located within the fimX locus [5,8,9,31–33]. Given a downstream potential use of fimbrial subunits as vaccine targets, and the high genetic diversity observed, the goal of this report was to comprehensively evaluate the genetic variation and phylogenic relationship of the fimX locus among B. bronchiseptica strains.
Materials and methods
Bacterial strains
B. bronchiseptica (taxon ID = 518) Reference Sequence (RefSeq) genome assemblies were obtained from the National Center for Biotechnology Information (NCBI) RefSeq database (https://www.ncbi.nlm.nih.gov/genbank/, accessed in October 2024). Genome assemblies for isolates MBORD624 and 00-P-2796 were subsequently excluded from our dataset due to poor quality. Genome assemblies from B. bronchiseptica multi-isolate studies were additionally included in the dataset [31,32]. Multi-locus sequence typing (MLST) was performed in silico utilizing the BIGSdb-Pasteur databases hosted by the Institut Pasteur (https://bigsdb.pasteur.fr). In total, 259 B. bronchiseptica genome assemblies were included in the dataset. All strains and strain-associated metadata are provided in S1 Table.
Analysis and categorization of fimX loci into cluster types
Genes flanking the fimX locus, tripartite ATP-independent periplasmic transporter (TRAP) and phenylacetate-CoA ligase (paaK), were used to locate and extract the fimX locus nucleotide sequence from all NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (versions 6.5–6.8) [37] annotated assemblies. When the fimX locus was not located within a single contiguous region, the partial regions were identified by proximity to one of the flanking genes and the presence of one or more fimbrial subunit genes. Nucleotide sequences containing these partial regions were examined to ensure that they did not contain the fimbrial subunit genes fim2, fim3, fimA, or fimbrial protein 2 (homolog to KM22 CJ015_08855), which are located in other chromosomal locations. The fimX loci were categorized into cluster types based on [1] the number of fimbrial subunit genes located within the fimX locus and [2] the fimbrial subunit gene types contained within the fimX locus. A reference strain sequence was chosen to represent each fimX cluster type with preference given to strains with closed whole-genome assemblies. For example, strain C2020-8 was chosen as reference strain for cluster type 5. When no closed whole-genome assemblies were available, preference was given to strains D16-037096 (cluster type 3d), D17-021385 (cluster type 3e), D16-050946 (cluster type 2b), and F-1 (cluster type 2c) because they are maintained in our laboratory isolate collection, which allowed for confirmatory Sanger sequencing and further evaluation. All twelve reference strains and their corresponding cluster types are provided in Fig 1 and S1 Table.
Genomic organization of fimX locus among B. bronchiseptica genome assemblies.Reference strain sequence and cluster type provided at left. Predicted fimbrial subunit genes are represented as arrows and color-coded by fimbrial subunit type with gene names, defined by nucleotide sequence identity. Dashes indicate break in the sequence.
Sequencing and analysis of fimX locus
The fimX locus region of strains D16-037096 (Cluster Type 3d), D17-021385 (Cluster Type 3e), D16-050946 (Cluster Type 2b), and F-1 (Cluster Type 2c) was amplified by PCR, sequenced, and further analyzed. Isolate F-1 was obtained from the USDA-ARS Culture Collection (NRRL) and isolates D16-037096, D17-021385, and D16-050946 have been previously described [31]. Strains were grown from frozen stocks (−80°C in 30% glycerol) on Bordet-Gengou agar (Difco, Sparks, MD) supplemented with 10% sheep’s blood and 40 µg/ml Streptomycin.
A single colony was inoculated into Stainer-Scholte broth containing 40 µg/ml Streptomycin and grown overnight at 37°C with shaking. Genomic DNA was isolated using the High Pure PCR Template Preparation Kit (Roche Diagnostics, Mannheim, Germany). The entire fimX locus was PCR amplified using primers TRAP_for and paaK_rev in a PCR reaction with Platinum SuperFi II PCR Master Mix (Invitrogen, Carlsbad, CA) as specified by the manufacturer. PCR products were purified using the Monarch Spin DNA Gel Extraction Kit (New England Biolabs, Ipswich, MA). Sanger sequencing was then performed on purified PCR products with appropriate primers (Table 1). Isolates evaluated were obtained from samples collected as part of previous studies and did not require Institutional Animal Care and Use Committee (IACUC) approval.
Table 1: Primers used in study.
