Resolution of severe dilated cardiomyopathy with significant arrhythmia burden using hydroquinidine, in addition to guideline-directed medical therapy, in a patient with a pathogenic SCN5A variant: a case report
Rebecca L M Griffiths, Peter J Cowburn, Catherine Mercer, Michael Papadakis, Elijah R Behr

TL;DR
A 17-year-old with severe heart disease and arrhythmia due to an SCN5A gene variant showed full recovery after treatment with hydroquinidine, a sodium channel blocker.
Contribution
Demonstrates long-term success of hydroquinidine in treating arrhythmia and heart function in a patient with a pathogenic SCN5A variant.
Findings
Hydroquinidine led to complete resolution of arrhythmia and improved heart function in a patient with SCN5A p.R814W variant.
Withdrawal of hydroquinidine caused recurrence of atrial arrhythmia, confirming its therapeutic role.
Early genetic testing and multidisciplinary care enabled personalized treatment success.
Abstract
Dilated cardiomyopathy has a diverse aetiology. Around 20% of cases have an underlying genetic cause. A subset of patients with dilated cardiomyopathy is prone to arrhythmia (‘arrhythmogenic’ cardiomyopathy). (Likely) Pathogenic variants of SCN5A, the gene coding for the alpha subunit of the main cardiac sodium voltage-gated channel, are a known cause of this subset. A 17-year-old male presents with new-onset severe left ventricular systolic dysfunction with atrial flutter and significant ventricular ectopy. Despite medical therapy, his management was challenging. A LifeVest was fitted to allow outpatient optimization of his medications whilst bridging to a decision about implantable cardioverter defibrillator implantation. Specialist genetic testing revealed a pathogenic variant in SCN5A (p.R814W) leading to gain of function. This prompted the use of a sodium channel blocker,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
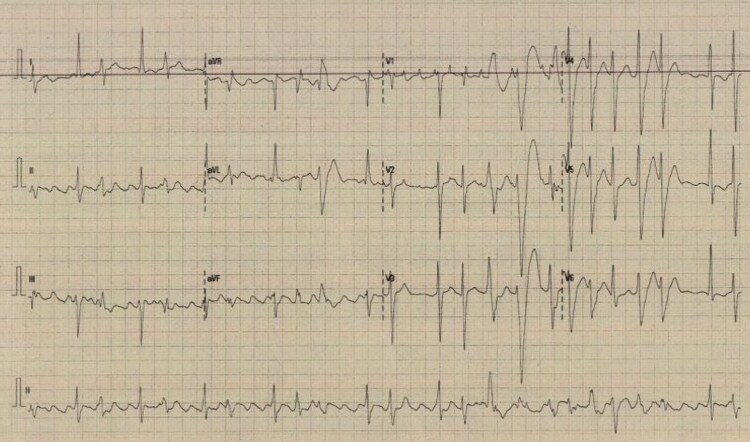 Figure 1
Figure 1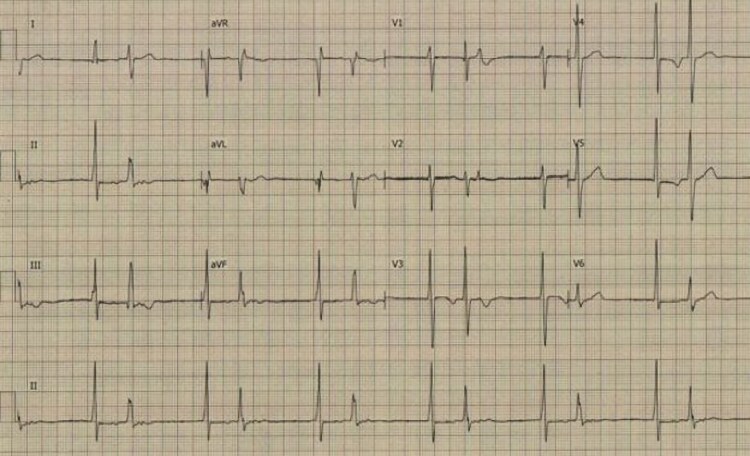 Figure 2
Figure 2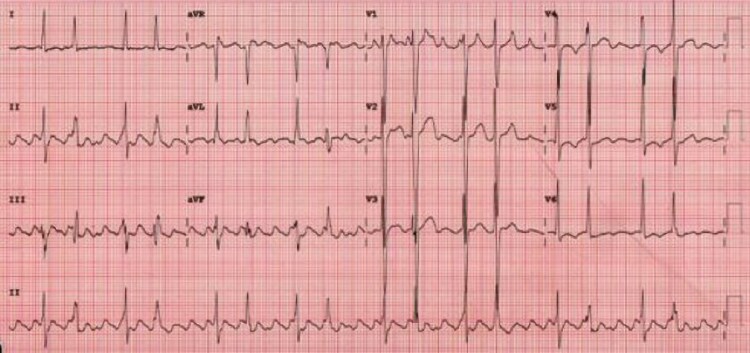 Figure 3
Figure 3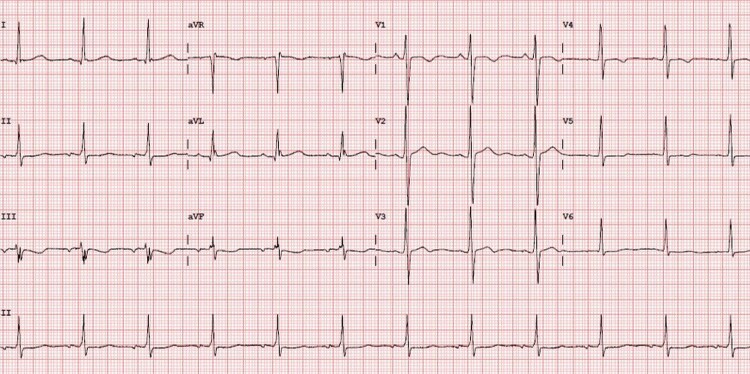 Figure 4
Figure 4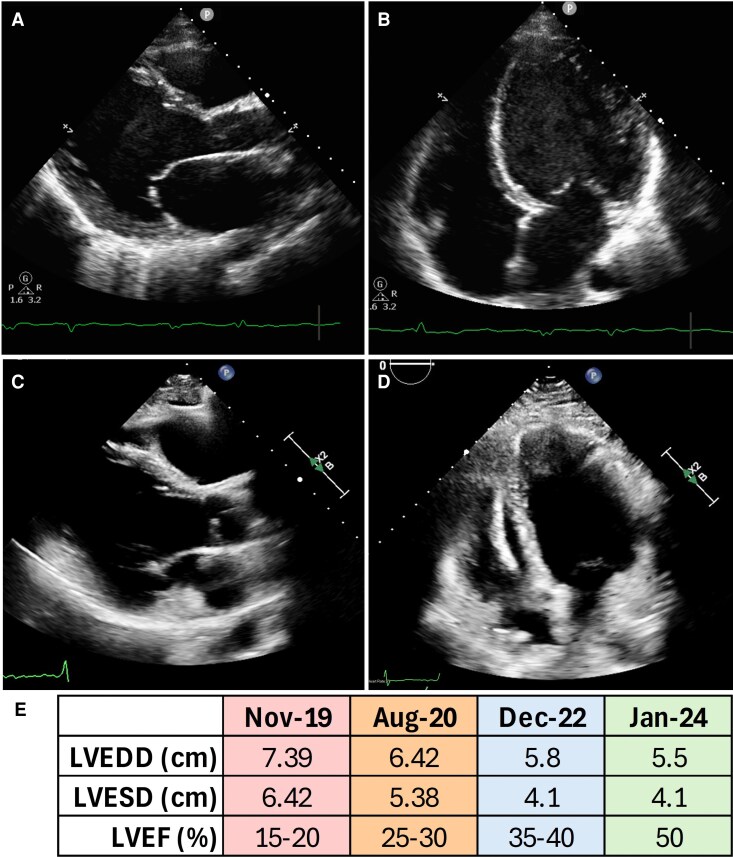 Figure 5
Figure 5| 13/11/2019 | Presentation to local district general hospital with heart failure and atrial arrhythmias |
| 15/11/2019 | Transoesophageal echocardiogram-guided direct current cardioversion (DCCV) |
| 21/11/2019 | Repeat DCCV on amiodarone |
