Characteristics of pediatric-onset anti-contactin1 antibody-associated autoimmune nodopathies with concomitant membranous nephropathy
Qiuyue Guan, Yi Xie, Yonghua He, Liru Qiu, Yan Liu

TL;DR
A rare autoimmune disorder affecting children, linked to nerve and kidney issues, is studied through a new case and literature review.
Contribution
First detailed report of pediatric-onset CNTN1-AN with membranous nephropathy and a global literature review of six confirmed pediatric cases.
Findings
Pediatric CNTN1-AN is rare, with renal involvement observed in 3/7 cases and MN confirmed in two.
Neurological symptoms improved with low-dose rituximab, but renal issues often require stronger treatment.
Pediatric and adult CNTN1-AN cases show similar clinical features but differ in incidence rates.
Abstract
Anti-contactin-1 antibody-associated autoimmune nodopathy (CNTN1-AN) is a rare disorder predominantly affecting older individuals, characterized by sensorimotor peripheral neuropathy, with over 50% of cases presenting with proteinuria and membranous nephropathy (MN). Pediatric-onset CNTN1-AN is exceptionally rare, and its clinical profile remains poorly characterized. We report a pediatric case of CNTN1-AN with MN and conduct a literature review to elucidate the pathogenesis and clinical features of CNTN1-AN combined with MN in children. The patient, an 11-year-old girl, presented with a one-month history of progressive lower limb weakness and a 10-day progression of gait instability. Physical examination demonstrated bilateral proximal lower limb weakness, restricted mobility, and sensory abnormalities.No facial or limb edema or frothy urine was observed. Elevated anti-CNTN1 antibody…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
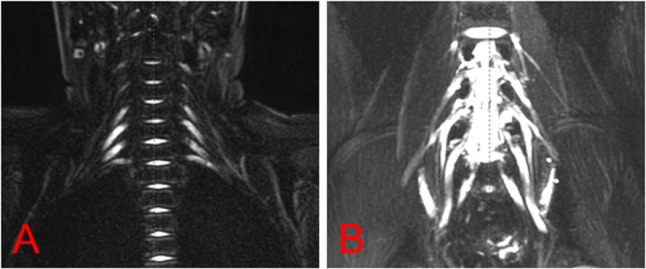 Figure 1
Figure 1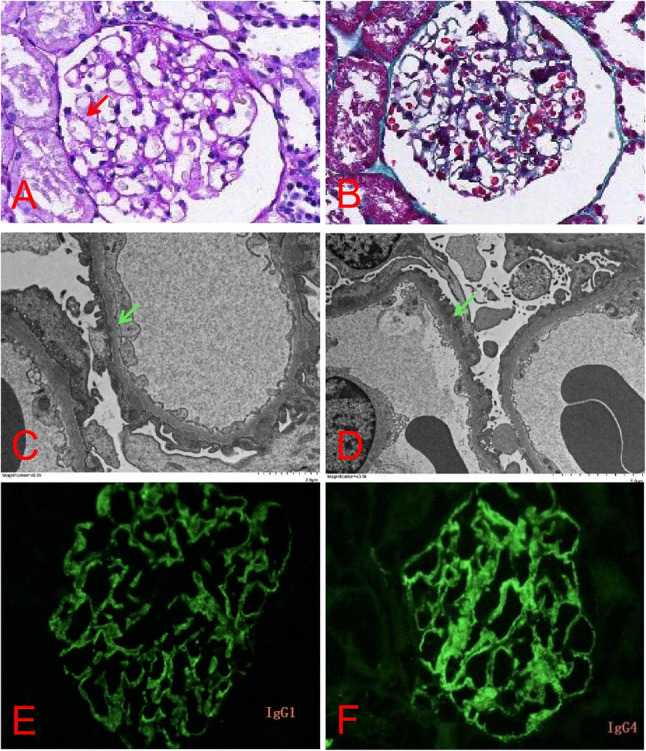 Figure 2
Figure 2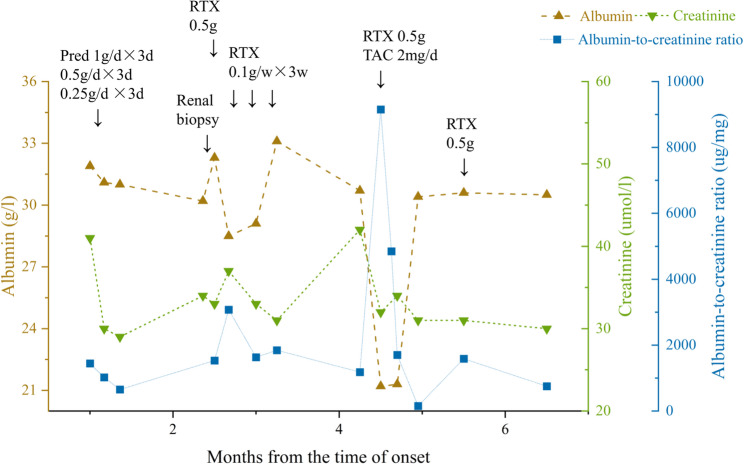 Figure 3
Figure 3 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPeripheral Neuropathies and Disorders · Autoimmune Neurological Disorders and Treatments · Peripheral Nerve Disorders
Introduction
Emerging evidence reveals distinct clinical characteristics in patients with autoantibodies against nodal/paranodal proteins (NF186, NF155, CNTN1, Caspr1), showing significantly different pathological, electrophysiological, and therapeutic patterns distinct from classic chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). According to the 2021 European Academy of Neurology/Peripheral Nerve Society (EAN/PNS) classification, CIDP patients with anti-nodal antibodies are now categorized under the newly defined entity of AN [1]. AN is characterized by acute, subacute, or chronic sensorimotor peripheral neuropathy and requires careful differentiation from Guillain-Barré syndrome (GBS) and CIDP.