Analysis and categorization of fimbrial subunit types
Nucleotide sequences encoding predicted fimbrial subunit genes were extracted from all fimX locus nucleotide sequences and identical sequences were identified and grouped into fimbrial subunit gene types using seqkit v.2.4.0 [38]. For each fimbrial subunit gene type consisting of identical nucleotide sequences, a single reference sequence was chosen to represent each fimbrial subunit gene type (S2 Table). Sequence alignment with MAFFT v.7.490 [39] was then used to classify the representative sequences by similarity into related gene types. This analysis resulted in the identification of six fimbrial subunit gene types: fimbrial protein 4, fimbrial protein 3, fimbrial protein 1, fimN, fim2-like, and fimX. Any fimbrial subunit gene sequence that could not be translated into a complete protein sequence, due to a truncation, frameshift, or incomplete sequence, was labeled a pseudogene and excluded from further analysis. In total, 1,145 predicted fimbrial subunit genes were identified, of which 5 were designated as pseudogenes, leaving 1,140 genes used for further analysis. The reference gene sequence from each fimbrial subunit gene type was translated and used to identify similar protein sequences from the unique protein sequences to group all protein sequences into a fimbrial subunit protein type. Eighty-one unique nucleotide sequences encoding predicted fimbrial subunit genes were identified from this analysis, which resulted in sixty-seven unique protein sequences after translation. The recombination detection program 5 (RDP5) was used for detection of recombination events within the 81 unique fimbrial subunit gene sequences using default settings [40]. Only the events that passed two out of seven implemented methods in RDP5 with a significance level (p) < 0.05 were considered true recombination events.
Phylogenetic analysis
Sequence alignments of both nucleotide and amino acid sequences were performed using MAFFT v.7.490 [39]. Maximum-likelihood phylogenetic trees were inferred using IQ-TREE v.2.4.0 using the automatic model selection process provided by ModelFinder Plus [41,42] and visualized with iTOL v.7.2 [43].
Results
fimX locus cluster designations
The fimX locus was located in the same genomic position flanked by tripartite ATP-independent periplasmic transporter (TRAP) and phenylacetate-CoA ligase (paaK) genes in all B. bronchiseptica genome assemblies analyzed. The diversity within the locus included both the number of genes harbored, as well as the type of predicted fimbrial genes present within the fimX locus. These two criteria were used to categorize the locus sequences from all B. bronchiseptica genome assemblies into twelve distinct fimX locus cluster types. A reference strain sequence was chosen to represent each fimX cluster type, which were named cluster type 5 through cluster type 2c (Fig 1 and S1 Table). Poly-cytosine or poly(C) tracts were identified upstream of the annotated start codon for all the predicted fimbrial genes harbored within the fimX locus for all the B. bronchiseptica genome assemblies evaluated. The metadata associated with all the B. bronchiseptica genome assemblies utilized, such as lineage, ST, and host, was evaluated for a correlation between a fimX locus cluster type and these attributes. However, no correlation was found (S1Table).
One hundred and forty-six B. bronchiseptica genome assemblies were observed to harbor five predicted fimbrial subunit gene types within the fimX locus. The five predicted fimbrial subunit gene types harbored by these isolates were fimbrial protein 3, fimbrial protein 1, fimN, fim2-like, and fimX (Fig 1 and S1 Table). Fimbrial protein 3 is a recently identified predicted fimbrial subunit gene type [31]. Genome assemblies harboring this configuration of predicted fimbrial gene types were categorized as cluster type 5, which was the most prevalent among all the B. bronchiseptica genome assemblies analyzed.
Fifty-six B. bronchiseptica genome assemblies harbored four fimbrial subunit gene types that included fimbrial protein 1, fimN, fim2-like, and fimX. Genome assemblies that harbored this configuration of predicted fimbrial subunit gene types were classified into cluster 4a (Fig 1 and S1 Table). Twenty-six B. bronchiseptica genome assemblies harbored four fimbrial subunit gene types that included fimbrial protein 3, fimN, fim2-like, and fimX. Genome assemblies harboring this configuration of predicted fimbrial gene types were grouped into cluster 4b. Four B. bronchiseptica genome assemblies harbored four fimbrial subunit gene types that included fimbrial protein 3, fimbrial protein 1, fim2-like, and fimX. These assemblies did not contain a fimN gene and were categorized as cluster type 4c.
B. bronchiseptica genome assemblies RB50 and FDAARGOS_176 contained three fimbrial subunit gene types that included fimbrial protein 3, fimN, and fimX, and were categorized as cluster type 3a (Fig 1 and S1 Table). Twelve B. bronchiseptica genome assemblies harbored three fimbrial subunit gene types that included fimN, fim2-like, and fimX, and were classified into the cluster type 3b. B. bronchiseptica genome assemblies D973 and D17-015854 harbored three fimbrial subunit gene types that included fimbrial protein 3, fim2-like, and fimX, and were categorized into cluster type 3c. Both fimX locus cluster types 4c and 3c did not contain a fimN gene. The genome assembly for strain D16-0377096 harbored three fimbrial subunit gene types that included fimbrial protein 1, fim2-like, and fimX, and was grouped into cluster type 3d. The genome assembly for strain D17-021385 harbored three fimbrial subunit gene types that included fimbrial protein 1, fimN, and fimX, and was placed into cluster type 3e.
Six B. bronchiseptica genome assemblies harbored two fimbrial subunit gene types, fim2-like and fimX, and were classified into the fimX locus cluster type 2a (Fig 1 and S1 Table). The genome assembly for strain D16-050946 harbored two fimbrial subunit gene types, fimbrial protein 4 and fimX, and was classified into cluster type 2b. The fimbrial protein 4 gene is a recently identified predicted fimbrial subunit gene type and the assembly for strain D16-050946 was the only assembly observed to harbor fimbrial protein 4. B. bronchiseptica genome assemblies F-1 and F2 harbored two fimbrial subunit gene types, fimbrial protein 1 and fimX, and these assemblies were categorized as cluster type 2c.
