Organoruthenium Glycomimetics Exhibit High Selectivity and Nanomolar Affinity for Human Galectin‑1
Vojtěch Hamala, Martin Kurfiřt, Lucie Červenková Št́astná, Filip Dvořák, Jana Bernášková, Adéla Sýkorová, Jaroslav Kozák, Martin Zavřel, Tatiana Staroňová, Peter Šebest, Veronika Ostatná, Jakub Červený, Pavla Bojarová, Jitka Holčáková, Tomáš Hrstka, Roman Hrstka, Jindřich Karban

TL;DR
This paper introduces new ruthenium-based compounds that strongly and selectively inhibit human galectin-1, a protein linked to tumor growth and immune suppression.
Contribution
The study presents highly selective and potent hGal-1 inhibitors with a novel organoruthenium glycomimetic scaffold.
Findings
The inhibitors show nanomolar affinity and 2-3 orders of magnitude selectivity for hGal-1 over hGal-3.
The most effective compound blocks hGal-1 binding to tumor cells and reduces cell viability.
It also suppresses hGal-1-induced phosphatidylserine exposure in Jurkat cells.
Abstract
Human galectin-1 (hGal-1) is an abundant β-galactoside-binding animal lectin that plays an essential role in promoting the immunosuppressive tumor microenvironment. Although hGal-1 has been identified as a promising target for pharmacological inhibition, developing potent and selective hGal-1 inhibitors has been complicated by the high degree of sequence similarity of the glycan-binding site across the galectin family. Herein, we present potent nanomolar hGal-1 inhibitors with unprecedented selectivity of 2 to 3 orders of magnitude over human galectin-3 (hGal-3). Their primary structural feature is the modification of a thiodigalactoside scaffold at the 3- and 3′-positions with a half-sandwich ruthenium(II) arene complex containing a bidentate 4-(2-pyridyl)-1H-1,2,3-triazol-1-yl ligand. The most potent inhibitor in the series efficiently blocked the binding of hGal-1 to the surface of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1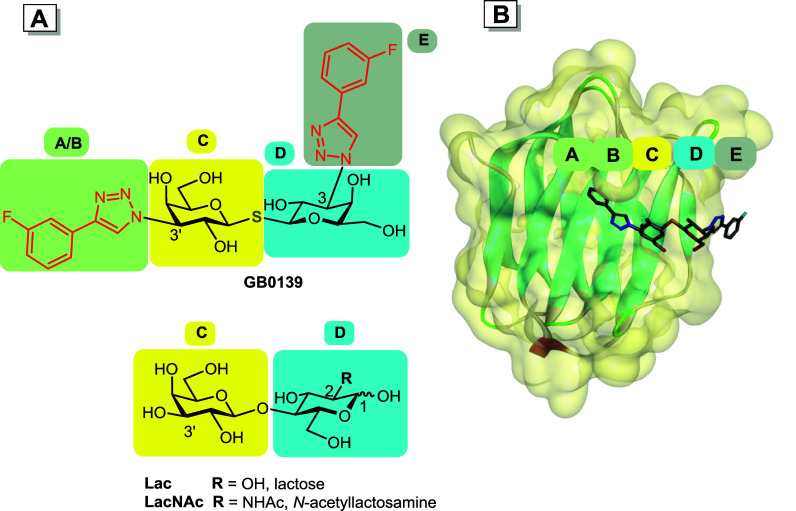 2
2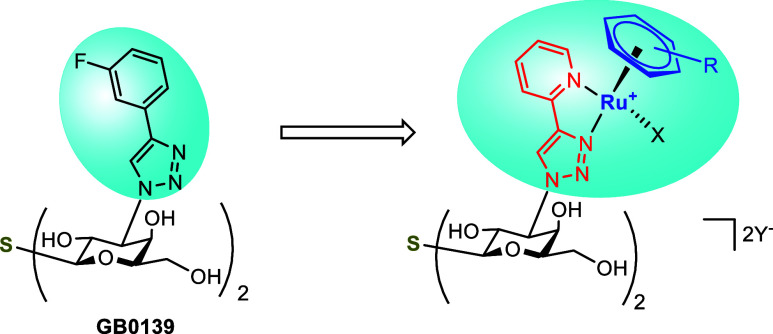 3
3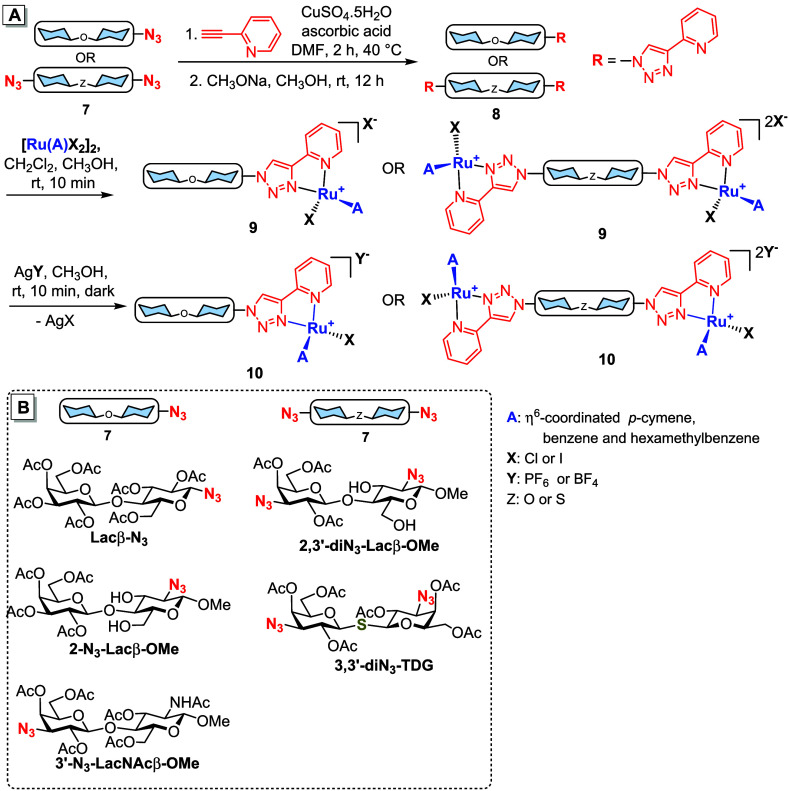 1
1 4
4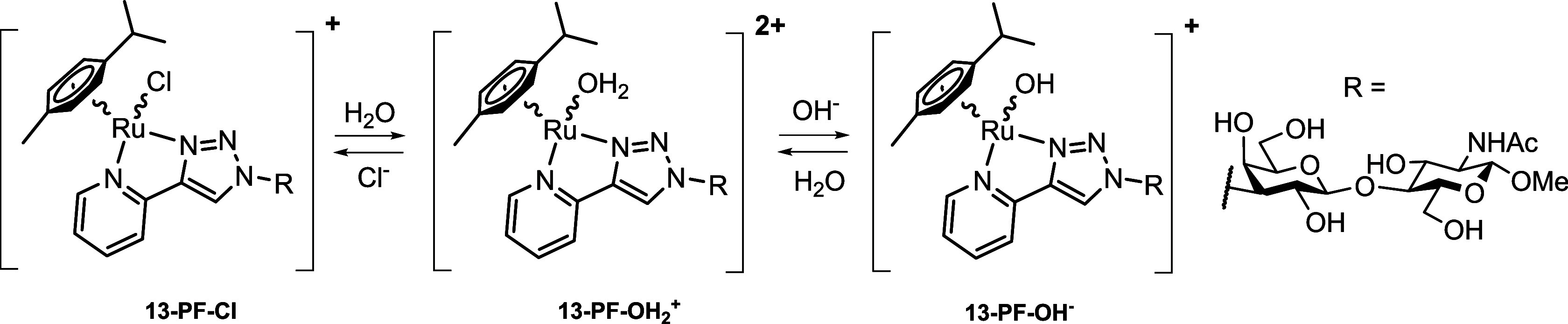 2
2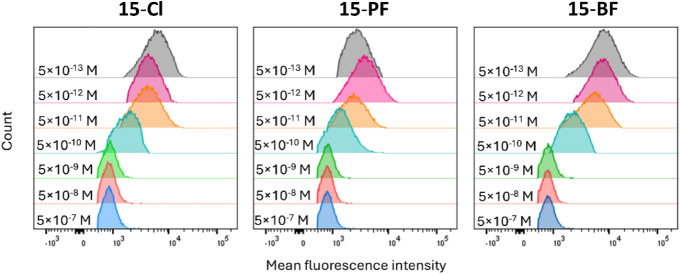 5
5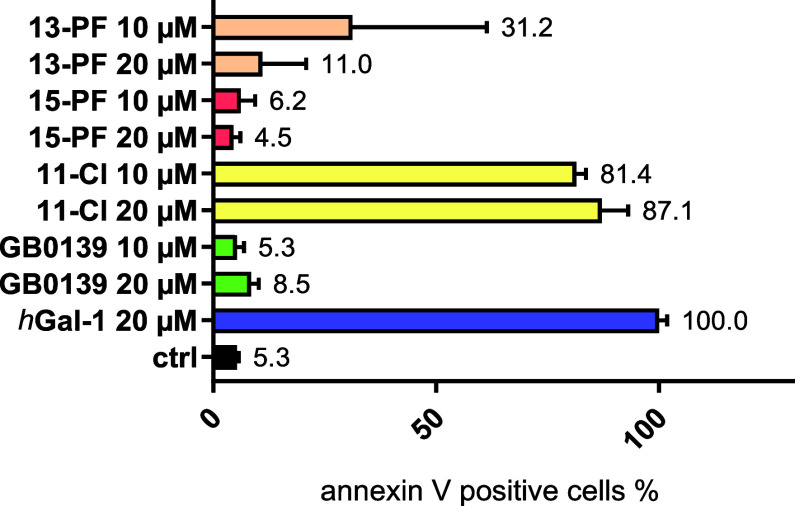 6
6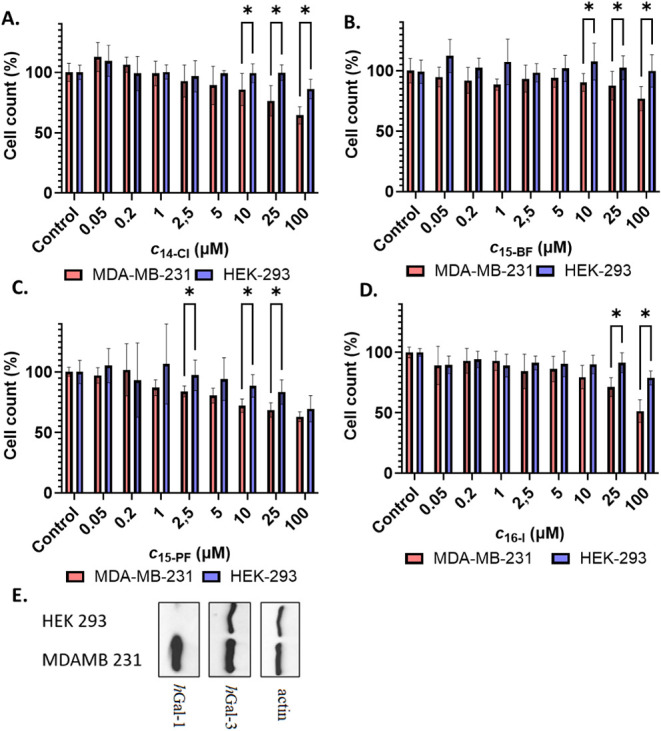 7
7| Compound | R |
|
|
|
| Selectivity
to |
|---|---|---|---|---|---|---|
|
| [3′-FPh] | 0.12 ± 0.01 | 1.0 | 0.008 ± 0.001 | 1.0 | 0.07 |
|
|
| 0.16 ± 0.05 | 0.7 | 0.25 ± 0.02 | 0.032 | 1.6 |
|
|
| 0.11 ± 0.01 | 1.1 | 30 ± 2 | 2.7 × 10–4 | 270 |
|
|
|
|
|
|
|
|
|
|
| 0.070 ± 0.025 | 1.7 | 14 ± 1 | 5.7 × 10–4 | 200 |
|
|
| 0.019 ± 0.011 | 6.3 | 17 ± 1 | 4.7 × 10–4 | 890 |
|
|
| 0.028 ± 0.007 | 4.3 | 2.2 ± 0.3 | 3.6 × 10–3 | 79 |
| 18-Cl |
| 0.23 ± 0.06 | 0.5 | 31 ± 4 | 2.6 × 10–4 | 130 |
| Compounds | R |
|
| Selectivity
to |
|---|---|---|---|---|
|
|
|
|
|
|
|
|
| 0.00094 ± 0.00007 | 3.4 ± 0.7 | 3 600 |
|
|
| 0.0028 ± 0.0002 | 2.8 ± 0.5 | 1 000 |
|
| [ | 0.08 ± 0.01 | 0.0021 ± 0.0004 | 0.03 |
|
|
| 0.0012 ± 0.0001 | 0.010 ± 0.002 | 8.3 |
|
|
| 0.010 ± 0.001 | 0.40 ± 0.10 | 40 |
| Entry | Compound |
|
|
| SS |
|---|---|---|---|---|---|
|
| |||||
| 1 |
| 68 000 ± 29 000 | 22 ± 10 | 0.032 ± 0.018 | 0.037 ± 0.009 |
| 2 |
| 2 052 000 ± 208 000 | 62 ± 15 | 0.003 ± 0.001 | 0.004 ± 0.001 |
| 3 |
| 1 200 000 ± 250 000 | 136 ± 42 | 0.011 ± 0.003 | 0.002 ± 0.002 |
| 4 |
| 1 670 000 ± 540 000 | 55 ± 36 | 0.003 ± 0.002 | 0.003 ± 0.002 |
|
| |||||
| 5 |
| 45 000 ± 12 000 | 1.2 ± 1.0 | 0.003 ± 0.002 | 0.013 ± 0.07 |
| 6 |
| 2590 ± 420 | 265 ± 7 | 10 ± 3 | 15 ± 6 |
| 7 |
| 60 ± 11 | 39 ± 13 | 65 ± 23 | 12 ± 5 |
| 8 |
| 4250 ± 250 | 43 ± 6 | 1 ± 0.1 | 16 ± 6 |
| Entry | Ligand |
|
|
|---|---|---|---|
|
| Lac (standard) | 4.3 ± 0.5 × 106 | 1 |
|
|
| 340 ± 170 | 12 700 |
|
|
| 64 ± 26 | 67 000 |
|
|
| 48 ± 15 | 90 000 |
- —Grantov? Agentura, Univerzita Karlova10.13039/100007543
- —Ministerstvo ?kolstv?, Ml?de?e a Telov?chovy10.13039/501100001823
- —Ministerstvo ?kolstv?, Ml?de?e a Telov?chovy10.13039/501100001823
- —Grantov? Agentura Cesk? Republiky10.13039/501100001824
- —Ministerstvo Zdravotnictv? Cesk? Republiky10.13039/501100003243
- —Ministerstvo Zdravotnictv? Cesk? Republiky10.13039/501100003243
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGalectins and Cancer Biology · Glycosylation and Glycoproteins Research · Brucella: diagnosis, epidemiology, treatment
Introduction
Lectins, a diverse group of glycan-binding proteins found ubiquitously in nature, act in vital processes in human biology, including participation in cell–pathogen interactions, modulation of the inflammatory response, autoimmune diseases, and cancer progression.? Despite their significance in biomedicine, the development of small-molecule inhibitors of lectins has been lagging behind other classes of proteins, ?−? ? ? partially due to the challenges associated with targeting of their shallow water-exposed binding cavity? and the difficulty in achieving selectivity among structurally homologous lectins.? However, recent advances in glycochemistry, aided by X-ray crystallography and NMR spectroscopy, have facilitated the design and synthesis of potent lectin inhibitors,? particularly those based on synthetically modified endogenous carbohydrate lectin ligands (glycomimetics). ?,?