The CNTN1/Caspr1 complex, localized at the axolemma of the paranodal region of the node of Ranvier, interacts with NF 155 at the myelin terminus to form an axo-glial septate-like junction. This junction stabilizes the myelin sheath attachment to the axolemma and maintains the spatial segregation of voltage-gated Na⁺ channels in the nodal region from voltage-gated K⁺ channels in the juxtaparanodal region. However, anti-CNTN1 IgG4 antibodies can infiltrate the paranodal region, disrupting the interaction between CNTN1 and other paranodal proteins. This disruption causes detachment of the myelin terminal loops from the juxtaparanodal axolemma, nodal widening, incr eased nodal capacitance, and current leakage. Consequently, reduced forward current and ectopic juxtaparanodal hyperpolarization occur due to the mislocalization of voltage-gated K⁺ channels. These pathological changes lead to slowed nerve conduction velocity, conduction block, and ultimately manifest as limb weakness and sensory deficits.
MN is an autoimmune disorder characterized by the subepithelial deposition of immune complexes, consisting of immunoglobulin G (IgG) and target antigens, along the glomerular basement membrane (GBM). This condition is primarily driven by immune system activation triggered by various podocyte membrane antigens.In 2021, Le Quintrec et al. [2] identified CNTN1 as a novel target antigen in CIDP-associated MN. Subsequent studies have reported a rising incidence of CNTN1-AN cases with concurrent MN, particularly among middle-aged and elderly male populations. In contrast, pediatric-onset CNTN1-AN with MN remains exceptionally rare, with limited documented cases, and its clinical characteristics remain to be explored.
Case presentation and literature review
Case presentation
Medical history and physical examination
An 11-year-old girl was admitted to Pediatric Neurology in September 2024, with progressive bilateral lower limb weakness over one month and gait instability for 10 days. Her medical, developmental, and family histories were unremarkable. Physical examination revealed no evident skin rash, no joint swelling or tenderness, and no oral ulcers. Frothy urine was not observed. Neurological examination revealed grade 5 strength in the distal upper and lower limbs, but only grade 3 strength in the proximal lower limbs. Romberg’s sign was negative, and finger-to-nose testing was stable. However, she could not walk in a straight line. Sensory testing showed decreased tactile, pain, and complex pattern sensation in the feet and lower legs. Her gait was abnormal with reduced foot lift, and she was unable to squat, jump, or climb stairs. She reported lumbago and foot numbness after walking.
Auxiliary examination
Routine laboratory investigations, including complete blood count, renal function tests, electrolyte panel, coagulation profile, blood ammonia, lactate, glucose, pyruvate, anti-phospholipase A2 receptor antibodies, folic acid, vitamin B12, complement levels (C3 0.3 g/L, reference 0.1–0.4 g/L; C4 1.48 g/L, reference 0.8–1.6 g/L), anti-dsDNA, and anti-Smith antibodies were all within normal ranges. Peripheral blood testing revealed a positive antinuclear antibody (ANA) titer of 1:100 (granular pattern), and an elevated erythrocyte sedimentation rate of 35 mm/h (reference range: 0–20). Serum analysis showed reduced immunoglobulin G (5.45 g/L; reference range: 8–16), hypoalbuminemia (31.9 g/L; reference range: 40–55), and hypoproteinemia (56 g/L; reference range: 66–87), along with hypercholesterolemia (6.56 mmol/L; reference range: <5.18), hyperuricemia (430 µmol/L; reference range: 142.8–339.2), and vitamin D deficiency (25-hydroxyvitamin D: 8.4 ng/mL). Urinalysis demonstrated hematuria(1+), significant proteinuria (4+), and an elevated urine albumin-to-creatinine ratio of 1445.1 µg/mg (reference range: <30). Urinary N-acetyl-β-D-glucosaminidase was elevated at 19.5 U/L (reference range: 0.3–11.5), with 24-hour urine total protein 2196.8 mg/L.