Fimbrial subunit types
Focusing on each fimbrial subunit gene type, analysis of all the fimbrial protein 3 genes identified among the B. bronchiseptica genome assemblies resulted in the detection of seven unique protein sequences (S2 Table). Fimbrial protein 3 sequences were highly conserved among each other, with a pairwise amino acid identity that ranged from 83.68% to 99.52%, and divergent from the other fimbrial subunit type protein sequences (Table 2). The Fimbrial protein 3 amino acid sequence from strain E013 contained a 35 amino acid internal insertion that was not present in any other Fimbrial protein 3 sequences. Aside from the 35 amino acid insertion, the Fimbrial protein 3 amino acid sequences from strains E013 and RB50 were identical (Fig 2). A conserved Ala residue was observed in all Fimbrial protein 3 amino acid sequences located at residue 20 for MBORD681 and at residue 27 for the other Fimbrial protein 3 amino acid sequences (Fig 2 and Table 5). The majority of the amino acid variations observed among all the Fimbrial protein 3 sequences were located within the N-terminal region, encompassing the start codon to the conserved Ala residue (Fig 2). When the N-terminal region was removed, so that the protein sequence used for comparisons began at the conserved Ala residue and extended to the C-terminal end residue, the upper range of the pairwise amino acid identity increased to 100% (Table 3).
Table 2: Pairwise amino acid identity (%) from the full-length protein sequences of fimbrial subunit types.
Table 3: Pairwise amino acid identity (%) from the protein sequences of fimbrial subunit types without the N-terminal region.
Table 5: N-terminal amino acid sequence groups.
Amino acid sequence alignment of Fimbrial protein 3 sequences.MAFFT alignment of the unique representative Fimbrial protein 3 protein sequences. B. bronchiseptica strain shown at left with blue numbers in parentheses indicating the number of genome assemblies found to encode the amino acid sequence. The most prevalent protein sequence was used as the reference in the alignment (top); amino acid residues that differ from the reference are highlighted by black bars and grey indicates amino acid residues that are identical to the reference sequence. The conserved A residue is highlighted in pink. The bar at the top of the alignment represents the mean pairwise identity over all pairs in the column, with green indicating 100% identity and yellow indicating at least 30% and less than 100% identity.
Examination of the proteins encoded by the fimbrial protein 1 genes identified among the B. bronchiseptica genome assemblies resulted in the identification of nineteen unique protein sequences (S2 Table). The Fimbrial protein 1 amino acid sequences were highly conserved among each other, with a pairwise amino acid percent identity that ranged from 81.68% to 99.52%, and divergent from the other fimbrial subunit type protein sequences (Table 2). All the unique Fimbrial protein 1 amino acid sequences were from strains belonging to B. bronchiseptica lineage I, except for strain I328, which is a B. bronchiseptica lineage II strain (S1 Table). The lowest pairwise amino acid percent identity was observed for the Fimbrial protein 1 sequence from strain I328 (S3 Table). This is consistent with previous reports demonstrating that virulence factors and vaccine antigens encoded by genes from B. bronchiseptica lineage II strains have a lower sequence similarity to factors encoded by paralogous genes from B. bronchiseptica lineage I strains [32,33]. When the Fimbrial protein 1 sequence from strain I328 was excluded, the pairwise amino acid identity range increased to 84.62% to 99.52% (Table 4).
Table 4: Pairwise amino acid identity (%) from the full-length protein sequences of fimbrial subunit types from Lineage I strains.
Similar to the Fimbrial protein 3 sequences, a conserved Ala residue was observed in all Fimbrial protein 1 sequences located at residues 13–27 (Table 5). The majority of the amino acid variations observed among all the Fimbrial protein 1 sequences were also located within the N-terminal region, encompassing the start codon up to the residue preceding the conserved Ala residue (Fig 3). When the N-terminal region was removed, so that the protein sequence used for comparisons began at the conserved Ala residue and extended to the C-terminal end residue, the amino acid identity range increased to 86.89% to 100% (Table 3).
Amino acid sequence alignment of Fimbrial protein 1 sequences.MAFFT alignment of the unique representative Fimbrial protein 1 protein sequences. B. bronchiseptica strain shown at left with blue numbers in parentheses indicating the number of genome assemblies found to encode the amino acid sequence. The most prevalent protein sequence was used as the reference in the alignment (top); amino acid residues that differ from the reference are highlighted by black bars and grey indicates amino acid residues that are identical to the reference sequence. The conserved A residue is highlighted in pink. The bar at the top of the alignment represents the mean pairwise identity over all pairs in the column, with green indicating 100% identity and yellow indicating at least 30% and less than 100% identity.