| 04/12/2019 | Discharged with LifeVest (bisoprolol withheld due concerns about junctional rhythm) |
| 23/12/2019 | Bisoprolol reintroduced with ongoing uptitration of medical therapy |
| 09/03/2020 | Sinus rhythm with left ventricular (LV) improvement on optimal medical therapy |
| 04/06/2020 | Holter monitor (off amiodarone)—very frequent atrial/ventricular ectopy and paroxysmal atrial flutter |
| 06/07/2020 | Pathogenic variant in |
| 15/07/2020 | Reviewed at St George’s Hospital—decision made to proceed with transvenous implantable cardioverter defibrillation (ICD) implantation with a trial of hydroquinidine |
| 21/08/2020 | Admitted for ICD and commenced hydroquinidine |
| 20/04/2021 | LV function improved, ectopics suppressed, NT-proBNP normalized |
| 04/01/2024 | Asymptomatic |
| 18/06/2024 | Device alert, frequent atrial arrhythmias—the patient had run out of hydroquinidine (difficulty sourcing medication) |
| 26/07/2024 | Repeat download 1 month following recommencement of hydroquinidine—no atrial arrhythmias detected |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac electrophysiology and arrhythmias · Cardiovascular Effects of Exercise · Cardiomyopathy and Myosin Studies
Introduction
Dilated cardiomyopathy (DCM) is diagnosed in patients with dilatation of their left ventricle (LV, defined as an LV end-diastolic diameter, EDD, >58 mm in males and >52 in females) and regional/global LV systolic impairment (defined as an LV ejection fraction, EF, <50%) that cannot be explained by loading conditions or coronary artery disease alone.^1,2^ In around 20% of cases, (likely) pathogenic genetic variants are responsible for its development.^3^
A subgroup of patients with DCM is predisposed to arrhythmias, so-called ‘arrhythmogenic’ cardiomyopathy. Several genes have been implicated in the pathophysiology of this phenotype, including SCN5A, the gene coding for the alpha subunit of the primary cardiac sodium voltage-gated channel, Na_V_1.5.^4^ We present the case of a 17-year-old gentleman with severe DCM and significant arrhythmic burden and demonstrate how early genetic testing personalized his care and produced an optimal clinical outcome.
Summary figure
**: **
Case presentation
A 17-year-old-male presented to his GP with increasing breathlessness, cough, palpitations, and chest pain. He had experienced a viral illness 1 week previously. He had no underlying medical problems and played county rugby. There was no family history of cardiac disease. He took no regular medication. He did not smoke, drank little alcohol, and took no recreational drugs. He was referred to his local hospital for further assessment.