Peripheral neuropathy antibody testing revealed elevated anti-CNTN1 IgG titers (serum: 1:320; CSF: 1:3.2) via cell-based assay (CBA), while other serum and CSF peripheral neuropathy antibodies were negative, including GQ1b, GM1, myelin-associated glycoprotein (MAG), sulfatide, NF155, NF186, and CASPR1. CSF analysis showed a nucleated cell count of 5 × 10^6/L, elevated total protein (2266 mg/L), albumin (1440 mg/L), and immunoglobulins (IgG: 169.0 mg/L; IgM: 11.4 mg/L; IgA: 44.1 mg/L). EMG indicated peripheral nerve involvement in both upper limbs and the right lower limb, affecting motor, sensory, distal, and proximal nerve roots, with evidence of both demyelinating and axonal damage, predominantly demyelinating motor fiber involvement (Table 1). MRI of the lumbosacral and brachial plexus revealed bilateral L4-5 nerve root swelling and thickening, and inflammatory changes in the bilateral brachial plexus (Fig. 1).
Table 1. Nerve conduction studies (NCS) resultsNormalCase presentationCase presentation after treatmentTimes from onset1 month3.5 monthsSideL/RL/RMedian nerveDL (ms)≤4.2Wrist 6.25/6.75Wrist 7.2/7.2Elbow 10.75/10.85Elbow 12.1/11.3MCV (m/s)≥50.045.8/48.344.8/51.2CMAP amplitude (mV)≥4.8Wrist 7.67/9.78Wrist 9.58/11.81Elbow 6.11/9.11Elbow 8.13/11.28F wave latency (ms)≤30.044.75/43.1545.1/41.9F wave response frequency≥79%100%/0%100%/100%SCV (m/s)≥50.839.0/41.232.0/33.9SNAP amplitude (µV)≥8.024.9/23.928.05/22.63Ulnar nerveDL(ms)≤3.1Wrist 3.80/4.85Wrist 5.0/4.0Elbow 11.35/13.80Elbow 11.1/9.7MCV (m/s)≥51.031.9/25.131.1/33.3CMAP amplitude (mV)≥5.5Wrist 3.16/2.68Wrist 4.06/3.01Elbow 0.39/1.17Elbow 3.32/3.12F wave latency (ms)≤30.046.35/44.55Not examinedF wave response frequency≥79%29%/0%Not examinedSCV (m/s)≥50.646.2/59.336.2/45.4SNAP amplitude (µV) ≥5.013.6/22.811.27/18.08Tibial nerveDL(ms)≤5.8R Ankle 4.85Ankle 7.4/6.6R Popliteal 13.15Popliteal 16.3/15.4MCV (m/s)≥39.4R 41.639.3/39.7CMAP amplitude (mV)≥5.0R Ankle 4.89Ankle 2.34/2.09R Popliteal 1.30Popliteal 1.45/1.92F wave latency (ms)≤60.083.9/79.673.2/71.4F wave response frequency≥79%0%/0%56.3%/100%PeronealDL (ms) ≤4.6 R Ankle 5.95Ankle 7.4/7.7R Head of fibula 13.15Head of fibula 14.7/15.2MCV (m/s)≥39.8R 40.736.9/38.6CMAP amplitude (mV)≥2.3R Ankle 1.58Ankle 0.49/1.13R Head of fibula 1.28Head of fibula 0.35/0.65Superficial peronealSCV (m/s)≥41.3R Not elicited48.0/47.3SNAP amplitude (µV)≥4.0R Not elicited10.88/6.99SuralSCV (m/s)≥41.9R Not elicited50.0/50.0SNAP amplitude (uV)≥7.0R Not elicited8.75/7.67μV microvolt, mV millivolt, m/s meter per second, ms millisecond, CMAP compound muscle action potential, DL distal motor latency, MCV motor nerve conduction velocity
Fig. 1. Neuroimaging. (A) MRI showed thickened bilateral brachial plexus nerves with T2 hyperintensity, without distortion or disruption. (B) revealed linear and patchy T1 and T2 hyperintensities in L1-L3 subcutaneous fat, with mild thickening and T2 hyperintensity in bilateral L4-5 nerve roots
Renal biopsy confirmed MN (stage I) on light microscopy. Immunofluorescence staining of frozen renal tissue demonstrated granular deposition of CNTN1 antibody IgG1 (++), IgG2 (+), IgG3 (+), and IgG4 (++/+++) along glomerular capillary loops. Paraffin immunofluorescence staining was negative for phospholipase A2 receptor (PLA2R), neural epidermal growth factor-like 1 (NELL-1), thrombospondin type 1 domain-containing 7 A (THSD7A) and etc. (Fig. 2). Additional imaging studies, including renal ultrasound, electrocardiogram, cranial MRI, and thoracic spine MRI, revealed no significant abnormalities.