Assessment of the proteins encoded by the fimN genes identified among the B. bronchiseptica genome assemblies resulted in the detection of sixteen unique protein sequences (S2 Table 2). The FimN protein sequences were highly conserved among each other, with a pairwise amino acid identity that ranged from 77.03% to 99.52%, and divergent from the other fimbrial subunit type protein sequences (Table 2). Similar to the Fimbrial protein 1 sequence, the lowest pairwise amino acid percent identity was observed for the FimN sequence from strain I328 and when the FimN sequence from strain I328 is excluded, the amino acid identity range increased to 80.38% to 99.52% (Table 4). A low pairwise amino acid percent identity was also observed for the FimN protein sequence from strain RB50, with a pairwise amino acid percent identity that ranged from 86.89% to 90.70%. The amino acid differences in FimN from RB50 were mainly located within a 40-residue region located at the C-terminus.
Similar to the other fimbrial subunit type protein sequences, a conserved Ala residue was observed in all FimN sequences at residue 16 for FimN from strain D16-046502 and at residue 27 for the other FimN sequences (Table 5). The majority of the amino acid variations observed among all the FimN sequences were located within the N-terminal region, encompassing the start codon to the conserved Ala residue (Fig 4). When the N-terminal region was removed, so that the protein sequence used for comparisons began at the conserved Ala residue and extended to the C-terminal end residue, the amino acid identity range increased to 83.06% to 100% (Table 3).
Amino acid sequence alignment of FimN sequences.MAFFT alignment of the unique representative FimN protein sequences. B. bronchiseptica strain shown at left with blue numbers in parentheses indicating the number of genome assemblies found to encode the amino acid sequence. The most prevalent protein sequence was used as the reference in the alignment (top); amino acid residues that differ from the reference are highlighted by black bars and grey indicates amino acid residues that are identical to the reference sequence. The conserved A residue is highlighted in pink. The bar at the top of the alignment represents the mean pairwise identity over all pairs in the column, with green indicating 100% identity, yellow indicating at least 30% and less than 100% identity, and red indicating less than 30% identity.
Investigation of the fim2-like genes identified among the B. bronchiseptica genome assemblies resulted in the identification of twelve unique protein sequences (S2 Table). Fim2-like protein sequences were highly conserved among each other, with a pairwise amino acid identity that ranged from 77.89% to 99.52%, and divergent from the other fimbrial subunit type protein sequences (Table 2). The lowest pairwise amino acid percent identity was observed the Fim2-like protein sequence from strains M2020-2, I328, and 99-R-0433, which belong to Bb lineage II. When these more diverse Fim2-like protein sequences from Bb lineage II isolates were excluded, the amino acid identity range increased to 88.94% to 99.52% (Table 4).
Similar to the protein sequences from the other fimbrial subunit type protein sequences, a conserved Ala residue was observed in all Fim2-like protein sequences at residue 27 for Fim2-like from strains 99-R-0433, M2020-2, D755, and RL57 and at residue 25 for the other Fim2-like protein sequences (Fig 5 and Table 5). The majority of the amino acid variations observed among all the Fim2-like protein sequences were located within the N-terminal region, encompassing the start codon to the conserved Ala residue (Fig 5). When the N-terminal region was removed, so that the protein sequence used for comparisons began at the conserved Ala residue and extended to the C-terminal end residue, the amino acid identity range increased to 84.78% to 99.46% (Table 3).
Amino acid sequence alignment of Fim2-like sequences.MAFFT alignment of the unique representative Fim2-like protein sequences. B. bronchiseptica strain shown at left with blue numbers in parentheses indicating the number of genome assemblies found to encode the amino acid sequence. The most prevalent protein sequence was used as the reference in the alignment (top); amino acid residues that differ from the reference are highlighted by black bars and grey indicates amino acid residues that are identical to the reference sequence. The conserved A residue is highlighted in pink. The bar at the top of the alignment represents the mean pairwise identity over all pairs in the column, with green indicating 100% identity, yellow indicating at least 30% and less than 100% identity, and red indicating less than 30% identity.
Analysis of the fimX genes identified among the B. bronchiseptica genome assemblies resulted in the detection of twelve unique protein sequences (S2 Table). FimX protein sequences were highly conserved among each other, with a pairwise amino acid identity that ranged from 70.53% to 99.50%, and divergent from the other fimbrial subunit type protein sequences (Table 2). The lowest pairwise amino acid percent identity was observed for the FimX protein sequence from strains M2020-2 and I328, which belong to Bb lineage II. When these FimX protein sequences were excluded, the amino acid identity range increased to 79.71% to 99.50% (Table 4).
A conserved Ala residue was observed in all FimX protein sequences at residue 25 for FimX from strain D17-021385 and at residue 21 for the other Fim2-like protein sequences (Fig 6 and Table 5). The majority of the amino acid variations observed among all the Fim2-like protein sequences were located within the N-terminal region, encompassing the start codon to the conserved Ala residue (Fig 6). When the N-terminal region was removed, so that the protein sequence used for comparisons began at the conserved Ala residue and extended to the C-terminal end residue, the amino acid identity range increased to 74.32% to 100% (Table 3).