On arrival, his vital signs indicated: an irregular, tachycardic pulse (130 b.p.m.); a low blood pressure (98/61 mmHg); and a raised respiratory rate (40/min) with oxygen saturations of 92% on air. He was afebrile.
Blood tests revealed a leucocytosis, deranged liver enzymes, and a normal C-reactive protein and troponin T. The patient’s presenting electrocardiogram (ECG) is shown in Figure 1. Heart rhythm monitoring revealed very frequent ventricular ectopy and short runs of non-sustained ventricular tachycardia. A chest X-ray showed cardiomegaly with mild pulmonary congestion. Bedside echocardiography revealed severe LV systolic dysfunction with elevated right ventricular systolic pressure.
The patient’s presenting electrocardiogram demonstrating typical atrial flutter and frequent multifocal ventricular ectopy.
Despite commencing appropriate medications for heart failure and atrial flutter, the patient did not improve. A transoesophageal echocardiogram-guided DC cardioversion (DCCV) was therefore performed, and his subsequent ECG is shown in Figure 2. Bisoprolol was uptitrated and amiodarone commenced for suppression of arrhythmia. He was transferred to his local tertiary cardiology centre for further management.
The patient’s electrocardiogram following transoesophageal echocardiogram-guided direct current cardioversion demonstrating junctional rhythm with bigeminy.
A cardiac MRI (CMR) on arrival confirmed significant LV dilatation and severely reduced function (see Supplementary material online, Videos S1–S3). There was no inflammation, myocardial oedema, or late enhancement (see Supplementary material online, Figures S1–S3).
A specialist genetics review was requested and a dilated and arrhythmogenic cardiomyopathy gene panel sent (see Appendix 1 for the list of genes assessed). He commenced regular furosemide; eplerenone and amiodarone were continued; and sacubitril/valsartan initiated. Bisoprolol was stopped due to junctional rhythm, with a subsequent relapse of atrial tachyarrhythmia (Figure 3). Direct current cardioversion was repeated leading to restoration of a junctional rhythm with bigeminy, as shown previously (Figure 2).
The patient’s electrocardiogram demonstrating recurrence of atrial tachyarrhythmia and frequent ectopy following bisoprolol withdrawal.