The challenges associated with lectin targeting also apply to galectins, a distinct family of lectins characterized by a high sequence homology and the ability to bind β-galactoside structural motifs in glycans. ?,? Galectins are implicated in pathologies including fibrotic diseases, inflammation, and cancer. ?−? ? In particular, human galectin-1 (hGal-1) and galectin-3 (hGal-3) have been the subject of extensive research due to their widespread tissue expression and biological impact. ?,? The bioactivity of galectins depends not only on their structure but also on their tissue and cellular localization. ?,?,? In addition, different galectins can exert opposing effects on disease progression,? underscoring the need of selective inhibitors of individual galectins.
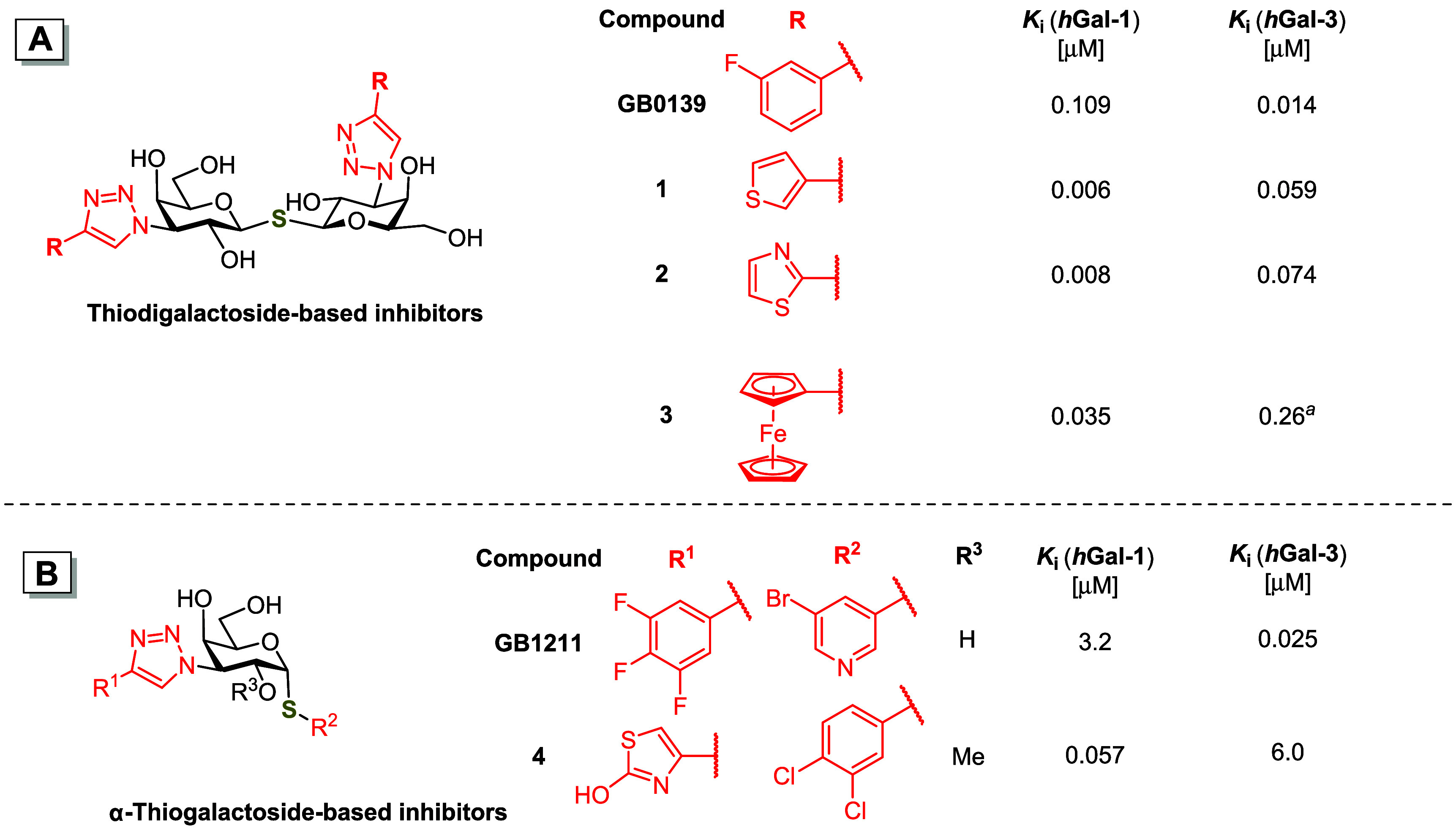
The design of small-molecule glycomimetic galectin inhibitors typically revolves around galactose-containing mono- and disaccharide glycomimetics modified with aromatic or heteroaromatic moieties to enhance interactions in the galectin binding site.? Notably, the inhibitors based on thiodigalactoside (TDG, FigureA) and α-thiogalactoside (FigureB) scaffolds achieved submicromolar affinities. The selective hGal-3 inhibitors GB0139 (also known as TD139)? and GB1211 ? (Figure) have advanced into clinical trials for the treatment of idiopathic pulmonary fibrosis (GB0139),? selected carcinomas (GB1211), and hepatic impairment (GB1211).? Despite significant progress in the development of hGal-3 inhibitors, achieving high potency and selectivity for hGal-1 remains a formidable challenge, notwithstanding the therapeutic promise of hGal-1 inhibition. ?−? ? ? ? ? ? ? ? ? ? The recently reported 3-thienyl and 2-thiazolyl thiodigalactoside analogs 1 and 2 (FigureA) ?,? and 1,3-disubstituted α-thiogalactoside 4 (FigureB)? demonstrated a nanomolar affinity and up to an over 100-fold selectivity for hGal-1 over hGal-3.
Structures and inhibitory activities of previously reported (A) thiodigalactoside-based galectin inhibitors GB0139, 1 , 2, and 3; and (B) α-thiogalactoside-based inhibitors GB1211 and 4 determined by the fluorescence polarization assay (FP) and expressed as inhibition constant K i; a hGal-3 carbohydrate recognition domain (CRD) instead of the full-length hGal-3 was used to determine K i.
To describe and model the interactions of galectins with inhibitors such as GB0139, the binding groove of galectins can be divided into five binding subsites A–E. ?,?,? The key interactions between the conserved subsites C and D and the endogenous disaccharide ligands lactose (Galβ1–4Glc, Lac) and N-acetyllactosamine (Galβ1–4GlcNAc, LacNAc, FigureA) are reproduced in the binding of the thiodigalactoside scaffold of GB0139 to these subsites, whereas the 3-fluorophenyl triazole moieties in GB0139 provide additional interactions with the flanking subsites A, B, and E (Figure), collectively resulting in the high affinities of GB0139 and of the related inhibitors to hGal-3. Assuming that the planarity of the decorating phenyl moieties is not critical for the high-affinity interactions of GB0139 with subsites A, B, and E, we propose that the aryl moieties in GB0139 and related inhibitors could be replaced with three-dimensional arene mimetics? including organotransition metal complexes. ?,? We hypothesized that the three-dimensional nature of organometallic complexes could more efficiently occupy galectin subsites A, B, and E, introducing additional interactions inaccessible to the planar 3-fluorophenyl in GB0139.? In addition, the sterically demanding organotransition metal complexes may better discriminate between the binding subsites of homologous galectins than the more easily accommodated fluorinated phenyl ring.
(A) Inhibitor GB0139 and endogenous ligands Lac and LacNAc divided into segments interacting with the respective galectin subsites A–E, with the 3-fluorophenyl-triazole moiety in red; the numbers indicate positions on the pyranose ring; (B) Subsites A–E shown for the complex of the carbohydrate recognition domain (CRD) of hGal-3 with GB0139 (PDB ID: 5H9P).
We synthesized and tested sandwich complexes of ruthenium and iron and found that the diferrocenyl analog 3 displayed an approximately 3-fold higher affinity to hGal-1 than GB0139, and a 7-fold selectivity for hGal-1 over hGal-3-CRD (FigureA).? These results confirmed the validity of our concept of designing galectin inhibitors, but the affinity and selectivity gains were moderate. Herein, we report that the incorporation of a half-sandwich (piano stool) ruthenium(II) arene complexes containing bidentate 4-(2-pyridyl)-1H-1,2,3-triazol-1-yl moiety (Figure) into the LacNAc or thiodigalactoside scaffolds tremendously improved the selectivity for hGal-1 while not compromising hGal-1 binding affinity. This concept has led to the discovery of potent small-molecule hGal-1 inhibitors with unprecedented selectivity for hGal-1 over hGal-3 that, in addition, could protect T cells against hGal-1-induced preaparesis and prevent hGal-1 from binding to the surface of cancer cells.