Amino acid sequence alignment of FimX sequences.MAFFT alignment of the unique representative FimX protein sequences. B. bronchiseptica strain shown at left with blue numbers in parentheses indicating the number of genome assemblies found to encode the amino acid sequence. The most prevalent protein sequence was used as the reference in the alignment (top); amino acid residues that differ from the reference are highlighted by black bars and grey indicates amino acid residues that are identical to the reference sequence. The conserved A residue is highlighted in pink. The conserved A residue is highlighted in pink. The bar at the top of the alignment represents the mean pairwise identity over all pairs in the column, with green indicating 100% identity and yellow indicating at least 30% and less than 100% identity.
The fimbrial protein 4 gene found in strain D16-050946 was the only fimbrial protein 4 gene found among the B. bronchiseptica genome assemblies evaluated. It was recently identified as a new predicted fimbrial subunit gene within the fimX locus due to its low nucleotide sequence identity to the other known predicted fimbrial subunit genes [31]. Pairwise comparisons of both the full-length and the trimmed portion in which the N-terminal region was removed, such that the protein sequences used for comparisons began at the conserved Ala residue and extended through the C-terminal end residue, of Fimbrial protein 4 sequence to the other fimbrial subunit type protein sequences demonstrated an overall low amino acid identity to other fimbrial subunit type protein sequences (Table 2 and Table 3). Further examination of the Fimbrial protein 4 amino acid sequence to other fimbrial subunit type protein sequences revealed that the Fimbrial protein 4 sequence shared regions with 100 percent amino acid identity with regions from three other fimbrial subunit type protein sequences. Specifically, residues 2–97 of Fimbrial protein 4 were 100% identical to 9–104 of RB50 Fimbrial protein 3, residues 90–161 of Fimbrial protein 4 were 100% identical to residues 97–168 of KM22 FimN, and residues 155–201 of Fimbrial protein 4 were 100% identical to162–208 of KM22 Fim2-like (Fig 7). Additionally, the N-terminal region encompassing, residues 1–20 of Fimbrial protein 4 were 100% identical to the N-terminal regions of the following fimbrial subunit types: MBORD681 Fimbrial protein 3, D17-007548 FimN, and Fimbrial protein 1 from strains D16-011513, D16-20785, B20-10725633, D16-041144, D16-014064, 253, MBORD678, and KM22. This data suggests that new fimbrial subunit genes are likely generated by homologous recombination between existing or ancestral fimbrial subunit genes located within the fimX locus.
Fimbrial protein 4 alignment.MAFFT alignment of Fimbrial protein 4 protein sequence with regions of RB50 Fimbrial protein 3, KM22 FimN, and KM22 Fim2-like are 100% identical. Amino acid residues 9 - 104 of RB50 Fimbrial protein 3 are 100% identical to amino acid residues 2 - 97 of Fimbrial protein 4. Amino acid residues 97 - 168 of KM22 FimN are 100% identical to amino acid residues 90 - 161 of Fimbrial protein 4. Amino acid residues 162 - 208 of KM22 Fim2-like are 100% identical to amino acid residues 155 - 201 of Fimbrial protein 4. The conserved A residue is highlighted in pink. The bar at the top of the alignment represents the mean pairwise identity over all pairs in the column, with green indicating 100% identity.
N-terminal region
To further evaluate the N-terminal region, the N-terminal amino acid sequences from all fimbrial subunit types were analyzed and placed into groups based on amino acid identity. This resulted in 15 N-terminal groups each consisting of two or more identical amino acid sequences and one group comprised of 10 unique N-terminal amino acid sequences (Table 5). The majority of the N-terminal sequence groups contained sequences from different fimbrial subunit types despite sharing 100% identity. For example, the N-terminal amino acid sequence of FimX from strain D17-021385 was 100% identical to the N-terminal amino acid sequence of Fim2-like from strains 980−2, A345, FDAARGOS_634, FDAARGOS_693, KM22, MBORD707, and MO149 (S5 Table and Table 5). The N-terminal amino acid sequence from all of these strains comprised N-terminal group 1 (Table 5).
Next, a representative amino acid sequence was chosen from each N-terminal amino acid sequence group and aligned along with each of the ten unique N-terminal amino acid sequences revealing several conserved sequence motifs. An overall pattern of hydrophobic residues followed by short stretches of several hydrophilic residues or neutral residues were observed throughout the N-terminal sequence alignment (Fig 8). Additionally, a conserved Leu at residues 15 and 19 were detected and observed to be part of a conserved sequence motif of Leu, Ala, Ala, Ala, Leu, and Ala at residues 15–20. Another conserved sequence motif consisted of Met, Lys, and Gln at residues 1–3 and a Pro, Ala, His, and Ala conserved sequence motif at residues 25–30 was additionally observed.