Fig. 2. Pathological results of renal biopsy. Light microscopy of 30 glomeruli revealed 2 with global sclerosis and 2 with segmental sclerosis. Focal mild mesangial hypercellularity and matrix expansion were noted. The glomerular basement membrane showed ribbon-like remodeling with early spike formation (PAS stain, red arrow). Tubulointerstitial changes included focal tubular epithelial degeneration and patchy interstitial fibrosis (A and B). Electron microscopy revealed diffuse podocyte foot process effacement with microvillous transformation, scattered subepithelial electron-dense deposits (green arrow), and segmental mild mesangial hypercellularity and matrix expansion (C and D). Renal tissue cryosection immunofluorescence showed CNTN1 antibody IgG1++(E),IgG2+,IgG3+,IgG4++/+++(F),with granular deposits in glomerular capillary loops
Diagnosis and treatment
Based on the above symptoms, signs, and diagnostic findings, the patient was diagnosed with CNTN1-AN. The patient underwent prednisone pulse therapy followed by oral prednisone for one month. Subsequently, she achieved independent stair climbing and improved tactile, pain, and complex sensory function in the lower limbs; however, proximal lower limb strength did not further improve. Persistent proteinuria (± to +++) prompted a renal biopsy, confirming concurrent MN. Given the predominant IgG4 pathology of CNTN1-AN and the documented poor response to intravenous immunoglobulin (IVIG) in such cases, we opted for B-cell depletion therapy with rituximab as the primary immunomodulatory strategy [3–5]. She then received rituximab therapy (initial dose:500 mg, followed by weekly doses of 100 mg). After four rituximab doses, she could independently squat and stand, with proximal muscle strength improving to grade 4 + in all four limbs. Sensation in the calves and feet normalized. Mild gait instability continued, with inability to walk in a straight line. At 3.5 months into the disease course, urine protein remained 2+, serum anti-CNTN1 IgG titers persisted at 1:320, and EMG revealed ongoing peripheral nerve involvement in both upper and lower limbs, affecting motor axons, myelin, and nerve roots (Table 1). At 4.5 months, the patient developed limb edema. A 7-day trial of irbesartan (2 mg/kg/day) was discontinued due to dizziness, hypotension, and urticarial rash. Follow-up tests showed urine protein (4+), elevated urine albumin-to-creatinine ratio (9156.2 µg/mg), decreased albumin (21.1 g/L), and creatinine (32 µmol/L). Immunoglobulin G (IgG) was reduced to 2.5 g/L, and complement C3 was elevated to 1.59 g/L. A 6-minute walk test showed a distance of 395 m. Given the patient’s demonstrated hypersensitivity to irbesartan with concern for potential cross-reactive adverse effects, along with the presence of severe, active nephrotic syndrome requiring more definitive intervention, we decided not to trial alternative RAASi and instead, in accordance with the KDIGO 2021 Glomerular Disease Guidelines [6], oral tacrolimus (2 mg/day) was initiated, with trough concentrations fluctuating between 4.01 and 7.84 ng/ml. Additionally, the dose of rituximab was increased. The patient received a fifth rituximab pulse (500 mg), followed by a sixth rituximab pulse (500 mg) one month later. Follow-up tests at 6.5 months showed reduced urine protein (2+) and a decreased urine albumin-to-creatinine ratio (752.9 µg/mg). Albumin was 30.5 g/L (Fig. 3). There was significant improvement in limb mobility, with proximal muscle strength graded at 5-. To monitor the biological response to rituximab, B-cell (CD19+) counts were serially assessed. The pre-treatment B-cell count was 743 cells/µL. Following rituximab administration, complete B-cell depletion (0 cells/µL) was achieved and sustained on subsequent measurements, confirming effective pharmacological targeting.
Fig. 3. The clinical course of the current case. RTX: rituximab; Pred: prednisone; TAC: tacrolimus
Literature review
Literature was identified from PubMed database, using the search terms “CIDP,” “anti-CNTN1,” “autoimmune neuropathy,” “MN,” and “membranous glomerulopathy.” The search encompassed studies and case reports published up to November 2024. After removing duplicates and merging relevant records, we identified over 100 documented cases of CNTN1-AN [3, 7–14]. Among these, only 9 cases involved patients with an onset age under 18 years. Among the 9 cases, 3 lacked detailed clinical descriptions. Of the remaining 6 cases with complete clinical data, only 1 was associated with concurrent MN (Table 2).