Replacement of the 3-fluorophenyl-triazole moiety in thiodigalactoside GB0139 by a half-sandwich ruthenium(II) complex. The 4-(3-fluorophenyl)-triazole and organoruthenium moieties are highlighted, the 4-(2-pyridyl)-1H-1,2,3-triazol-1-yl moiety in red, the π-coordinated arene in blue, R = alkyl(s), X = halogen, Y = counteranion.
Results
Synthesis of hGal-1 Inhibitors
The ruthenium half-sandwich complex was attached to the 1- and 2-positions of disaccharide Lac, the 3′-position of disaccharide LacNAc, and the 3- and 3′-positions of the thiodigalactoside (TDG) scaffold. These positions were selected because their modifications do not interfere with galectin binding. ?,?−? ? ? ? ? First, the 4-(2-pyridyl)-1H-1,2,3-triazole moiety was easily introduced by copper-catalyzed cycloaddition of an acetylated monoazido- or diazido-disaccharide of general structure 7 with 2-ethynylpyridine (Scheme). Base-catalyzed Zemplén deacetylation yielded a disaccharide of general structure 8 that was subjected to complexation with a dihalogenido(arene)ruthenium(II) dimer [Ru(A)X_2_]2 (A = arene, X = halogen) to afford target organoruthenium-modified disaccharide 9 with a chloride or iodide counteranion. A counteranion exchange by the reaction of chloride 9 with AgPF_6_ or AgBF_4_ afforded the corresponding hexafluorophosphate or tetrafluoroborate salts 10 (see Supporting Information for synthetic details). The starting peracetylated azido-disaccharides (SchemeB) were synthesized as previously described, ?,?−? ? ? except for the acetylated methyl 2,3′-diazido-β-lactoside 2,3′-diN _ 3 _ -Lacβ-OMe prepared by chemical glycosylation (see the Supporting Information for details).
(A) General Synthesis of Ruthenium Piano-Stool Glycomimetics (Only One Diastereomer at the Ru Center Is Shown for Clarity); (B) Structures of the Starting Azido-Disaccharides
The target ruthenium complexes 11–13 (Figure) were obtained as a mixture of two diastereomers due to the introduction of a new stereogenic center at the ruthenium atom. Bisruthenium complex 14 was obtained as a mixture of four diastereomers because two new stereogenic centers were introduced to the molecule. Only three diastereomers were formed in the case of bisruthenium complexes 15–18, because the R,S and S,R combinations at the two ruthenium centers were identical due to the C 2 axis of symmetry. These diastereomers could be detected by ^1^H and ^13^C NMR spectroscopy. LacNAc-, Lac-, and TDG-based precursors 19–21 with one or two 4-(2-pyridyl)-1H-1,2,3-triazole moieties uncoordinated to ruthenium were used as reference compounds in affinity evaluation, along with methyl N-acetyl-β-lactosaminide LacNAcβ**-OMe** ? (Figure) and lactose.
Structures of ruthenium glycomimetics (only one diastereomer at the Ru center is shown for clarity), and reference compounds 19–21 and LacNAcβ-OMe. The values in parentheses are the yields of the complexation reaction (including counterion exchange, where applicable).
The organoruthenium complexes were stable in air and well soluble in water, methanol, or DMSO. The most potent inhibitor 15-PF (vide infra) was stable in an aqueous solution containing chloride Cl^–^ ions at standard saline concentrations (c NaCl = 154 mM), including phosphate-buffered saline (PBS buffer) and human blood serum (see the Supporting Information for spectra). The complexes generally undergo an exchange reaction of the halide ligand in aqueous solutions ?,? that were studied in detail for compound 13-PF (Scheme and the Supporting Information, Figure S1). Complex 13-PF undergoes partial aquation-dechlorination in an aqueous solution leading to a dynamic equilibrium between chloro- and aqua-ruthenium species 13-PF-Cl and 13-PF-OH _ 2 _ ^ + ^. Under alkaline conditions, the hydroxy-ruthenium species 13-PF-OH ^–^ predominates (see the Supporting Information, Section B for details). The irreversible substitution of the weakly bound halide ligand with a nucleophilic group from biomolecules can compromise the galectin binding affinity. However, our ^1^H NMR monitoring of complex 15-PF in blood serum suggests that stable coordination bonds with serum proteins do not form to a significant extent.
Equilibrium between Chloro-, Aqua-, and Hydroxy-Ruthenium Species
Affinity to Galectins
Screening of All Galectin Inhibitors by Competitive
Fluorescence Polarization Assay
The affinities of ruthenium glycomimetic inhibitors to hGal-1 and to the carbohydrate recognition domain of hGal-3 (denoted here as hGal-3-CRD) were first determined using a competitive fluorescence polarization (FP) assay, in which a fluorescently labeled high-affinity probe competes with the inhibitor for the galectin binding site.? This assay allows for rapid affinity screening with minimal consumption of the inhibitor and galectin. The inhibition constant K i was calculated and used to estimate the inhibitory activity of compounds (see the Supporting Information, Section D, for details).
The introduction of an organoruthenium moiety at the 1- and 2-positions of Lac-derived compounds (11-Cl, 11-PF, 12-Cl) enhanced the binding affinity to hGal-1 considerably more (up to 20-fold) than to hGal-3-CRD (up to 4.5-fold) compared with the parent unmodified disaccharides, but the found affinities still remained in the micromolar range (Supporting Information, Table S2). In contrast, the attachment of the organoruthenium complex to the 3′-position of LacNAc significantly improved the binding to hGal-1 and selectivity over hGal-3-CRD (Table). Thus, complexes 13-Cl and 13-PF bound hGal-1 approximately 200- and 240-fold more strongly, respectively, than parent LacNAcβ-OMe, while their affinity to hGal-3-CRD was reduced approximately 5-fold, resulting in a selectivity for hGal-1 of 2 orders of magnitude. Both 13-Cl (K i = 0.9 μM) and 13-PF (Ki = 0.75 μM) by far outperformed the corresponding uncoordinated compound 19, previously reported ferrocenyl-trizole complex 22, and 3-fluorophenyl-triazole complex 23 in terms of hGal-1 selectivity.? Counterion exchange (Cl^–^ → PF_6_ ^–^) had only a marginal effect on galectin affinity (cf. 13-PF vs 13-Cl, Table). One additional organoruthenium complex attached to the 2-position of 13-Cl to give 2,3′-bisruthenium complex 14-Cl further increased the binding affinity to both galectins approximately 4-fold, reaching an astounding 200 nM K i for hGal-1 and a 290-fold selectivity over hGal-3. These excellent selectivities are clearly due to the presence of the organoruthenium complex, since 20, an immediate precursor of 14-Cl without the ruthenium complex, was a submicromolar binder of both galectins, moderately selective for hGal-3. The present results demonstrate a noteworthy observation that the attachment of the organoruthenium complex to the 3′-position of LacNAc is by far more beneficial for binding affinity and selectivity for hGal-1 compared to the 1- and 2-positions of the closely related Lac scaffold.
1: Inhibition Constant K i, Relative Potency rp , and Selectivity of Inhibitors Derived from LacNAc Determined by FP
Even much more striking affinity results were achieved in the series of organoruthenium-modified thiodigalactosides (Table). The replacement of the 3- and 3′-fluorophenyl moieties in the state-of-the-art inhibitor GB0139 with 2-pyridyl to give precursor 21 significantly reduced the affinity to hGal-3-CRD, but only slightly to hGal-1. The decrease in hGal-3-CRD affinity is most likely due to the elimination of the orthogonal multipolar fluorine interactions with the polypeptide backbone of hGal-3-CRD.? The complexation of 21 with ruthenium to give the piano-stool complex 15-Cl afforded a similar affinity to hGal-1 as GB0139, but a drastically reduced affinity to hGal-3-CRD by 3 orders of magnitude, producing a highly selective hGal-1 inhibitor (K i = 110 nM, 270-fold selectivity over hGal-3). Obviously, the ruthenium half-sandwich complex strongly discriminates hGal-3-CRD but favors hGal-1 (cf. 15-Cl vs 21).
2: Inhibition Constant K i, Relative Potency rp , and Selectivity of Thiodigalactoside Analogs Determined by FP
Introduction of iodide as a ligand and counteranion instead of chloride (compounds 16-I), or counteranion exchange of chloride for hexafluorophosphate (15-PF), produced approximately a 5-fold increase in the affinity to hGal-1, resulting in an exceptional hGal-1 selectivity of 950-fold for 15-PF over hGal-3. Introducing the tetrafluoroborate counteranion (15-BF) produced affinity and selectivity similar to those of chloride 15-Cl. Replacing the p-cymene ligand in 15-Cl with the sterically less demanding benzene in 17-Cl diminished the selectivity by improving the affinity to hGal-3-CRD more than to hGal-1. The same overall effect was reached in 18-Cl where the bulky hexamethylbenzene substantially reduced binding to hGal-1 in contrast to that of hGal-3-CRD. This may suggest that balancing the steric clashes with the binding site plays an important role in modulating the inhibitory potency. In summary, the substitution of the fluorophenyl moiety in GB0139 with the half-sandwich organoruthenium complexes resulted in potent hGal-1 inhibitors with an outstanding selectivity over hGal-3. The highest selectivity in the series was obtained for compound 15-PF, reaching a K i value of 22 nM with hGal-1. This compound was much more selective than the recently reported small-molecule hGal-1 inhibitors 1–4 (Figure), ?,?−? ? making it the most selective and one of the most potent hGal-1 inhibitors reported to date.
Assessment
of the Affinity of the Best Inhibitors by Intrinsic Tryptophan Fluorescence Assay
In parallel, we determined the binding affinity of the potent hGal-1 inhibitors 15-PF, 15-BF, and 17-Cl and the reference compounds GB0139, 1, and 3 by a complementary method based on ligand-induced changes in the intrinsic fluorescence of tryptophan residues (ITF).? We expressed the affinity as the dissociation constant K D (Table). The K D values obtained by ITF generally indicated stronger hGal-1 binding than the K i values from the FP assay. This discrepancy may be due to the nature of the assays themselves, as the ITF assay is a direct binding assay, unlike the competitive FP assay. The tendency of hGal-1 to form dimers under ITF assay conditions may also play a role.? In any case, the ITF assay confirmed the high affinity of the bisruthenium thiodigalactosides 15-PF, 15-BF, and 17-Cl for hGal-1, and their exceptional selectivity compared to the recently reported inhibitors 1 and 3. ?,? Consistent with the FP assay, the selectivity of the complexes increased in the order 17-Cl < 15-BF < 15-PF. The increased affinity observed for tetrafluoroborate and hexafluorophosphate complexes may reflect their increasing chaotropic character.? In addition, the recently reported tendency of anions to form tight ion pairs with p-cymene ruthenium complexes may also affect the binding affinity of the associated ruthenium cation complex.?
3: Dissociation Constant K D and Selectivity Determined by ITF
Assessment of the Best
Inhibitors by Biolayer Interferometry
To gain further insight into the inhibitory potential of the best-performing inhibitors, the binding affinity of compounds 15-Cl/BF/PF was assessed by biolayer interferometry (BLI, Table). This label-free solid-phase-based technique provides real-time measurements of molecular interactions and, in addition to the dissociation constant K D, it also determines the kinetic parametersassociation rate constant (k a) and dissociation rate constant (k d). The respective galectin carrying a 15-aa AVI-tag (GLNDIFEAQKIEWHE) labeled with a single molecule of biotin is immobilized on a streptavidin-coated biosensor via biotin–streptavidin interactions. To avoid compromising lectin activity, a short peptide linker was inserted between the AVI-tag and hGal-1 sequence as previously published.? In the case of hGal-3, the AVI-tag was placed on the flexible N-terminus. The AVI-tag is monobiotinylated during protein expression by coexpressed BirA biotin ligase. All measurements were evaluated using the one-to-one kinetic model, the simplest model for describing ligand–receptor binding, and were cross-validated with steady-state analysis to ensure data accuracy. Unlike the previous two methods, this method analyzes binding to full-length hGal-3 instead of hGal-3-CRD.