N-terminal amino acid sequence alignment.Alignment includes a single representative N-terminal sequence from each N-terminal group designation [12–15] and all unique N-terminal sequences listed in Table 5. Residues are colored by hydrophobicity values [59] in a graduated color scheme from strongly hydrophobic (dark blue) to strongly hydrophilic (bright yellow), with glycine as the midpoint in white. B. bronchiseptica strain shown at left with fimbrial subunit type name. Fp1 is used to indicate Fimbrial protein 1 and Fp3 is used to indicate Fimbrial protein 3. The bar at the top of the alignment represents the mean pairwise identity over all pairs in the column, with green indicating 100% identity, yellow at least 30% and less than 100% identity, and red less than 30% identity.
Phylogenic and recombination analysis
A phylogenetic tree inferred from the alignment of all the full-length representative fimbrial subunit type protein sequences resulted in an overall tree topology in which all fimbrial subunit
protein sequences from each fimbrial subunit type clustered together (Fig 9). Fim2-like protein sequences were the exception to the overall tree topology. The Fim2-like proteins shared a common ancestor with the remainder of the subunit protein types, but were polyphyletic, with some singletons more closely related to different proteins.
Maximum-likelihood phylogenetic tree inferred from the full-length protein sequences of fimbrial subunit types.B. bronchiseptica strain names colored according to the legend shown at the left of tree.
When the N-terminal region was removed from all representative fimbrial subunit type protein sequences and the more conserved region of the proteins that begins at the conserved Ala residue and extends to the C-terminal end residue was used to infer the phylogenetic relationships, the resulting tree topology extensively aligned with the distribution of fimbrial gene types (Fig 10). Notably, the Fim2-like protein sequences of strains D755 and RL57 were placed in the same cluster as other Fim2-like protein sequences, demonstrating that the separation of these protein sequences from other Fim2-like protein sequences, when whole protein sequences were compared, was likely due to the N-terminal region (Fig 9).
Maximum-likelihood phylogenetic tree inferred from the protein sequences of fimbrial subunit types without the N-terminal region.The N-terminal region was removed for all protein sequences, such that the protein sequences used for comparisons began at the conserved A residue and extended through the C-terminal end residue.
When the N-terminal regions, beginning at the start codon up to and including the conserved Ala residue, from all representative fimbrial subunit type protein sequences were used to infer a phylogeny, the resulting tree topology did not align with the distribution of fimbrial gene types (Fig 11). In fact, a high degree of variation was observed with the N-terminal sequences from the fimbrial subunit types interspersed throughout the tree. The exception to this distribution was the FimX and FimN N-terminal sequences, in which most of the sequences grouped together based on fimbrial subunit type (Fig 11). However, while the tree topology did not align with the distribution of fimbrial gene types, the N-terminal sequences did group together based on N-terminal group designations. This resulting distribution was likely, given that the N-terminal group designations are based on amino acid identity. Collectively, the detailed phylogenic inferences suggest that diversity among the fimbrial subunit genes located within the fimX locus was likely the result of recombination events.
Maximum-likelihood phylogenetic tree inferred from the N-terminal amino acid sequences of fimbrial subunit types.The N-terminal region used included the start codon up to and including the conserved A residue. B. bronchiseptica strain names colored according to the legend shown at the left of tree. N-terminal group designations, 1-15 and unique indicated by U, provided alongside strain names on the right side.
To further explore the likelihood that genetic diversity within the fimX locus was driven by local recombination events, all unique fimbrial subunit gene sequences were evaluated for local recombination events. Fifteen separate recombination events were detected using at least two statistics-based methods (S6 Table). The recombinant genes were the result of recombination between parent genes from different gene families. For example, recombination between a fimN gene and a fim2-like gene resulted in a new fimN gene sequence. The majority of the detected recombination events were located at the 5-prime end of the gene sequences, consistent with the observation that the phylogenetic tree inferred from N-terminal sequences was topologically distinct from the phylogenetic tree inferred from the fimbrial protein sequences with the N-terminus removed (Fig 10 and Fig 11). Two distinct recombination events were detected in the fimbrial protein 4 gene found in strain D16-050946, consistent with the observation that the protein sequence contains regions similar to three different fimbrial proteins (S6 Table and Fig 7). Combined, the data from phylogenic and recombination analysis suggests that diversity among the fimbrial subunit genes located within the fimX locus was likely the result of homologous recombination.
Discussion
Comparative genomic analysis of B. bronchiseptica strains to date have shown limited genomic variability [8,9,31–33,44]. One of the few regions in which genomic variability has been reported is within the fimX locus. In this report, we analyzed 259 B. bronchiseptica genome assemblies to evaluate the genetic variability located within the fimX locus. Despite the sequence diversity within the fimX locus, the locus was located in the same genomic location flanked by the TRAP transporter and paaK genes in all B. bronchiseptica genome assemblies analyzed. The genomic organization of the fimX locus in B. bronchiseptica differs from the genomic organization of the fimX locus in B. pertussis strains, in which the locus only contains the fimX gene and the ends of the locus are chromosomally separated. In B. pertussis assemblies, a transposase gene is co-located next to the 3-prime end of the TRAP gene with approximately 60 kb or more sequence, depending on the B. pertussis assembly, separating those genes from another transposase gene co-located next to fimX, followed by the paaK gene. This difference in gene order is likely the result of genomic rearrangements induced by insertion sequence elements harbored by B. pertussis isolates [45,46]. In fact, while B. bronchiseptica genomes rarely contain any insertion sequence elements, genomes of B. pertussis isolates contain numerous insertion sequence elements, which have resulted in genome reduction and genome rearrangements [5,9,45–47].