Table 2. Clinical features of pediatric CNTN1-AN and CNTN1-AN with MNPatient 1Patient 2, 3Patient 4Patient 5Patient 6Patient 7Year/author2019/Carrera-García L et al. [10]2020/De Simoni et al. [11]2023/Merel C Broers et al. [12]2024/Chen J et al. [13]2023/Tang Y et al. [14]2025/ this articleAge of onset(years)/sex2.75/M7.9(mean age)/NAToddler/F12/M14/M11/FAge of diagnosis(years)7.75NANA20NA11Mode of onset/ course of diseaseActue, relapsingChronicActueChronicActue, relapsingActuePreceding infection-NANA-++Initial symptomsLower limb weakness and frequent fallsNABilateral lower limb weakness and pain, complete dependence on a wheelchairLimb tremors, slurred speech, and mild limb weaknessLower limb weakness and numbnessProgressive weakness in the lower limbsSequence of manifestationsNo renal involvementNANAAN→Renal involvement, 8 yearsConcurrentConcurrentTremble-+/+++--Ataxia++/+NA+++WeaknessLD > LP > UP = UDNAL > UL > ULP > LD > UP = UDLP > LD > UP = UDNumbness-NANA++-Pain-+/++-++Autonomic symptomsDiarrheaNA-Orthostatic dizziness+HyperhidrosisDysarthria-NANA+--Cranial nerve damage-One case of optic neuritis----Nystagmus-NANA+--Sensory disturbances-NANADistal lower limb pain with impaired vibration sense.Hyperalgesia with impaired vibration sense.Impaired touch, pain, and vibration sense in bilateral calves and feet.Muscle atrophy-NANA+--CNTN1 antibody++Serum 1:6400, IgG1 ±, IgG2 ±, IgG3 -, and IgG4 +Serum 1:100,CSF 1:10+Serum 1:320,CSF 1:3.2AutoantibodiesNANANAANA 1:200NAANA 1:100Serum anti-PLA2R antibodiesNANANANA--24 h-UTP (g/24 h)NANANA5.15.462.2Serum creatinine(µmol/l)NANANANA4241CSF protein(g/l)1.482.924(mean value)Elevated CSF protein3.0092.842.266CSF cells(*10^6/L)Normal4.6(mean value)NA1215Protein-cell dissociation++/+NA+++Neurological MRINANANADiffuse thickening of bilateral cervical, lumbosacral nerve roots and cauda equina, with multiple intercostal root sheath cysts.Lumbosacral nerve MRI was normalBilateral L4-5 nerve root hypertrophy with brachial plexus inflammatory changes.Electrophysiological studiesProlonged distal latencies, slowed conduction velocities, reduced CMAP amplitudes, and chronic denervation with fibrillations and positive sharp waves in multiple motor nerves.NAConsistent with EFNS/PNS 2010 diagnostic criteriaSevere demyelination and axonal injuryMultiple motor conduction blocks without temporal dispersion, prolonged distal latencies, slowed velocities, and reduced CMAP amplitudes (lower limb predominance).Demyelination and axonal damage were observed in the peripheral nerves of both upper limbs and the right lower limb, with predominant demyelination of motor fibersNerve biopsyNANANANADemyelination combined with axonal damageNARenal biopsyNANANANAMN (stage I)MN (stage I)TreatmentIVIG、GCIVIG、GC、RTXIVIG、GCIVIG、GC、PLEX、RTXGCGC、RTX、TACPrognosisInitial IVIG (2 g/kg) yielded transient improvement, followed by clinical progression confirmed by CSF and EMG. A second IVIG course was ineffective, but GC (30 mg/kg/day × 5 days) achieved significant response. Disease relapsed at 1 year but responded to IVIG retreatment.Patient 2 worsened after 4 months of IVIG + GC but rapidly improved post-RTX. Patient 3 showed partial IVIG response but relapsed post-infection; RTX induced significant improvement, achieving mRS 2 at final follow-up.Following IVIG failure, Patient 4 achieved GBS disability score 2 with RTX + GC, ambulating with braces.Patient 5 showed progressive deterioration despite 9-year intermittent IVIG + GC therapy. Three PLEX sessions were ineffective, but single-dose RTX (500 mg) induced marked clinical improvement.NAGC and low-dose RTX improved motor function but not renal progression. RTX dose escalation significantly reduced proteinuria.F female, M male, NA unknown, LP lower proximal, LD lower distal, UP upper proximal, UD upper distal, UTP urinary total protein, mRS modified Rankin Scale, ANA antinuclear antibody, IVIG Intravenous Immunoglobulin, GC Glucocorticoids, RTX rituximab, TAC tacrolimus, PLEX Plasma Exchange
Discussion
Comparative clinical characteristics between pediatric and adult populations with CNTN1-AN