4: Dissociation Constant (K D) and Kinetic Constants k a and k d of Selected Inhibitors Determined by BLI
For hGal-1, all tested complexes exhibited low nanomolar affinity by BLI, with slight variations in K D values depending on the counterion (K _D,15‑Cl _ = 3 nM, K _D,15‑BF _ = 11 nM, K D,15‑PF _ = 3 nM, Table) while GB0139 was up to an order of magnitude weaker inhibitor (K D = 32 nM). For hGal-3, BLI confirmed the previously published high binding affinity of GB0139 and its status as one of the leading compounds (K D = 3 nM). Among the ruthenium-based compounds, 15-PF (with a PF_6 ^–^ counterion) showed the highest affinity to hGal-3 and was therefore less selective than complexes 15-Cl/BF, but all of them had at least 2 orders of magnitude selectivity over hGal-3. Steady-state analysis, however, revealed comparable hGal-3 affinity (K D = 12–16 μM) and 3 orders of magnitude hGal-1 selectivity for all three complexes. While GB0139 had a comparable association rate constant k a for both hGal-1 and hGal-3, the organoruthenium complexes exhibited association rate constants for hGal-1 that were several orders of magnitude higher than those for hGal-3. This suggests that the complex of hGal-3 with the organoruthenium inhibitors forms more slowly because hGal-3 must undergo a conformational change in order to recognize them,? consistent with the presumed steric constraints of the hGal-3 binding site that disfavor binding of organoruthenium inhibitors.
Inhibition
of Galectin Binding to Cancer Cells
Complexes 15-Cl/BF/PF, which demonstrated a high inhibitory potential in galectin-binding assays, were evaluated for their ability to scavenge hGal-1 and prevent its binding to the cell surface. The inhibition of extracellular hGal-1 binding to the cell surface mimics the in vivo behavior of inhibitors and suggests their ability to interfere with hGal-1-mediated signaling pathways. We used the breast cancer cell line MDA-MB-231, which endogenously expresses several galectins, namely hGal-1, hGal-3, and hGal-8. This cell line has been widely reported as a suitable model for in vitro assays investigating galectin-related interactions. ?−? ? For the competitive binding assay, we employed a previously published? hGal-1 protein construct carrying a monobiotinylated AVI-tag, hGal-1-AVI. The biotinylated hGal-1-AVI was selectively detected on the cell surface by flow cytometry using streptavidin–phycoerythrin conjugates.
In the absence of glycomimetics, direct binding assays with increasing concentrations of hGal-1-AVI (Supporting Information, Figure S19A) revealed a clear interaction with the MDA-MB-231 cell surface: The binding curve had a nonsaturated character, which indicates a relatively weak binding of hGal-1-AVI to the natural glycocalyx on MDA-MB-231 cells. Consequently, the IC 50 values obtained for the glycomimetics in this assay were notably lower compared to other techniques, including lactose as the standard inhibitor, as it was easier to detach hGal-1-AVI from the cell surface. All tested glycomimetics exhibited remarkable potency in inhibiting hGal-1 binding, thus preventing it from interacting with the cell surface (Table and Figure). Among the compounds tested, the most effective inhibitors were 15-BF and 15-PF, which exhibited virtually the same inhibition potential with their IC 50 values within the experimental error (IC 50 = 48 ± 15 and 64 ± 26 pM, respectively), while 15-Cl with chloride counterion showed weaker inhibition (IC 50 = 340 ± 170 pM). These results highlight the potential of ruthenium-based glycomimetics to efficiently inhibit the binding of hGal-1 to the cancer cell surface, offering a promising approach for targeting hGal-1-mediated signaling in therapeutic applications.
5: Ability of Thiodigalactoside Analogs 15-Cl/BF/PF to Inhibit Binding of hGal-1-AVI to the Surface of MDA-MB-231 Cells
Overlay of representative flow cytometry histograms of competitive inhibition of the binding of the in vivo biotinylated hGal-1-AVI to the MDA-MB-231 cell surface in the presence of increasing concentrations of 15-Cl/BF/PF complexes. The residual galectins bound to the cell surface were stained by the streptavidin-phycoerythrin conjugate. The corresponding inhibition curves are shown in the Supporting Information, Figure S19B.
Inhibition of hGal-1-Induced Preaparesis in
Jurkat Cells
The hGal-1 protein promotes tumor escape from T cell-dependent antitumor immunity by inducing phosphatidylserine exposure in T cells, a process referred to as preaparesis, that occurs independently of apoptosis. ?−? ? ? We tested the ability of the complexes 11-Cl, 13-PF, 15-PF, 16-I, 17-Cl, and the benchmark inhibitor thiodigalactoside GB0139 to inhibit hGal-1-induced preaparesis in Jurkat cells, a human T-lymphoblasticleukemia cell line,? at molar ratios of inhibitor to hGal-1 of 1:1 and 1:2 (Figure and Figure S20 in the Supporting Information). The commercially available inhibitor GB0139 fully suppressed preaparesis at both tested ratios. The weak inhibitor 11-Cl based on C-1-substituted lactose demonstrated almost no ability to attenuate hGal-1-induced preaparesis. In contrast, the more potent LacNAc-based complex 13-PF almost completely prevented preaparesis at an equimolar ratio with hGal-1 and significantly inhibited it at a ratio of 1:2. The strong TDG-based inhibitors 15-PF, 16-I, and 17-Cl fully blocked preaparesis at both 1:1 and 1:2 ratios. These results demonstrate that organoruthenium inhibitors effectively block propreaparetic signaling in T cells and highlight their potential to counteract tumor immune escape.
Inhibition of hGal-1 (20 μM)-induced preaparesis in Jurkat T cells by glycomimetic inhibitors. Preaparesis features were assessed by flow cytometry after Annexin V-Alexa Fluor 488 staining of phosphatidylserine externalization. The data represent the mean ± standard deviation (SD) of at least three independent experiments and are normalized to the preparesis level caused by hGal-1 (20 μM) alone. The results for complexes 16-I and 17-Cl are shown in Figure S20 in the Supporting Information. The organoruthenium inhibitors and GB0139 alone induced a normalized increase in the Annexin V-positive cell population by a maximum of 8% (Figure S21 in the Supporting Information). Representative histograms of individual samples, including negative controls, are shown in Figures S22 and S23 in the Supporting Information.
Cytotoxicity
The cytotoxicity of all synthesized organometallic compounds and the bispyridyl precursor 21 was evaluated using the MTT assay in the breast cancer cell line MDA-MB-231 and in noncancerous HEK-293 cells, which do not express detectable levels of hGal-1 as confirmed by Western blot analysis (Figure and Figures S24–S32 in the Supporting Information). Compounds 11-Cl/PF, 12-Cl, 13-Cl/PF, 17-Cl, and 21 exhibited negligible cytotoxicity for both cell lines at concentrations up to 100 μM. In contrast, the bisruthenium LacNAc analog 14-Cl and the three most potent hGal-1 inhibitors, 15-BF/PF, and 16-I, reduced the viability of the hGal-1-expressing MDA-MB-231 cells by up to 50% at concentrations typically of 25–100 μM (Figure). Cytotoxicity against HEK-293 cells was negligible for most of the compounds. Only inhibitor 15-Cl induced a slight decrease in the viability of both cell lines (Figure S29 in the Supporting Information). Surprisingly, the hexamethylbenzene inhibitor 18-Cl exhibited a distinct cytotoxicity profile with a higher toxicity for HEK-293 cells than for MDA-MB-231 cells (Figure S31 in the Supporting Information). The higher cytotoxicity of potent hGal-1 inhibitors toward hGal-1-expressing MDA-MB-231 tumor cells might indicate that inhibition of hGal-1, which is overexpressed in these cells, affects signaling or redox homeostasis, thereby slightly increasing the sensitivity to ruthenium complexes. This hypothesis is supported by the negligible cytotoxicity of compound 21 (Supporting Information, Figure S32), which has hGal-1 inhibitory activity comparable to the cytotoxic 14-Cl complex but lacks the ruthenium complex (K _i,14‑Cl _ = 0.20 ± 0.04 μM, K _i,21 _ = 0.16 ± 0.05 μM, see FigureA for cytotoxicity of 14-Cl). Moreover, the application of inhibitors to the hGal-1-negative HEK-293 cell line did not result in any significant cytotoxic effect, except for complex 18-Cl, which further supports our hypothesis. However, other cellular differences between MDA-MB-231 and HEK-293 cells may also influence the compound uptake or susceptibility. Therefore, while our results do not demonstrate a direct mechanistic role of hGal-1 in ruthenium-induced cytotoxicity, they are consistent with the hypothesis that modulation of Gal-1-dependent pathways may influence cellular susceptibility to metal-based compounds.?
(A–D) Cytotoxicity of complexes 14-Cl, 15-BF/PF, and 16-I for the breast cancer MDA-MB-231 cell line and noncancerous HEK-293 cell line after 72 h of incubation. (E) Western blots of hGal-1 and hGal-3 expression in MDA-MB-231 and HEK-293 cell lines. Asterisks indicate statistically significant differences determined by one-way ANOVA (P < 0.01).
Discussion
An important breakthrough in the development of potent selective small-molecule glycomimetic hGal-3 inhibitors was the introduction of an arene-triazole moiety at the 3-position of the galactopyranoside moieties of the LacNAc and thiodigalactoside scaffolds, ?,?,? which led to the discovery of the selective hGal-3 inhibitors, including GB0139.? On the basis of X-ray crystallography and molecular dynamics simulations, ?,? we hypothesized that substituting the arene moieties in GB0139 with a three-dimensional organometallic fragment may improve selectivity and the binding affinity to hGal-1 due to steric constraints of the hGal-3 binding site. Investigation of diferrocenylthiodigalactoside 3 (FigureA)? confirmed this hypothesis, but as its hGal-1 selectivity was still moderate, we sought for another stable and inert organotransition metal complex. Since pseudo-octahedral ruthenium(II) complexes are characterized by high inertness and kinetic stability, ?−? ? we prepared a series of analogs, in which the arene-triazolyl moieties in LacNAc and thiodigalactoside-based inhibitors were substituted with half-sandwich ruthenium(II) arene complexes. The bidentate 4-(2-pyridyl)-1H-1,2,3-triazol-1-yl coordinating ligand in these half-sandwich organoruthenium complexes enhances their stability and inertness,? and mimics the arene-triazole substituents found in potent galectin inhibitors? while the ruthenium atom serves as an inert center that precisely organizes the coordinating ligands in space.
Three different types of binding assays independently revealed that the introduction of the half-sandwich organoruthenium substituent at the 3-position of the galactopyranose unit of both LacNAc and TDG increased the hGal-1 selectivity by at least 2 orders of magnitude: it strongly reduced the inhibitory activity to hGal-3 while improving the hGal-1 inhibitory activity. Comparison with the respective uncoordinated 2-pyridyl-triazole-modified disaccharide analogs (cf. 20 and 21 with 14-Cl and 15-Cl, respectively) shows that while the hGal-1 binding affinity is moderately (14-Cl) or slightly (15-Cl) improved upon complexation with ruthenium, the hGal-3 binding affinity decreases dramatically upon coordination with ruthenium (Tables and ?). The ruthenium complex probably effectively mimics the binding interactions between hGal-1 and 2-pyridyl-triazole moieties found in precursors 20 and 21; however, more importantly, it strongly disfavors binding to hGal-3, which correlates with the three- to four-orders-of-magnitude lower association rate of TDG-based organoruthenium inhibitors to hGal-3 (Table) compared with hGal-1 in BLI. We hypothesize that the three-dimensional organoruthenium complex does not readily fit into the hGal-3 binding site, which must undergo a conformational change to accommodate this complex,? resulting in low association rates and low binding affinity. The decrease in affinity to hGal-3 observed for the 3′-substituted LacNAc analogs 13-Cl and 13-PF compared to their uncoordinated precursor 19 suggests that the A and B subsites in the binding groove particularly discriminate between the two galectins because the modification at the 3′-position of the LacNAc scaffold positions the organoruthenium complex in galectin binding subsites A and B. ?,?,?,?
Counterion exchange with more lipophilic anions further positively modulated the hGal-1 binding affinity in the TDG series; inhibitor 15-PF achieved an unprecedented combination of hGal-1 binding affinity and selectivity, highlighting the potential of organometallic glycomimetics in lectin inhibition. Inert metal complexes have previously been used as protein binders,? notably in a series of potent and selective protein kinase inhibitors based on staurosporin mimetics obtained by substitution of the tetrahydropyran moiety in staurosporin with octahedral ruthenium(II) complexes. ?−? ? ? However, with the exception of our ferrocene-based hGal-1 inhibitors,? to our knowledge, organometallic structures have never been used in the design of lectin inhibitors, though ferrocene moieties in multivalent glycomimetics have been tested as electroactive probes. ?−? ?