The genetic variability identified within the fimX locus included both the number of genes harbored, as well as the type of predicted fimbrial subunit genes within the locus. A total of six different fimbrial subunit gene types were identified among the B. bronchiseptica genome assemblies analyzed in this study. Poly(C) tracts were identified upstream of all the predicted fimbrial gene types harbored within the fimX locus for all the B. bronchiseptica genome assemblies evaluated, suggesting that they are regulated by phase-variation, a reversible switching ON or OFF of gene expression [48,49]. Different configurations consisting of the type of predicted fimbrial genes harbored and the number of predicted fimbrial genes present were used to categorize the locus sequence from all B. bronchiseptica genome assemblies into twelve distinct fimX locus cluster types.
Six different fimbrial subunit gene types were identified among the B. bronchiseptica genome assemblies evaluated in this study. The protein sequences, encoded from each specific fimbrial subunit gene type, were highly conserved among each other and divergent from the other protein sequences encoded by the different fimbrial subunit gene types located within the fimX locus. This was demonstrated in the phylogenetic tree inferred from the alignment of all the full-length representative fimbrial subunit type protein sequences, which resulted in an overall tree topology in which protein sequences from each fimbrial subunit type tended to form monophyletic groups. The protein sequences encoded from any specific fimbrial subunit gene type from strains belonging to B. bronchiseptica lineage II strains had a lower sequence similarity to paralogous genes from B. bronchiseptica lineage I strains. This is consistent with previous studies documenting that proteins encoded by genes from B. bronchiseptica lineage II strains have a lower sequence similarity to factors encoded by paralogous genes from B. bronchiseptica lineage I strains [32,33]. A conserved Ala residue located toward the N-terminal region was identified in every fimbrial protein sequence analyzed. Most of the amino acid variations observed among all the fimbrial protein sequences were located between the start codon and the conserved Ala residue, which was subsequently referred to as the N-terminal region. For all fimbrial protein sequences, the region starting from the Ala residue and extending to the C-terminal end residue was more conserved.
When the N-terminal amino acid sequences from all fimbrial subunit types were analyzed, 15 N-terminal groups consisting of at least two identical amino acid sequences and one group comprised of 10 unique N-terminal amino acid sequences were identified. The majority of the N-terminal sequence groups contained amino acid sequences from different fimbrial subunit types despite sharing 100% identity. This was similarly reflected in the tree topology observed from the phylogenetic analysis using the N-terminal sequences. Specifically, the tree topology did not align with the distribution of fimbrial gene types, but instead the N-terminal sequences clustered together based on N-terminal group designations.
The Bordetella fimbriae are members of the type I pili family and are assembled and exported by the chaperone–usher pathway, which has been extensively investigated in UPEC [24–30]. UPEC pilus subunits have a highly conserved N-terminal extension containing a conserved motif of alternating hydrophobic residues, which serve a key role in the donor strand exchange mechanism during pilus assembly. While the precise alternating hydrophobic residues categorized for UPEC pilus subunits were not found in the B. bronchiseptica N-terminal sequences, a pattern of hydrophobic residues followed by short stretches of several hydrophilic residues or neutral residues was found, along with two other conserved sequence motifs. These conserved motifs located within the N-terminal region of all fimbrial subunit type protein sequences suggest a functional role in the assembly and export pathway.
Data from both phylogenic and recombination analyses suggest that the diversity within the fimX locus was likely driven by recombination. For instance, data showing that the majority of the detected recombination events were located at the 5-prime end of the gene sequences supports the tree topology resulting from the N-terminal sequences, in which the N-terminal sequences grouped together based on N-terminal group designations instead of aligning with the distribution of fimbrial gene types. Additionally, two distinct recombination events were detected in the fimbrial protein 4 gene, consistent with the Fimbrial protein 4 amino acid sequence containg regions with 100 percent identity with regions from Fimbrial protein 3, FimN, and Fim2-like protein sequences. Similar observations have been documented previously for the Staphylococcus aureus enterotoxin genes located within the enterotoxin gene cluster (egc), in which new enterotoxin genes have been generated by recombination [50].