Patients with CNTN1-AN typically present with an older age of onset, a male predominance, and acute or subacute onset, often accompanied by a preceding infection [14]. Some cases may initially be misdiagnosed as acute GBS [3, 4]. The clinical manifestations primarily include sensory ataxia, symmetric limb weakness, paresthesia, neuropathic pain, tremors, numbness, and autonomic dysfunction. Some patients may also exhibit involvement of the respiratory system and cranial nerves(predominantly manifesting as facial palsy, ophthalmoplegia, and diplopia) [13, 15–17]. Over half of the patients exhibit proteinuria, renal involvement, and MN, with renal biopsy revealing deposition of IgG4 along the GBM [3, 4, 18–20]. The diverse clinical spectrum of CNTN1-AN is linked to the expression pattern of CNTN1, which is detectable in the retina, spinal cord, cerebral cortex, hippocampus, cerebellum, and glomeruli. However, it remains to be definitively confirmed whether CNTN1 is expressed by podocytes [5, 21]. Sensory ataxia is associated with the widespread expression of CNTN1 in the dorsal root ganglia of sensory nerves, where CNTN1 antibodies can cross the relatively permeable blood-nerve barrier. CNTN1 antibodies may disrupt CNTN1-PTPα interactions, inhibiting Fyn and Src kinase activity and contributing to cerebellar dysfunction [13].
Nerve biopsy findings typically show nodal elongation, paranodal disorganization, and myelin loop detachment. Notably, characteristic pathological features of classic CIDP are absent, such as segmental demyelination and onion-bulb formations resulting from Schwann cell proliferation [22]. EMG reveal prolonged distal motor latency (DML), reduced nerve conduction velocity (NCV), and the presence of conduction blocks without waveform dispersion. F-wave latency is either prolonged or absent, compound muscle action potential (CMAP) amplitude is significantly reduced, sensory conduction velocity (SCV) is slowed, and sensory nerve action potential (SNAP) amplitude is diminished [23]. EMG findings in this pediatric case align with those reported in previous studies, including prolonged DML and reduced CMAP amplitude in the median and ulnar nerves of both upper limbs, as well as markedly prolonged F-wave latency and decreased occurrence rates in the median, ulnar, and tibial nerves bilaterally.
Patients with CNTN1-AN demonstrate significantly elevated CSF protein levels, accompanied by a distinct protein-cell dissociation. To date, there is no evidence supporting intrathecal synthesis of CNTN1 antibodies. Neuroimaging findings in CNTN1-AN patients commonly show contrast enhancement or hypertrophy of nerve roots in both lumbosacral and brachial plexus regions [24]. Experimental studies have shown that CNTN1 antibodies bind to hippocampal neurons and cerebellar tissue in rats. Some researchers hypothesize that the elevated CSF protein levels may result from the relatively permeable blood-nerve barrier at the nerve roots, facilitating the infiltration of peripheral circulating proteins into the central nervous system [16].
Reports on pediatric CNTN1-AN remain limited in the literature. In 2019, Carrera-García et al. [10] reported the first pediatric case of CNTN1 antibody-associated CIDP. Subsequently, in 2020,De Simoni D et al. [11] conducted a CBA screening for CNTN1 antibodies in 54 children diagnosed with GBS or CIDP, identifying two CIDP cases positive for CNTN1 antibodies. A 2024 study cohort of 31 CNTN1-AN patients revealed that 3 (9.35%) were pediatric cases, and 11 (35.5%) had concurrent renal involvement, including 5 (16.1%) diagnosed with membranous glomerulonephritis [3].
Through a literature review, we identified over 100 reported cases of CNTN1-AN, with only 9 exhibiting pediatric onset, suggesting a higher incidence in adults. We retrospectively analyzed six pediatric CNTN1-AN cases with complete data, supplemented by the case presented in this report (Table 2). The findings revealed that limb weakness was the most common initial symptom (five cases, 71.4%), with a subset of patients reporting preceding infections (two cases, 28.5%). Renal involvement was observed in three cases (Patients 5–7), of which 66.7% were diagnosed with stage I membranous nephropathy. Neurological and renal symptoms frequently occurred concurrently. The most prevalent clinical manifestations included: ataxia (six cases, 85.7%); limb weakness (five cases, 71.4%), predominantly affecting the lower limbs and proximal muscles; pain (five cases, 71.4%); tremor (four cases, 57.1%); autonomic symptoms (four cases, 57.1%), such as hyperhidrosis and diarrhea; and sensory disturbances, primarily involving vibration and pain sensation (three cases, 42.9%). Additionally, some patients exhibited symptoms including numbness, dysarthria, cranial nerve involvement (e.g., optic neuritis), and muscle atrophy. CSF analysis in six pediatric patients demonstrated protein-cell dissociation. NCS indicated more significant involvement of motor fibers compared to sensory fibers. In the early disease stages, partial responsiveness to intravenous immunoglobulin (IVIG) and glucocorticoids (GC) was observed; however, rituximab demonstrated superior efficacy overall. In summary, beyond a potentially lower incidence, CNTN1-AN shows no substantial differences between pediatric and adult populations.