The patient improved, and prior to discharge, a cardiopulmonary exercise test was performed. He achieved a peak workload of 216 W with a peak heart rate and blood pressure of 148 b.p.m. and 152/80 mmHg, respectively. Low-level exercise induced frequent ventricular ectopy and bigeminy, settling at peak exercise but returning quickly in recovery. His exercise capacity was severely impaired (VO_2_peak 16.9 mL/min/kg, 34% of predicted) by his cardiac disease evidenced by a lack of rise in oxygen pulse at higher exercise intensities. The anaerobic threshold was 11.4 mL/min/kg. A multidisciplinary team discussion concluded that the patient’s arrhythmic risk was high, and he should be discharged with a LifeVest whilst his medications were optimized and ICD implantation considered.
The patient continued to improve symptomatically with medication optimization and gradual reintroduction of bisoprolol. This improvement was reflected in his investigations (resolution of ectopy on ECG, improvement of ventricular size and function on echocardiography and CMR). Amiodarone was stopped (in view of its long-term adverse effects) and the LifeVest retained.
A follow-up Holter off amiodarone showed a resurgence of arrhythmia with frequent junctional and ventricular ectopy, episodes of bigeminy, and periods of atrial flutter with very little sustained normal sinus rhythm. His clinical picture was therefore one of DCM attributable, at least in part, to atrial, Purkinje, and ventricular arrhythmias.
The patient’s genetic test revealed a pathogenic (Class V) variant in SCN5A (c.2440C>T (p.Arg814Trp)), p.R814W. He was thereafter referred for specialist input. Subsequently, he was admitted in August 2020 for introduction of oral hydroquinidine (300 mg BD Modified-Release) and implantation of a transvenous ICD.
The patient was a keen sportsman prior to diagnosis and was therefore reviewed by a sports cardiology specialist. He was advised to avoid high-intensity competitive sport but encouraged to exercise to maintain physical fitness and mental well-being. His school was written to with specifics of what activities were reasonable.
Subsequent ECG and Holter monitoring on hydroquinidine showed neither arrhythmia nor ectopy (Figure 4). His latest echocardiogram showed resolution of ventricular dilatation with only mild LV impairment (EF 50%; Figure 5).
The patient’s electrocardiogram whilst taking hydroquinidine. Note the absence of ectopy or atrial arrhythmia.
Visual and numeric demonstration of the improvement of the patient’s left ventricular structure and function over time. Parasternal long axis (A and C) and apical four-chamber (B and D) views from the patient’s initial and most recent echocardiograms, performed in November 2019 (A and B) and January 2024 (C and D), respectively. There is clear visual improvement in left ventricular dilatation. Panel (E) presents measurements of the patient’s left ventricular end-diastolic diameter, left ventricular end-systolic diameter, and left ventricular ejection fraction from serial imaging. An improvement of these parameters is observed over time.
In May 2024 due to a misunderstanding about how much medication he had remaining, the patient ran out of hydroquinidine and remained off this medication for 3 weeks due to supply issues. His other medications continued. An ICD download reported 35 episodes of atrial tachyarrhythmia with a maximum duration of 58 min, correlating to symptoms of weakness and feeling ‘jittery’. On resumption of hydroquinidine, his symptoms improved and the atrial tachyarrhythmias resolved, with none reported on his latest ICD download.
Discussion
Early genetic testing in our patient’s case facilitated the use of a personalized genomics-based therapy to produce a favourable clinical response. The p.R814W variant in SCN5A has been observed both de novo, through analysis of probands within a DCM cohort, as well as in a family with a history of sudden cardiac death and DCM.^4,5^ Of note, in our case, the p.R814W variant was not detected in our patient’s parents, suggesting a de novo finding. A group in Chicago, USA, have also shown that the p.R814W variant co-segregates with DCM, sudden death, and the need for transplantation.
The variant affects the fourth, voltage-sensing segment (S4) of the second domain of Na_v_1.5 with replacement of a conserved arginine residue with tryptophan.^6^ The p.R814W variant predominantly causes gain of function through mechanisms of altered activation, increase in window current, and slower deactivation of Na_v_1.5, resulting in enhanced sodium current and supraventricular and ventricular hyperexcitability, thereby causing frequent arrhythmia.^5,6^ Contractile impairment may result from this frequent arrhythmia, or through other mechanisms such as blunting/reversal of the force–frequency relationship, abnormalities in intracellular calcium levels (caused by a higher intracellular sodium), ineffective regulation of pH (caused by abnormal sodium and calcium homeostasis), and effects on mitochondria (e.g. swelling and dysfunction, thereby leading to metabolic disruption of cardiomyocytes).^5–7^ The variant’s functional effect justified use of hydroquinidine, a sodium channel antagonist leading to a striking improvement in LV size and function, as well as his arrhythmic burden.