B. pertussis isolates are known to produce two fimbrial serotypes, referred to as Fim2 and Fim3 [51]. Both are included as components of current acellular pertussis vaccines [52] and several studies have advocated for their inclusion in new vaccine designs [53–58]. Due to the medical importance of whooping cough, the vast majority of studies investigating the expression of fimbrial subunit genes and subsequent production of fimbrial serotypes have focused on B. pertussis isolates. Given that B. bronchiseptica genomes contain more fimbrial subunit genes than genomes of B. pertussis isolates, studies investigating which fimbrial serotypes B. bronchiseptica isolates produce are needed. The functional role of B. bronchiseptica fimbriae in adherence to respiratory epithelium and any potential role as a protective antigen has come from studies utilizing a B. bronchiseptica strain that is incapable of producing any fimbrial serotypes due to an in-frame deletion mutation of the fimBCD genes. [35,55]. Vaccines targeting UPEC fimbrial subunits and therapies targeting the blocking of binding and subsequent function of UPEC fimbrial subunits have been successful and are currently undergoing human clinical trials [17–21]. Focusing on veterinary applications, variations of a vaccine targeting multiple fimbrial subunits harbored by enterotoxigenic Escherichia coli was demonstrated to be broadly protective against porcine post‑weaning diarrhea [22,23]. Given the success of therapies and vaccines targeting fimbrial antigens in both human and veterinary applications, future efforts targeting the fimbrial antigens harbored by B. bronchiseptica offer a promising avenue for veterinary therapies needed to decrease the B. bronchiseptica disease burden among animals.
Supporting information
S1 TableDataset information.The table lists all strains, RefSeq accession numbers, and strain-associated metadata.(XLSX)
S2 TableRepresentative nucleotide and protein sequence information.The table lists the representative nucleotide and protein sequences in the format “strain name: locus_tag number: fimbrial subunit type name”; “fp3” = fimbrial protein 3, “fp1” = fimbrial protein 1, “fp4” = fimbrial protein 4, “UG” = unique group.(XLSX)
S3 TablePairwise amino acid identity (%) for all full-length protein sequences of fimbrial subunit types.The table lists the pairwise amino acid identity (%) from MAFFT multi-sequence alignments of all representative full-length protein sequences of fimbrial subunit types.(XLSX)
S4 TablePairwise amino acid identity (%) for all protein sequences of fimbrial subunit types without the N-terminal region.The table lists the pairwise amino acid identity (%) from MAFFT multi-sequence alignments of all representative full-length protein sequences in which the N-terminal region was removed, such that the protein sequences used for comparisons began at the conserved A residue and extended through the C-terminal end residue.(XLSX)
S5 TablePairwise amino acid identity (%) for all N-terminal amino acid sequences of fimbrial subunit types.The table lists the pairwise amino acid identity (%) from MAFFT multi-sequence alignments of all N-terminal region sequences that include the start codon up to residue preceding the conserved A residue.(XLSX)
S6 TableDetected recombination events.The table lists the recombination events detected within the 81 unique fimbrial subunit gene sequences using RDP5 [40]. Fimbrial subunit genes with a recombination event (Recombinant) are listed along with the contributing genes (Major Parent, Minor Parent). The representative nucleotide and protein sequence names, and N-terminal region sequence group (see Supplemental Table 2, Table 5) are listed for each recombination event. The “Found In” column lists the number of times the identified recombination event occurred in the gene sequences analyzed. The “+” in the “Detection Methods” column indicates the evaluation method that detected the recombination event (R = RDP, G = GENECONV, B = BOOTSCAN, M = MAXCHI, C = CHIMERA, S = SISCAN, T = 3SEQ).(XLSX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mattoo S, Cherry JD. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin Microbiol Rev. 2005;18(2):326–82. doi: 10.1128/CMR.18.2.326-382.2005 15831828 PMC 1082800 · doi ↗ · pubmed ↗
- 2Brockmeier SL, Register KB, Nicholson TL, Loving CL. Bordetellosis. Diseases of Swine. Wiley. 2019. 767–77. doi: 10.1002/9781119350927.ch 49 · doi ↗
- 3Chambers JK, Matsumoto I, Shibahara T, Haritani M, Nakayama H, Uchida K. An Outbreak of Fatal Bordetella bronchiseptica Bronchopneumonia in Puppies. J Comp Pathol. 2019;167:41–5. doi: 10.1016/j.jcpa.2018.12.002 30898296 PMC 7094580 · doi ↗ · pubmed ↗
- 4Goodnow RA. Biology of Bordetella bronchiseptica. Microbiol Rev. 1980;44(4):722–38. doi: 10.1128/mr.44.4.722-738.1980 7010115 PMC 373201 · doi ↗ · pubmed ↗
- 5Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 2003;35(1):32–40. doi: 10.1038/ng 1227 12910271 · doi ↗ · pubmed ↗
- 6Preston A, Parkhill J, Maskell DJ. The bordetellae: lessons from genomics. Nat Rev Microbiol. 2004;2(5):379–90. doi: 10.1038/nrmicro 886 15100691 · doi ↗ · pubmed ↗
- 7Diavatopoulos DA, Cummings CA, Schouls LM, Brinig MM, Relman DA, Mooi FR. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. P Lo S Pathog. 2005;1(4):e 45. doi: 10.1371/journal.ppat.0010045 16389302 PMC 1323478 · doi ↗ · pubmed ↗
- 8Park J, Zhang Y, Buboltz AM, Zhang X, Schuster SC, Ahuja U, et al. Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens. BMC Genomics. 2012;13:545. doi: 10.1186/1471-2164-13-545 23051057 PMC 3533505 · doi ↗ · pubmed ↗