The role of CNTN1 antibodies and therapeutic approaches
CNTN1-AN antibodies are predominantly of the IgG4 subclass, with IgG3 detectable during acute disease progression.In our case, renal immunofluorescence showed predominant IgG4 and IgG1 anti-CNTN1 deposition along the glomerular basement membrane, though serum antibody subclasses were not tested. Numerous studies have demonstrated that IgG4 subclass CNTN1 antibodies exhibit high affinity for the node of Ranvier and are directly pathogenic [25]. The most pathogenic IgG4 subclass is generated through subclass switching in the sequence of IgG3, IgG1, IgG2, and IgG4 [26]. Norito Kokubun et al. [27] reported antibody subclass switching in four AN patients, noting that IgG1 disappeared as the disease stabilized, suggesting that IgG1 may serve as a potential biomarker for disease activity. Other researchers have proposed that antibody titers, serum neurofilament light chain (sNfL), and serum CNTN1 levels could be useful for monitoring disease activity [3, 28]. IgG3 and IgG1 can activate complement and contribute to acute conduction block and axonal degeneration, correlating with clinical severity during the acute phase [29], while IgG2 shows little C1q binding and IgG4 completely lacks these Fc-mediated effector functions.
IVIG exerts its therapeutic effect by blocking the Fc receptor region of immunoglobulin antibodies, facilitating antibody clearance. However, CNTN1 antibodies, primarily IgG4 subclass, demonstrate poor IVIG response due to limited Fc binding and complement activation. Early-stage patients may show better responses, correlating with antibody profiles. Activated B lymphocytes generate pathogenic anti-CNTN1 antibodies, making rituximab’s B-cell depletion mechanism an effective therapeutic strategy. While early intervention often leads to sustained remission, chronic cases with established axonal degeneration typically demonstrate poorer clinical outcomes [3–5]. Corticosteroids and plasma exchange are also effective treatment modalities.
The standard rituximab regimen (375 mg/m²/week for 4 weeks) is recommended for membranous nephropathy, with approximately 60–70% of patients achieving long-term clinical remission [30]. However, this regimen is costly and associated with a higher incidence of adverse effects. Low-dose rituximab exhibits limited efficacy in PMN, with remission rates below 50%, whereas it demonstrates rapid and sustained therapeutic benefits in anti-CNTN1 nodopathy [31–33]. In our case, considering the predominant neurological involvement with subclinical renal manifestations (proteinuria 4 + but no edema/frothy urine), we initiated low-dose rituximab. This approach resulted in marked neurological improvement but suboptimal renal response. Importantly, this dissociation between neurological and renal improvement occurred despite confirmed complete B-cell depletion (CD19 + count of 0 cells/µL), underscoring that the persistent renal disease was likely driven by pre-existing antibodies and slow tissue repair rather than an ongoing B-cell response. This observation is consistent with existing reports. In accordance with the KDIGO 2021 Glomerular Disease Guidelines [6], we escalated to a dual-target strategy: tacrolimus for rapid podocyte stabilization via calcineurin inhibition [30], complemented by intensified rituximab to eradicate pathogenic B-cell clones. Upon dose intensification with rituximab and the addition of tacrolimus, proteinuria showed further improvement. Scant literature documents the long-term prognosis of CNTN-AN with MN. Notably, earlier MN presentation correlates with better proteinuria remission [8]. Our case aligns with published evidence demonstrating earlier neurological versus renal recovery post-immunotherapy, with MN remission typically requiring high-dose rituximab and/or combination immunosuppression [4, 28, 31, 34].
CNTN1-AN with concomitant MN
Our patient is an 11-year-old girl presenting with concurrent CNTN1-AN and MN. A study reviewing the clinical features of 35 cases of CNTN1-AN with MN reported up to April 2023 found a mean age of onset of 57.45 ± 2.728 years, with only one case presenting in a patient under 18 years of age [14]. Subsequent studies have reported a rising incidence of CNTN1-AN cases with concurrent MN, with proteinuria identified as a potential biomarker for CNTN1-AN [20]. Therefore, regular monitoring of proteinuria is essential in patients with CNTN1-AN. No statistically significant differences in clinical features or therapeutic outcomes were observed between CNTN1-AN patients with and without MN, except for a higher proportion of acute/subacute onset in the former group. Notably, MN neither exacerbates nor alleviates peripheral neurological symptoms [4].