A cellular in vitro assay demonstrated that the most potent thiodigalactoside-based organoruthenium inhibitors effectively inhibit the binding of extracellular hGal-1 to the surface of MDA-MB-231 cancer cells in a dose-dependent manner, a prerequisite to counteract protumor hGal-1-mediated signaling. Moreover, inhibitor 15-PF was able to completely suppress hGal-1-induced preaparesis in Jurkat cells, highlighting its potential to block this important mechanism of tumor immune escape. The biological relevance of hGal-1 inhibition extends beyond the preaparesis induction in T cells, because hGal-1 is a key regulator of immune tolerance in the tumor microenvironment, where it promotes T cell exhaustion and expansion of regulatory T cells while excluding cytotoxic lymphocytes from the tumor niche. Inhibiting extracellular hGal-1 binding to glycan structures on T cells or endothelial cells may therefore not only restore T cell viability but also enhance their trafficking and persistence in tumors. Given the capacity of 15-PF to prevent hGal-1-induced preaparesis in vitro, it is plausible that this compound could reprogram the immune landscape in vivo, ultimately potentiating antitumor immunity.
Conclusion
Motivated by the emerging significance of selective potent galectin inhibition in medicinal chemistry research and the urgent need to develop small-molecule inhibitors selective for individual galectins, we have developed an operationally simple synthesis of potent nanomolar inhibitors of hGal-1 that are by 2 to 3 orders of magnitude selective over hGal-3. The unprecedented selectivity was achieved by installing a half-sandwich ruthenium(II) p-cymene complex containing the 4-(2-pyridyl)-1H-1,2,3-triazol-1-yl substituent as a bidentate ligand at the 3- and 3′-positions of the TDG scaffold. The most potent inhibitors not only displayed strong binding to hGal-1 but also effectively prevented the binding of hGal-1 to the tumor cell surface and suppressed hGal-1-induced preaparesis in Jurkat cells, demonstrating the potential of these inhibitors to interfere with protumorigenic hGal-1 signaling pathways. It is envisaged that introducing inert organometallic complexes into glycomimetics will promote the exploration of new dimensions of the glycochemical space and accelerate the discovery of novel potent, selective inhibitors of therapeutically relevant lectins.
Experimental
Section
General Chemistry Methods
Chemicals were used as received. Petroleum ether fraction (with boiling point 40–65 °C) was distilled before use. TLC was carried out with Sigma-Aldrich TLC Silica gel 60 F_254_, and spots were detected by UV detection at 254 nm or visualized with an anisaldehyde solution (EtOH/anisaldehyde/AcOH/H_2_SO_4_ 340:15:4:12.5, v/v). Column chromatography was performed with silica gel 60 (70–230 mesh, Material Harvest). If not stated otherwise, solutions were concentrated under reduced pressure at temperatures below 45 °C. Anhydrous sodium sulfate was used to dry solutions after aqueous workup. NMR spectra were recorded using Bruker Avance 400 (^1^H at 400.1 MHz, ^19^F at 376.4 MHz, ^13^C at 100.6 MHz) at 25 °C. The ^1^H and ^13^C NMR spectra were referenced to the solvent (δ/ppm; δH/δC: CDCl_3_, 7.26/77.16, CD_3_OD, 3.31/49.00, and DMSO-d 6, 2.50/39.52). The ^19^F NMR spectra were referenced to the line of the internal standard hexafluorobenzene (δ/ppm; −163.00 in CDCl_3_, −166.62 in CD_3_OD, and −163.86 in DMSO-d 6). The structural assignment of proton and carbon NMR spectra was made by a combination of 1D and 2D NMR measurements: ^1^H–^1^H gCOSY, ^1^H–^13^C gHSQC, ^1^H–^13^C gHMBC, and ^1^H–^13^C gHSQC TOCSY. HRMS analyses were done using a Bruker MicrOTOF-QIII, using ESI ionization in the positive mode. The purities of the compounds were determined by NMR and HPLC, and they were ≥95%. The copies of HPLC chromatograms for the most potent inhibitors are in the Supporting Information. The ruthenium, copper, silver, phosphor, and boron contents in the final compounds were analyzed using an atomic emission spectrometer with microwave-induced plasma (MP-OES Agilent Technologies, Inc., USA) equipped with an autosampler model SPS 3 (Agilent Technologies, Inc., USA) in a 1% HNO_3_ solution. In all cases, the copper and silver content was below the spectrometer’s detection limit, while the ruthenium, boron, and phosphor content corresponded with the calculated values. See Supporting Information, Schemes S1–S5 for the structures of synthetic intermediates S1–S7.
General Procedure for an
Azide–Alkyne Cycloaddition
Copper(II) sulfate pentahydrate (12 mg, 0.05 mmol), ascorbic acid (8 mg, 0.05 mmol), and 2-ethynylpyridine (1.2–1.5 equiv per reacting azido group) were added into a solution of azido sugar (1 equiv) in DMF (c = 0.1–0.2 M) and the reaction mixture was stirred at 40 °C for 2 h. The reaction mixture was allowed to cool to room temperature; then, it was diluted with DCM (30 mL) and washed with water (30 mL), and the aqueous phase was extracted with DCM (2 × 20 mL). The combined organic phases were dried and concentrated, and the crude product was purified by column chromatography.
General Procedure for Deacetylation
The acetylated compound was dissolved or dispersed in the mixture of DCM and MeOH 1:1 (c ≈ 0.01 M), and a 0.5 M solution of MeONa in MeOH was added dropwise until pH = 9–10. The reaction mixture was stirred for 12–72 h at rt until TLC (DCM/MeOH) indicated the end of the reaction and the presence of only one polar product. The reaction was neutralized with DOWEX 50W(H^+^) ion-exchange resin, filtered, and concentrated. The crude product was purified by column chromatography if necessary.
General Procedure
for the Complexation Reaction
A dark red solution of dihalogenido(arene)ruthenium(II) dimer (0.5 equiv) in DCM (c = 0.05 M) was added to a solution of sugar ligand (1 equiv) in a 1:1 mixture of MeOH/DCM (c = 0.05 M). Almost immediately, a color change from red to yellow occurred. The mixture was stirred at room temperature for 10 min. The reaction mixture was concentrated, and the residue was dissolved in a minimal amount of methanol. The organoruthenium complex precipitated by slow addition of methyl tert-butyl ether as a yellow solid mixture of diastereomers. A sample free from the traces of residual solvents was obtained by dissolution of the product in a small amount of methanol and removal of the residual solvents by coevaporation with chloroform under reduced pressure. The target ruthenium complexes 11–13 were obtained as a mixture of two diastereomers due to the introduction of a new stereogenic center at ruthenium. Bisruthenium complex 14 was obtained as a mixture of four diastereomers because two new stereocenters were introduced into the molecule. Three diastereomers were formed in the case of bisruthenium complexes 15–18, because the R,S and S,R combinations at the two ruthenium centers are identical molecules due to the C 2 axis of symmetry. However, the nuclei related by the C 2 symmetry operation are diastereotopic in this R,S (= S,R) diastereomer, although they are not always distinguishable in NMR spectra. Consequently, some nuclei in samples 15-18, typically the triazole proton, can produce up to four signals at very close but distinct frequencies. Whenever possible, we estimated and reported the ratio of stereoisomers of a given product from the analysis of NMR spectra.
General Procedure for Counterion Exchange
A solution of AgPF_6_ (1.05 equiv) or AgBF_4_ (1.05 equiv) in MeOH (c = 0.02 M) was added dropwise into a solution of organoruthenium-chloride complex in MeOH (c = 0.01 M), and the reaction mixture was stirred at rt for 10 min in the dark, filtered through a syringe filter (VWR Syringe Filter, Nylon, 25 mm, 0.45 μm), and concentrated. The crude product was dissolved in a minimal amount of methanol, and the organoruthenium complex precipitated by slow addition of methyl tert-butyl ether as a yellow solid mixture of diastereomers. A sample free from traces of residual solvents was obtained by the dissolution of the product in a small amount of methanol and removal of the residual solvents by coevaporation with chloroform under reduced pressure.
1-[4-O-(β-d-Galactopyranosyl)-β-d-glucopyranosyl]-4-(pyridin-2-yl)-1H-1,2,3-triazole
(S2)
Copper(II) sulfate pentahydrate (12 mg, 0.05 mmol), ascorbic acid (8 mg, 0.05 mmol), 2-ethynylpyridine (73 μL, 0.72 mmol), and β-lactosyl azide peracetate? (400 mg, 0.60 mmol) were reacted according to the general procedure of azide–alkyne cycloaddition. Column chromatography in EtOAc/PE 2:1 provided O-acetylated 1-(β-lactosyl)-4-(pyridin-2-yl)-1H-1,2,3-triazole (S1, 372 mg, 80%, see the Supporting Information, Scheme S1, for the structure) as a white amorphous solid. ^1^H NMR (CD_3_OD, 400 MHz, H–H COSY): δ 8.62 (s, 1H, CH Tria), 8.59 (ddd, 1H, J = 4.8, 1.8, 1.2 Hz, CH Py), 8.09 (dt, 1H, J = 7.9, 1.2 Hz, CH Py), 7.93 (ddd, 1H, J = 7.9, 7.6, 1.8 Hz, CH Py), 7.19 (ddd, 1H, J = 7.6, 4.8, 1.2 Hz, CH Py), 6.17 (d, 1H, J = 9.2 Hz, H-1), 5.61 (dd, 1H, J = 9.6, 9.2 Hz, H-2), 5.50 (dd, 1H, J = 9.6, 8.2 Hz, H-3), 5.39 (d, 1H, J = 3.4 Hz, H-4′), 5.15 (dd, 1H, J = 10.4, 3.4 Hz, H-3′), 5.06 (dd, 1H, J = 10.4, 7.8 Hz, H-2′), 4.78 (d, 1H, J = 7.8 Hz, H-1′), 4.59 (dd, 1H, J = 11.9, 0.8 Hz, H-6a), 4.23–4.11 (m, 6H, H-4, H-5, H-6b, H-5′, H-6′a, H-6′b), 2.15, 2.10, 2.08 (3 × s, 3 × 3H, Me), 2.07 (2 × s, 2 × 3H, Me), 1.94, 1.85 (2 × s, 2 × 3H, Me). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC): δ 172.4, 172.1, 172.0, 171.7, 171.5, 171.2, 170.6 (7 × CO), 150.6 (CH_Py_, C q(Py)), 149.1 (C q(Tria)), 139.0, 124.8 (2 × CH_Py_), 123.2 (CH_Tria_), 121.7 (CH_Py_), 102.1 (C-1′), 86.6 (C-1), 77.1 (C-4, C-5), 74.2 (C-3), 72.5 (C-3′), 72.2 (C-2), 71.9 (C-5′), 70.7 (C-2′), 68.6 (C-4′), 63.5 (C-6), 62.4 (C-6′), 21.1, 20.71, 20.65, 20.6 (4 × Me), 20.5 (2 × Me), 20.1 (Me). HRMS-ESI (m/z): [M
- H]^+^ calculated for C_33_H_41_N_4_O_17_, 765.2461; found 765.2460.