Improvement of LV dynamics and arrhythmias with quinidine monotherapy has previously been observed in SCN5A gain-of-function variants.^8–10^ The p.R222Q SCN5A variant which, like p.R814W, is in a voltage-sensing segment of Na_V_1.5 was shown in a case report to reduce arrhythmia and improve LV function.^8^ Of note, unlike in the present case, complete resolution of arrhythmia and restoration of LV function was not observed.^8^ This may be due to the different biophysical effects of disparate SCN5A variants, a shorter follow-up period of the case (2.5 years), dose of quinidine used, or due to confounding effects from concurrent coronary artery disease.^8^
Zakrzewska-Koperska et al.^5^ studied a family of patients with arrhythmogenic DCM and the p.R814W variant. Interestingly, quinidine monotherapy was met with mixed success. Combinations of Class I antiarrhythmics were required to restore clinical stability in this family.^5^ Furthermore, the follow-up period in this case series was relatively short (1.5 years) meaning sustained improvement could not be fairly assessed. Contrastingly, in our case, hydroquinidine monotherapy led to sustained improvement of symptoms, LV size and function, and arrhythmic burden over a 4-year follow-up period—to the authors’ knowledge, the first description of such.
Recent ESC guidelines on the management of cardiomyopathies and ventricular arrhythmias recommend that genetic testing is undertaken in patients with cardiomyopathies where genetics will help to establish a diagnosis, prognosticate, stratify therapy or assist in reproductive management (Class IB Level of Evidence).^2,11^ In our patient’s case, prompt genetic testing facilitated the use of personalized therapy, producing an optimal and sustained clinical improvement.
Lead author biography
Rebecca graduated from Cardiff University Medical School in 2017, having previously achieved a First Class Honours in her Intercalated BSc in Biomedical Sciences (Neuroscience). As an undergraduate, she developed a strong interest in cardiology, later earning a distinction in an MSc in Translational Cardiovascular Medicine at the University of Bristol. Her academic training, combined with experience as a cardiology registrar, prompted a focused interest in inherited cardiac conditions. She currently works a Clinical Research Fellow at City St George’s, University of London, and is undertaking a PhD focussing on the inherited arrhythmia syndrome, Brugada Syndrome.
Supplementary Material
ytag100_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J 2007;29:270–276.17916581 10.1093/eurheartj/ehm 342 · doi ↗ · pubmed ↗
- 2Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, et al 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J 2023;44:3503–3626.37622657 10.1093/eurheartj/ehad 194 · doi ↗ · pubmed ↗
- 3Verdonschot JAJ, Hazebroek MR, Krapels IPC, Henkens MTHM, Raafs A, Wang P, et al Implications of genetic testing in dilated cardiomyopathy. Circ Genom Precis Med 2020;13:476–487.32880476 10.1161/CIRCGEN.120.003031 · doi ↗ · pubmed ↗
- 4Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, et al Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005;293:447–454.15671429 10.1001/jama.293.4.447PMC 2039897 · doi ↗ · pubmed ↗
- 5Zakrzewska-Koperska J, Bilińska ZT, Truszkowska GT, Franaszczyk M, Elikowski W, Warmiński G, et al A combination of quinidine/mexiletine reduces arrhythmia in dilated cardiomyopathy in two patients with R 814W SCN 5A mutation. ESC Heart Fail 2020;7:4326–4335.33084224 10.1002/ehf 2.12993 PMC 7754730 · doi ↗ · pubmed ↗
- 6Nguyen TP, Wang DW, Rhodes TH, George AL. Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res 2008;102:364–371.18048769 10.1161/CIRCRESAHA.107.164673 · doi ↗ · pubmed ↗
- 7Beckermann TM, Mc Leod K, Murday V, Potet F, George AL. Novel SCN 5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy. Heart Rhythm 2014;11:1446–1453.24815523 10.1016/j.hrthm.2014.04.042PMC 4108519 · doi ↗ · pubmed ↗
- 8Zakrzewska-Koperska J, Franaszczyk M, Bilińska Z, Truszkowska G, Karczmarz M, Szumowski Ł, et al Rapid and effective response of the R 222Q SCN 5A to quinidine treatment in a patient with purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: a case report. BMC Med Genet 2018;19:94.29871609 10.1186/s 12881-018-0599-4PMC 5989373 · doi ↗ · pubmed ↗