Anti-CNTN1 Antibody is a potential cause of neuro-renal syndrome. Liu et al. demonstrated that CNTN1 IgG eluted from renal specimens binds to CNTN1 at the node of Ranvier, with IgG4 being the predominant subclass. In contrast, IgG eluted from PLA2R-MN specimens does not exhibit this binding, suggesting that CNTN1 serves as a shared target antigen in both AN and MN [4]. In patients with CNTN1-AN and MN, peripheral nerve symptoms typically precede or coincide with renal manifestations, although rare cases present with MN occurring earlier than AN [8]. The temporal relationship between the onset of these two conditions may depend on whether glomerular damage is primarily mediated by complement activation or results from rapid antibody-mediated disruption of physiological pathways essential for maintaining podocyte foot process integrity. The former mechanism often requires several months to progress to significant proteinuria and nephropathy [35].
The potential mechanisms of CNTN1-mediated renal injury include:①In situ immune complex formation: CNTN1 antibodies bind to CNTN1 expressed on podocyte foot processes, disrupting their normal biological functions.②Exogenous CNTN1 deposition: Circulating CNTN1 implants in the subepithelial space, becoming a target for anti-CNTN1 antibodies.③Circulating immune complex deposition: Immune complexes formed by CNTN1 and anti-CNTN1 antibodies deposit in the subepithelial space. Complement system activation through these pathways induces podocyte injury and glomerular filtration impairment [5]. However, the pathogenesis of CNTN1 antibody-induced neuro-renal syndrome remains incompletely understood. Key unresolved questions include the initial site of CNTN1 exposure to the immune system and the reasons why CNTN1-positive patients develop specific peripheral nerve and/or renal damage.
Conclusion
CNTN1-AN is rare, with MN comorbidity being relatively common. Regular renal monitoring is crucial in CNTN1-AN patients. Pediatric CNTN1-AN with MN is exceptionally uncommon. We present a pediatric case and characterize this entity to enhance clinical recognition and management. Notably, neurological recovery with low-dose rituximab precedes and surpasses renal improvement. Further studies should investigate the differential efficacy of low-dose rituximab in AN versus MN, optimize treatment protocols (including dosing and combination regimens) for CNTN1-AN with MN.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Caballero-Ávila M, Martín-Aguilar L, Pascual-Goñi E et al. Long-Term follow up in Anti-Contactin-1 autoimmune nodopathy [J]. Ann Neurol, 202410.1002/ana.2714210.1002/ana.2714239601182 · doi ↗ · pubmed ↗
- 2Cortese A, Lombardi R, Briani C, et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: clinical relevance of Ig G isotype [J]. Neurol Neuroimmunol Neuroinflamm. 2020;7(1). 10.1212/nxi.0000000000000639.10.1212/NXI.0000000000000639 PMC 693583731753915 · doi ↗ · pubmed ↗
- 3Carrera-García L, Natera-de Benito D, Lleixà C, et al. Chronic inflammatory demyelinating polyneuropathy associated with contactin-1 antibodies in a child [J]. Neurol Neuroimmunol Neuroinflamm. 2019;6(5). 10.1212/nxi.0000000000000602.10.1212/NXI.0000000000000602 PMC 670562331454775 · doi ↗ · pubmed ↗
- 4De Simoni D, Ricken G, Winklehner M, et al. Antibodies to nodal/paranodal proteins in paediatric immune-mediated neuropathy [J]. Neurol Neuroimmunol Neuroinflamm. 2020;7(4). 10.1212/nxi.0000000000000763.10.1212/NXI.0000000000000763 PMC 728665832487720 · doi ↗ · pubmed ↗
- 5Hashimoto Y, Ogata H, Yamasaki R, et al. Chronic Inflammatory Demyelinating Polyneuropathy With Concurrent Membranous Nephropathy: An Anti-paranode and Podocyte Protein Antibody Study and Literature Survey [J]. Front Neurol. 2018;9:997. 10.3389/fneur.2018.00997.10.3389/fneur.2018.00997 PMC 627769930538665 · doi ↗ · pubmed ↗
- 6Grüner J, Stengel H, Werner C, et al. Anti-contactin-1 antibodies affect surface expression and sodium currents in dorsal root ganglia [J]. Neurol Neuroimmunol Neuroinflamm. 2021;8(5). 10.1212/nxi.0000000000001056.10.1212/NXI.0000000000001056 PMC 840715034429341 · doi ↗ · pubmed ↗