Compound S2 (see the Supporting Information, Scheme S1, for the structure) was prepared according to the general procedure for deacetylation starting from S1 (200 mg, 0.26 mmol). The reaction mixture was stirred at room temperature overnight. Neutralization with ion-exchange resin DOVEX 50W(H^+^), filtration, and solvent evaporation yielded S2 (122 mg, quantitative, 80% over two steps) as a yellowish amorphous solid, R _ f _ 0.40 (DCM/MeOH 5:1), −21 (c 0.84, MeOH).^1^H NMR (CD_3_OD, 400 MHz, H–H COSY): δ 8.66 (s, 1H, CH Tria), 8.59 (br s, 1H, CH Py), 8.10 (d, 1H, J = 7.7 Hz, CH Py), 7.93 (dd, 1H, J = 7.9, 7.7 Hz, CH Py), 7.40 (br s, 1H, CH Py), 5.75 (d, 1H, J = 9.2 Hz, H-1), 4.46 (d, 1H, J = 7.6 Hz, H-1′), 4.06 (dd, 1H, J = 9.2, 9.0 Hz, H-2), 3.94–3.85 (m, 2H, H-6a, H-6b), 3.83–3.76 (m, 5H, H-3, H-4, H-5, H-4′, H-6′a), 3.74 (dd, 1H, J = 11.5, 4.5 Hz, H-6′b), 3.65 (ddd, 1H, J = 7.4, 4.5, 0.9 Hz, H-5′), 3.61 (dd, 1H, J = 9.8, 7.6 Hz, H-2′), 3.53 (dd, 1H, J = 9.8, 3.3 Hz, H-3′). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 150.6 (CH_Py_), from HMBC 148.6 (C q(Tria)), not detected (C q(Py)), 139.1 (CH_Py_), from HSQC 124.7 (CH_Py_), 123.4 (CH_Tria_), 121.8 (CH_Py_), 105.1 (C-1′), 89.5 (C-1), 79.61/79.58 (C-4/5), 77.1 (C-5′), 76.8 (C-3), 74.8 (C-3′), 73.7 (C-2), 72.5 (C-2′), 70.3 (C-4′), 62.5 (C-6′), 61.5 (C-6). HRMS-ESI (m/z): [M + H]^+^ calculated for C_19_H_27_N_4_O_10_, 471.1722; found 471.1724.
Complex 11-Cl
Complex 11-Cl was prepared according to the general complexation procedure starting from dichloro(p-cymene)ruthenium(II) dimer (26 mg, 0.042 mmol) in DCM (1 mL) and the precursor S2 (40 mg, 0.085 mmol) in a 1:1 MeOH/DCM mixture (2 mL). The product 11-Cl (49 mg, 74%) precipitated from MeOH solution by the addition of methyl tert-butyl ether as a yellow solid mixture of two diastereomers in a ratio of 1:0.8 (^1^H NMR). ^1^H NMR (CD_3_OD, 400 MHz, H–H COSY): δ 9.42 (dd, 2 × 1H, J = 7.4, 1.4 Hz, CH Py), 9.26, 9.24 (2 × s, 2 × 1H, CH Tria), 8.19 (ddd, 2 × 1H, J = 8.1, 5.7, 1.4 Hz, CH Py), 8.12 (dd, 2 × 1H, J = 8.1, 3.4 Hz, CH Py), 7.68 (ddd, 2 × 1H, J = 7.4, 5.7, 1.5 Hz, CH Py), 6.12, 6.07 (2 × m, 2 × 2H, CH _ p‑cym_), 5.95, 5.92 (2 × d, 2 × 1H, J = 9.2 Hz, H-1), 5.90, 5.85 (2 × m, 2 × 2H, CH _ p‑cym_), 4.45 (2 × d, 2 × 1H, J = 7.6 Hz, H-1′), 4.05, 3.98 (2 × dd, 2 × 1H, J = 9.2, 7.8 Hz, H-2), 3.96 (br s, 2 × 2H, H-6a, H-6b), 3.85–3.80 (m, 5 × 2H, H-3, H-4, H-5, H-4′, H-6′a), 3.74 (2 × dd, 2 × 1H, J = 11.4, 4.5 Hz, H-6′b), 3.65–3.60 (m, 2 × 1H, H-5′), 3.61 (dd, 2 × 1H, J = 9.7, 7.6 Hz, H-2′), 3.52 (dd, 2 × 1H, J = 9.7, 3.3 Hz, H-3′), 2.76, 2.74 (2 × hept, 2 × 1H, J = 6.9 Hz, CH _ i‑Pr_), 2.23 (2 × s, 2 × 3H, Me _ p‑cym_), 1.16, 1.15, 1.10, 1.09 (4 × d, 4 × 3H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 156.8 (2CH Py), 149.63, 149.62 (2 × C q(Py)), 148.2, 148.0 (2 × C q(Tria)), 141.5, 127.6 (2 × 2CH_Py_), 125.7, 125.1 (2 × CH_Tria_), 123.6 (2CH_Py_), 106.7, 106.6 (2 × C q_CH i‑Pr_), 105.2, 105.1 (2 × C-1′), 104.1, 103.8 (2 × C q_Me), 91.0, 90.9 (2 × C-1), 87.4 (2CH p‑cym_), 86.2, 86.0, 85.55, 85.49, 84.9, 84.7 (6 × CH_ p‑cym_), 80.1, 80.0 (2 × C-5), 79.5 (2 × C-4), 77.24, 77.23 (2 × C-5′), 76.64, 76.59 (2 × C-3), 74.86, 74.85 (2 × C-3′), 74.1, 73.9 (2 × C-2), 72.5 (2 × C-2′), 70.3 (2 × C-4′), 62.6 (2 × C-6), 61.6, 61.3 (2 × C-6′), 32.31, 32.26 (2 × CH_ i‑Pr_), 22.60, 22.56, 21.93, 21.87 (2 × Me _ i‑Pr_), 18.8, 18.7 (2 × Me _ p‑cym_). HRMS-ESI (m/z): [M – Cl]^+^ calculated for C_29_H_40_ClN_4_O_10_Ru, 741.1476; found 741.1486.
Complex 11-PF
Complex 11-PF was prepared according to the general procedure for counterion exchange starting from complex 11-Cl (30 mg, 0.039 mmol) and AgPF_6_ (10 mg, 0.039 mmol). The product 11-PF precipitated by slow addition of methyl tert-butyl ether as a yellow solid mixture of two diastereomers in a ratio of 1:0.9 (26 mg, 76%). ^1^H NMR (CD_3_OD, 400 MHz, H–H COSY): δ 9.41 (ddd, 2 × 1H, J = 5.7, 1.3, 1.6 Hz, CH Py), 9.23, 9.21 (2 × s, 2 × 1H, CH Tria), 8.19 (ddd, 2 × 1H, J = 7.9, 7.6, 1.3 Hz, CH Py), 8.11 (ddd, 2 × 1H, J = 7.9, 3.2, 1.6 Hz, CH Py), 7.68 (ddd, 2 × 1H, J = 7.4, 5.7, 1.5 Hz, CH Py), 6.12, 6.06 (2 × m, 2 × 2H, CH _ p‑cym_), 5.94, 5.91 (2 × d, 2 × 1H, J = 9.2 Hz, H-1), 5.90, 5.84 (2 × m, 2 × 2H, CH _ p‑cym_), 4.46 (2 × d, 2 × 1H, J = 7.6 Hz, H-1′), 4.04, 3.98 (2 × dd, 2 × 1H, J = 9.2, 8.4 Hz, H-2), 3.96 (br s, 2 × 2H, H-6), 3.86–3.80 (m, 4 × 2H, H-3, H-4, H-5, H-4′), 3.82 (2 × dd, 2 × 1H, J = 11.4, 7.2 Hz, H-6′a), 3.74 (2 × dd, 2 × 1H, J = 11.4, 4.5 Hz, H-6′b), 3.65–3.60 (m, 2 × 1H, H-5′), 3.61 (dd, 2 × 1H, J = 9.7, 7.6 Hz, H-2′), 3.53 (dd, 2 × 1H, J = 9.7, 3.3 Hz, H-3′), 2.76, 2.74 (2 × hept, 2 × 1H, J = 6.9 Hz, CH _ i‑Pr_), 2.22 (2 × s, 2 × 3H, Me _ p‑cym_), 1.16, 1.15 (2 × d, 2 × 3H, J = 6.9 Hz, Me _ i‑Pr_), 1.09 (2 × d, 2 × 3H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 156.8 (2 × CH Py), 149.6, 149.5 (2 × C q(Py)), 148.2, 148.0 (2 × C q(Tria)), 141.5, 127.7 (2 × 2CH_Py_), 125.6, 125.1 (2 × CH_Tria_), 123.6 (2 × CH_Py_), 106.7, 106.6 (2 × C q_CH i‑Pr_), 105.13, 105.11 (2 × C-1′), 104.0, 103.8 (2 × C q_Me), 90.9, 90.8 (2 × C-1), 87.33, 87.32 (2 × CH p‑cym_), 86.2, 86.0, 85.6, 85.5, 84.9, 84.7 (6 × CH_ p‑cym_), 80.04, 79.99 (2 × C-5), 79.46, 79.45 (2 × C-4), 77.20, 77.18 (2 × C-5′), 76.6, 76.5 (2 × C-3), 74.8 (2 × C-3′), 74.1, 73.9 (2 × C-2), 72.5 (2 × C-2′), 70.3 (2 × C-4′), 62.58, 62.56 (2 × C-6), 61.34, 61.31 (2 × C-6′), 32.29, 32.25 (2 × CH_ i‑Pr_), 22.58, 22.55, 21.93, 21.88 (2 × Me _ i‑Pr_), 18.8, 18.7 (2 × Me _ p‑cym_). ^19^F NMR (CD_3_OD, 376 MHz): δ −76.03 (d, ^1^ J (F–P) = 707.6 Hz). ^31^P NMR (CD_3_OD, 162 MHz): δ −161.48 (hept, ^1^ J (F–P) = 707.6 Hz). HRMS-ESI (m/z): [M – PF_6_]^+^ calculated for C_29_H_40_ClN_4_O_10_Ru, 741.1476; found 741.1479.
Methyl 2-Deoxy-4-O-(β-d-galactopyranosyl)-2-[4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl]-β-d-glucopyranoside
(S3)
Copper(II) sulfate pentahydrate (12 mg, 0.048 mmol), ascorbic acid (8 mg, 0.045 mmol), 2-ethynylpyridine (15 μL, 0.15 mmol), and methyl 2-azido-2-deoxy-4-O-(2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl)-β-d-glucopyranoside? (65 mg, 0.12 mmol) reacted according to the general procedure for azide–alkyne cycloaddition. The acetyl-protected intermediate obtained after aqueous workup and concentration was not chromatographically purified but immediately subjected to deacetylation according to the general procedure. Column chromatography in DCM/MeOH 6:1 afforded product S3 (45 mg, 79%, see the Supporting Information, Scheme S2, for the structure) as a colorless gel, R f 0.15 (DCM/MeOH 7:1), −144 (c 0.30, MeOH). ^1^H NMR (CD_3_OD, 400 MHz, COSY): δ 8.58 (ddd, 1H, J = 4.7, 1.8, 1.2 Hz, CH Py), 8.49 (s, 1H, CH Tria), 8.08 (dt, 1H, J = 8.0, 1.2 Hz, CH Py), 7.92 (ddd, 1H, J = 8.0, 7.6, 1.8 Hz, CH Py), 7.37 (ddd, 1H, J = 7.6, 4.7, 1.8 Hz, CH Py), 4.99–4.97 (m, 1H, H-1), 4.44 (d, 1H, J = 7.5 Hz, H-1′), 4.36–4.34 (m, 2H, H-2, H-3), 4.01 (dd, 1H, J = 12.3, 2.7 Hz, H-6a), 3.97 (dd, 1H, J = 12.3, 3.8 Hz, H-6b), 3.83–3.79 (m, 1H, H-4), 3.80 (dd, 1H, J = 3.3, 1.0 Hz, H-4′), 3.74 (dd, 1H, J = 11.4, 7.7 Hz, H-6′a), 3.69 (ddd, 1H, J = 10.0, 3.8, 2.7 Hz, H-5), 3.66 (dd, 1H, J = 11.4, 4.5 Hz, H-6′b), 3.60 (ddd, 1H, J = 7.7, 4.5, 1.0 Hz, H-5′), 3.57 (dd, 1H, J = 9.9, 7.5 Hz, H-2′), 3.50 (dd, 1H, J = 9.9, 3.3 Hz, H-3′), 3.42 (s, 3H, OMe). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC): δ 151.1 (C q(Py)), 150.5 (CH_Py_), 148.1 (C q(Tria)), 138.9 (CH_Py_), 125.4 (CH_Tria_), 124.5, 121.5 (2 × CH_Py_), 105.3 (C-1′), 102.7 (C-1), 81.0 (C-4), 77.2 (C-5), 76.8 (C-5′), 74.8 (C-3′), 74.1 (C-3), 72.6 (C-2′), 70.3 (C-4′), 67.9 (C-2), 62.5 (C-6′), 61.7 (C-6), 57.4 (OMe). HRMS-ESI (m/z): [M + H]^+^ calculated for C_20_H_29_N_4_O_10_, 485.1878; found 485.1877.
Complex 12-Cl
Complex 12-Cl was prepared according to the general complexation procedure starting from dichloro(p-cymene)ruthenium(II) dimer (20 mg, 0.033 mmol) in DCM (1 mL) and the precursor S3 (31 mg, 0.064 mmol) in a 1:1 MeOH/DCM mixture (2 mL). The product 12-Cl (43 mg, 85%) precipitated from MeOH solution by addition of methyl tert-butyl ether as a yellow solid mixture of two diastereomers (denoted here as “a” and “b”) in a ratio of 3:2. ^1^H NMR (CD_3_OD, 400 MHz, H–H COSY): δ 9.41–9.40 (m, 2 × 1H, CH Py), 9.06 (2 × s, 2 × 1H, CH Tria), 8.19–8.14, 8.11–8.08, 7.68–7.63 (3 × m, 3 × 2 × 1H, CH Py), 6.20–6.14, 6.02–6.01, 5.97–5.93, 5.79–5.76 (4 × m, 4 × 2H, CH _ p‑cym_), 5.18 (d, 1H, J = 8.3 Hz, H-1a), 4.99–4.97 (m, 1H, H-1b), 4.55–4.50 (m, 3H, H-2a, H-2b, H-3b), 4.47 (d, 1H, J = 7.6 Hz, H-1′a/b), 4.46 (d, 1H, J = 7.5 Hz, H-1′a/b), 4.36 (dd, 1H, J = 10.6, 8.2 Hz, H-3a), 4.06–3.97 (m, 4H, H-6aa, H-6ab, H-6ba, H-6bb), 3.90–3.81 (m, 4H, H-4a, H-4b, H-4′a, H-4′b), 3.79–3.50 (m, 12H, H-5a, H-5b, H-2′a, H-2′b, H-3′a, H-3′b, H-5′a, H-5′b, H-6′aa, H-6′ba, H-6′ab, H-6′bb), 3.48, 3.41 (2 × s, 2 × 3H, OMe), 2.76–2.65 (m, 2H, CH _ i‑Pr_), 2.25, 2.23 (2 × s, 2 × 3H, Me _ p‑cym_), 1.17, 1.15, 1.05, 1.01 (4 × d, 4 × 3H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 156.8, 156.7 (2 × CH Py), 149.72, 149.67 (2 × C q(Py)), 147.6, 147.5 (2 × C q(Tria)), 141.4, 127.4 (2 × 2CH_Py_), 127.3, 127.2 (2 × CH_Tria_), 123.56, 123.55 (2 × CH_Py_), 106.4, 106.0 (2 × C q_CH i‑Pr_), 105.4, 105.3 (C-1′a, C-1′b), 104.8, 104.2 (2 × C q_Me), 102.2 (C-1b), 101.9 (C-1a), 87.3 (2 × CH p‑cym_), 86.1, 86.0, 85.4, 85.2, 84.6, 84.3 (6 × CH_ p‑cym_), 81.0 (C-4a), 80.9 (C-4b), 77.2, 77.1 (C-5′a, C-5′b), 76.9 (C-5a, C-5b), 74.8 (C-3′a, C-3′b), 74.0 (C-3a), 73.5 C-3b), 72.61, 72.59 (C-2′a, C-2′b), 70.23, 70.20 (C-4′a, C-4′b), 69.80, 69.78 (C-2a, C-2b), 62.5 (C-6′a, C-6′b), 61.6, 61.5 (C-6a, C-6b), 57.6 (OMe-b), 57.4 (OMe-a), 32.29, 32.27 (2 × CH_ i‑Pr_), 22.64, 22.56, 21.84, 21.80 (2 × Me _ i‑Pr_), 18.9, 18.8 (2 × Me _ p‑cym_). HRMS-APCI (m/z): [M – Cl]^+^ calculated for C_30_H_42_N_4_O_10_Cl Ru, 755.1633; found 755.1632.
Methyl 4-O-{3-[4-(Pyridin-2-yl)-1H-1,2,3-triazol-1-yl]-3-deoxy-β-d-galactopyranosyl}-2-acetamido-2-deoxy-β-d-glucopyranoside (19)
Ac_2_O (5.0 mL, 52.9 mmol) was added to a solution of methyl 4-O-(3-azido-3-deoxy-β-d-galactopyranosyl)-2-acetamido-2-deoxy-β-d-glucopyranoside? (135 mg, 0.32 mmol) in pyridine (7 mL), and the mixture was stirred at room temperature overnight. The reaction mixture was concentrated, and the residual volatiles were removed by coevaporation with toluene (3×). The residue was then dissolved in DMF (2 mL), copper(II) sulfate pentahydrate (12 mg, 0.05 mmol), ascorbic acid (8 mg, 0.05 mmol), and 2-ethynylpyridine (39 μL, 0.39 mmol) were added, and the reaction mixture was stirred at 40 °C for 2 h. The reaction mixture was allowed to cool to rt, and then it was diluted with DCM (30 mL) and washed with water (30 mL); the aqueous phase was extracted twice with DCM (20 mL). The combined organic phases were dried and concentrated. Column chromatography in EtOAc provided acetylated intermediate S4 (209 mg, 89%, see the Supporting Information, Scheme S3, for the structure) as a white amorphous solid, R _ f _ 0.35 (EtOAc). ^1^H NMR (CDCl_3_, 400 MHz, H–H COSY): δ 8.54 (d, 1H, J = 4.7 Hz, CH Py), 8.22 (s, 1H, CH Tria), 8.09 (d, 1H, J = 7.9 Hz, CH Py), 7.74 (dd, 1H, J = 7.9, 7.6 Hz, CH Py), 7.20 (dd, 1H, J = 7.6, 4.7 Hz, CH Py), 5.95 (d, 1H, J = 9.4 Hz, NH), 5.64 (dd, 1H, J = 11.5, 7.6 Hz, H-2′), 5.52 (d, 1H, J = 3.3 Hz, H-4′), 5.16 (dd, 1H, J = 11.5, 3.3 Hz, H-3′), 5.12 (dd, 1H, J = 9.4, 8.6 Hz, H-3), 4.70 (d, 1H, J = 7.6 Hz, H-1′), 4.50 (dd, 1H, J = 11.9, 2.7 Hz, H-6a), 4.40 (d, 1H, J = 7.6 Hz, H-1), 4.17 (dd, 1H, J = 11.9, 5.2 Hz, H-6b), 4.13 (br s, 3H, H-5′, H-6′a, 6′b), 4.07 (td, 1H, J = 9.4, 7.6 Hz, H-2), 3.85 (dd, 1H, J = 9.0, 8.6 Hz, H-4), 3.67 (ddd, 1H, J = 9.0, 5.2, 2.7 Hz, H-5), 3.45 (s, 3H, OMe), 2.11, 2.09, 2.08, 2.05, 1.97, 1.87 (6 × s, 6 × 3H, Me Ac). ^13^C{^1^H} NMR (CDCl_3_, 101 MHz, HSQC, HMBC): δ 170.9, 170.6, 170.54, 170.46 (4 × CO), 169.2 (2CO), 149.8 (C q), 149.6 (CH_Py_), 148.7 (C q(Tria)), 137.0, 123.2 (2 × CH_Py_), 121.6 (CH_Tria_), 120.3 (CH_Py_), 101.9 (C-1), 101.4 (C-1′), 75.8 (C-4), 72.7 (C-5), 72.5 (C-3), 72.1 (C-5′), 68.3/68.2.6 (C-2′/4′), 62.5 (C-6), 62.1 (C-3′), 61.2 (C-6′), 56.8 (OMe), 53.2 (C-2), 23.4, 21.00, 20.97, 20.8, 20.5, 20.4 (6 × Me Ac). HRMS-ESI (m/z): [M + H]^+^ calculated for C_32_H_42_N_5_O_15_, 736.2672; found 736.2669.
Compound 19 was prepared according to the general procedure for deacetylation starting from intermediate S4 (209 mg, 0.28 mmol). The reaction mixture was stirred at room temperature overnight. Neutralization with acidic ion-exchange resin DOVEX 50W(H^+^), filtration, and solvent evaporation yielded compound 19 (149 mg, quantitative yield for this step, 89% overall yield over both steps) as a yellowish amorphous solid, R _ f _ 0.35 (DCM/MeOH 3:1), −11 (c 0.42, MeOH). ^1^H NMR (CD_3_OD, 400 MHz, COSY): δ 8.56 (br s, 2H, CH Py, CH Tria), 8.10 (d, 1H, J = 6.0 Hz, CH Py), 7.93 (dd, 1H, J = 6.0, 8.4 Hz, CH Py), 7.38 (br s, 1H, CH Py), 4.90 (dd, 1H, J = 11.1, 3.2 Hz, H-3′), 4.68 (d, 1H, J = 7.5 Hz, H-1′), 4.35 (d, 1H, J = 8.2 Hz, H-1), 4.22 (dd, 1H, J = 11.1, 7.5 Hz, H-2′), 4.09 (d, 1H, J = 3.1 Hz, H-4′), 3.95 (dd, 1H, J = 12.2, 2.5 Hz, H-6a), 3.91–3.87 (m, 2H, H-6b, H-5′), 3.80 (dd, 1H, J = 11.4, 7.1 Hz, H-6′a), 3.76 (dd, 1H, J = 10.4, 8.2 Hz, H-2), 3.75–3.64 (m, 3H, H-3, H-4, H-6′b), 3.47 (s, 3H, OMe), 3.43 (ddd, 1H, J = 9.4, 4.2, 2.5 Hz, H-5), 1.98 (s, 3H, Me Ac). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC): δ 173.6 (CO), from HMBC 151.3 (C q(Py)), 150.4 (CH_Py_), from HMBC 148.1 (C q(Tria)), 139.0 (CH_Py_), from HSQC 124.4 (CH_Py_), 123.8 (CH_Tria_), from HSQC 121.7 (CH_Py_), 105.3 (C-1′), 103.6 (C-1), 80.7 (C-4), 77.9 (C-5′), 76.6 (C-5), 74.4 (C-3), 69.5/69.4 (C-2′/4′), 67.7 (C-3′), 62.3 (C-6′), 61.7 (C-6), 57.1 (OMe), 56.6 (C-2), 22.9 (Me Ac). HRMS-ESI (m/z): [M + Na]^+^ calculated for C_22_H_31_N_5_O_10_Na, 548.1963; found 548.1971.
Complex 13-Cl
Complex 13-Cl was prepared according to the general complexation procedure starting from dichloro(p-cymene)ruthenium(II) dimer (18 mg, 0.029 mmol) in DCM (1 mL) and disaccharide 19 (31 mg, 0.059 mmol) in a 1:1 MeOH/DCM mixture (2 mL). The product 13-Cl (42 mg, 86%) precipitated from the MeOH solution by the addition of methyl tert-butyl ether as a yellow solid mixture of two diastereomers in a ratio of 1:0.6. Major diastereomer: ^1^H NMR (CD_3_OD, 400 MHz, COSY): δ 9.42–9.40 (m, 1H, CH Py), 9.13 (s, 1H, CH Tria), 8.20–8.14 (m, 2H, CH Py), 7.68–7.64 (m, 1H, CH Py), 6.12, 6.03, 5.92, 5.81 (4 × m, 4 × 1H, CH _ p‑cym_), 5.16 (dd, 1H, J = 11.1, 3.1 Hz, H-3′), 4.74 (d, 1H, J = 7.6 Hz, H-1′), 4.38 (d, 1H, J = 8.2 Hz, H-1), 4.27 (dd, 1H, J = 11.1, 7.6 Hz, H-2′), 4.15 (dd, 1H, J = 3.1, 1.1 Hz, H-4′), 3.97–3.88 (m, 3H, H-6a, H-6b, H-5′), 3.85–3.65 (m, 5H, H-2, H-3, H-4, H-6′a, H-6′b), 3.47 (s, 3H, OMe), 3.45 (dt, 1H, J = 9.6, 3.3 Hz, H-5), 2.78–2.71 (m, 1H, CH _ i‑Pr_), 2.23 (s, 3H, Me _ p‑cym_), 1.99 (s, 3H, Me Ac), 1.16, 1.06 (2 × d, 2 × 3H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 172.3 (CO), 155.32 (CH py), 148.60 (C q(Py)), 146.3 (C q(Tria)), 140.01, 125.9 (2 × CH_Py_), 124.5 (CH_Tria_), 122.0 (CH_Py_), 105.02 (C q_CH i‑Pr_), 103.68 (C-1′), 102.53 (C q_Me), 102.15 (C-1), 85.9, 84.5, 84.1, 83.3 (4 × CH p‑cym_), 79.3 (C-4), 76.4 (C-5′), 75.18 (C-5), 72.9 (C-3), 68.2 (C-3′), 67.9 (C-2′), 67.7 (C-4′), 60.73 (C-6′), 60.4 (C-6), 55.7 (OMe), 55.5 (C-2), 30.8 (CH_ i‑Pr_), 21.5 (Me Ac), 21.2, 20.4 (2 × Me _ i‑Pr_), 17.3 (Me _ p‑cym_). Minor diastereomer: ^1^H NMR (CD_3_OD, 400 MHz, H–H COSY): δ 9.42–9.40 (m, 1H, CH Py), 9.09 (s, 1H, CH Tria), 8.20–8.14 (m, 2H, CH Py), 7.68–7.64 (m, 1H, CH Py), 6.11, 6.06, 5.89, 5.83 (4 × m, 4 × 1H, CH _ p‑cym_), 5.09 (dd, 1H, J = 11.0, 2.9 Hz, H-3′), 4.73 (d, 1H, J = 7.5 Hz, H-1′), 4.37 (d, 1H, J = 8.2 Hz, H-1), 4.34 (dd, 1H, J = 11.0, 7.5 Hz, H-2′), 4.20 (dd, 1H, J = 2.9, 1.1 Hz, H-4′), 3.97–3.88 (m, 3H, H-6a, H-6b, H-5′), 3.85–3.65 (m, 5H, H-2, H-3, H-4, H-6′a, H-6′b), 3.47 (s, 3H, OMe), 3.44 (dt, 1H, J = 9.6, 3.3 Hz, H-5), 2.78–2.71 (m, 1H, CH _ i‑Pr_), 2.22 (s, 3H, Me _ p‑cym_), 1.99 (s, 3H, Me Ac), 1.15, 1.09 (2 × d, 2 × 3H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 172.3 (CO), 155.29 (CH Py), 148.63 (C q(Py)), 146.2 (C q(Tria)), 140.00, 125.9 (2 × CH_Py_), 124.8 (CH_Tria_), 122.0 (CH_Py_), 105.01 (C q_CH i‑Pr_), 103.70 (C-1′), 102.46 (C q_Me), 102.16 (C-1), 86.0, 84.6, 84.0, 83.2 (4 × CH p‑cym_), 79.2 (C-4), 76.2 (C-5′), 75.20 (C-5), 73.0 (C-3), 68.5 (C-3′), 68.0 (C-4′), 67.5 (C-2′), 60.69 (C-6′), 60.3 (C-6), 55.7 (OMe), 55.4 (C-2), 30.9 (CH_ i‑Pr_), 21.5 (Me Ac), 21.2, 20.5 (2 × Me _ i‑Pr_), 17.4 (Me _ p‑cym_). HRMS-ESI (m/z): [M – Cl]^+^ calculated for C_32_H_45_ClN_5_O_10_Ru, 796.1899; found 796.1904.
Complex 13-PF
Complex 13-PF was prepared according to the general procedure for counterion exchange starting from 13-Cl (21 mg, 0.025 mmol) and AgPF_6_ (7 mg, 0.028 mmol). The product 13-PF precipitated by the slow addition of methyl tert-butyl ether as a yellow solid (20 mg, 84%) mixture of two diastereomers in a ratio of 1:0.6. Major diastereomer: ^1^H NMR (DMSO-d 6, 400 MHz, COSY): δ 9.47–9.46 (m, 1H, CH Py), 9.39 (s, 1H, CH Tria), 8.30–8.22 (m, 2H, CH Py), 7.84 (d, 1H, J = 7.5 Hz, NH), 7.70–7.66 (m, 1H, CH Py), 6.14, 6.07, 6.02, 5.86 (4 × m, 4 × 1H, CH _ p‑cym_), 5.86 (d, 1H, J = 6.2 Hz, OH-2′), 5.73 (d, 1H, J = 6.5 Hz, OH-4′), 5.18 (dd, 1H, J = 11.0, 3.1 Hz, H-3′), 4.89 (t, 1H, J = 5.0 Hz, OH-6′), 4.76 (t, 1H, J = 6.2 Hz, OH-6), 4.67 (d, 1H, J = 7.5 Hz, H-1′), 4.61 (d, 1H, J = 2.1 Hz, OH-3), 4.28 (d, 1H, J = 7.6 Hz, H-1), 4.14–4.04 (m, 1H, H-2′), 3.98 (dd, 1H, J = 6.5, 3.1 Hz, H-4′), 3.85 (t, 1H, J = 5.6 Hz, H-5′), 3.80–3.76 (m, 1H, H-6a), 3.71–3.65 (m, 1H, H-6b), 3.60–3.46 (m, 5H, H-2, H-3, H-4, H-6′a, H-6′b), 3.35 (s, 3H, OMe), 3.33–3.29 (m, 1H, H-5), 2.70–2.61 (m, 1H, CH _ i‑Pr_), 2.14 (s, 3H, Me _ p‑cym_), 1.81 (s, 3H, Me Ac), 1.07, 0.93 (2 × d, 2 × 3H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (DMSO-d 6, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 168.9 (CO), 155.7 (CH py), 148.1 (C q(Py)), 145.6 (C q(Tria)), 140.2, 125.9 (2 × CH_Py_), 125.4 (CH_Tria_), 122.0 (CH_Py_), 104.1 (C q_CH i‑Pr_), 103.5 (C-1′), 102.0 (C-1), 101.7 (C q_Me), 85.5, 84.5, 83.8, 82.9 (4 × CH p‑cym_), 80.7 (C-4), 76.0 (C-5′), 75.0 (C-5), 72.4 (C-3), 67.61/67.56 (C-2′/3′), 66.8 (C-4′), 60.2 (C-6), 60.0 (C-6′), 55.8 (OMe), 54.7 (C-2), 30.4 (CH_ i‑Pr_), 23.1 (Me Ac), 22.0, 21.2 (2 × Me _ i‑Pr_), 18.1 (Me _ p‑cym_). ^19^F NMR (DMSO-d 6, 376 MHz): δ −71.43 (d, ^1^ J (F–P) = 711.4 Hz). ^31^P NMR (DMSO-d 6, 162 MHz): δ −161.10 (hept, ^1^ J (P–F) = 711.4 Hz). Minor diastereomer: ^1^H NMR (DMSO-d 6, 400 MHz, COSY): δ 9.47–9.46 (m, 1H, CH Py), 9.37 (s, 1H, CH Tria), 8.30–8.22 (m, 2H, CH Py), 7.84 (d, 1H, J = 7.5 Hz, NH), 7.70–7.66 (m, 1H, CH Py), 6.17, 6.13, 5.98, 5.92 (4 × m, 4 × 1H, CH _ p‑cym_), 5.86 (d, 1H, J = 6.2 Hz, OH-2′), 5.65 (d, 1H, J = 6.4 Hz, OH-4′), 5.15 (dd, 1H, J = 11.2, 3.0 Hz, H-3′), 4.96 (t, 1H, J = 4.9 Hz, OH-6′), 4.75 (dd, 1H, J = 6.2, 4.1 Hz, OH-6), 4.67 (d, 1H, J = 7.5 Hz, H-1′), 4.60 (d, 1H, J = 2.0 Hz, OH-3), 4.27 (d, 1H, J = 7.3 Hz, H-1), 4.14–4.04 (m, 2H, H-2′, H-4′), 3.86 (dd, 1H, J = 5.9, 4.7 Hz, H-5′), 3.80–3.76 (m, 1H, H-6a), 3.71–3.65 (m, 1H, H-6b), 3.60–3.46 (m, 5H, H-2, H-3, H-4, H-6′a, H-6′b), 3.35 (s, 3H, OMe), 3.33–3.29 (m, 1H, H-5), 2.70–2.61 (m, 1H, CH _ i‑Pr_), 2.12 (s, 3H, Me _ p‑cym_), 1.81 (s, 3H, Me Ac), 1.07, 1.00 (2 × d, 2 × 3H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (DMSO-d 6, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 168.9 (CO), 155.7 (CH Py), 148.2 (C q(Py)), 145.3 (C q(Tria)), 140.2, 125.9 (2 × CH_Py_), 125.4 (CH_Tria_), 121.9 (CH_Py_), 104.0 (C q_CH i‑Pr_), 103.4 (C-1′), 101.9 (C-1), 101.7 (C q_Me), 85.7, 85.6, 83.6, 82.8 (4 × CH p‑cym_), 80.5 (C-4), 75.7 (C-5′), 75.0 (C-5), 72.4 (C-3), 67.61/67.56 (C-2′/3′), 67.0 (C-4′), 60.2 (C-6), 59.7 (C-6′), 55.8 (OMe), 54.7 (C-2), 30.4 (CH_ i‑Pr_), 23.1 (Me Ac), 22.0, 21.3 (2 × Me _ i‑Pr_), 18.1 (Me _ p‑cym_). ^19^F NMR (DMSO-d 6, 376 MHz): δ −71.43 (d, ^1^ J (F–P) = 711.4 Hz). ^31^P NMR (DMSO-d 6, 162 MHz): δ −161.10 (hept, ^1^ J (P–F) = 711.4 Hz). HRMS-ESI (m/z): [M – PF_6_]^+^ calculated for C_32_H_45_ClN_5_O_10_Ru, 796.1899; found 796.1897.
Methyl 4-O-(2,4,6-Tri-O-acetyl-3-azido-3-deoxy-β-d-galactopyranosyl)-2-azido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (S5)
Methyl 2-azido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside? (255 mg, 0.64 mmol) and phenyl 2,4,6-tri-O-acetyl-3-azido-3-deoxy-1-thio-β-d-galactopyranoside? (324 mg, 0.77 mmol) were codistilled with toluene (3×) and dried under vacuum using an oil pump for 15 min. Anhydrous dichloromethane (6 mL) and 3 Å molecular sieves (1 g) were added under an argon atmosphere, and the mixture was stirred at room temperature for 1 h and then cooled to 0 °C. N-Iodosuccinimide (314 mg, 1.40 mmol) and TfOH (20 μL, 0.23 mmol, added dropwise) were added, and the mixture was stirred at 0 °C for 1 h. The reaction mixture was diluted with DCM (30 mL); a saturated solution of NaHCO_3_ was added to quench the reaction and neutralize an excess of acid, and a 10% solution of Na_2_S_2_O_3_ (5 mL) was added to decolorize the reaction mixture. The reaction mixture was filtered and washed with water (30 mL). The aqueous phase was extracted with dichloromethane (2 × 30 mL). The combined organic phases were dried and concentrated. Column chromatography in EtOAc/PE 1:2 provided compound S5 (390 mg, 86%, see the Supporting Information, Scheme S4, for the structure) as a colorless gel. R _ f _ 0.30 (EtOAc/PE 1:2), −36 (c 0.60, CHCl_3_). ^1^H NMR (CDCl_3_, 400 MHz, H–H COSY): δ 7.42–7.27 (m, 10H, CH Ph), 5.27 (dd, 1H, J = 3.4, 1.2 Hz, H-4′), 5.03 (dd, 1H, J = 10.6, 7.9 Hz, H-2′), 4.95 (d, 1H, J = 10.6 Hz, CHH O-3Bn), 4.81 (d, 1H, J = 12.1 Hz, CHH O-6Bn), 4.73 (d, 1H, J = 10.6 Hz, CHH O-3Bn), 4.49 (d, 1H, J = 7.9 Hz, H-1′), 4.44 (d, 1H, J = 12.1 Hz, CHH O-6Bn), 4.13–4.11 (m, 1H, H-1), 3.99 (dd, 1H, J = 9.8, 8.9 Hz, H-4), 3.97 (dd, 1H, J = 11.3, 7.6 Hz, H-6′a), 3.80 (dd, 1H, J = 11.3, 6.1 Hz, H-6′b), 3.79 (dd, 1H, J = 10.9, 3.1 Hz, H-6a), 3.72 (dd, 1H, J = 10.9, 1.8 Hz, H-6b), 3.56 (s, 3H, MeO), 3.52 (dd, 1H, J = 7.6, 6.1, 1.2 Hz, H-5′), 3.41–3.35 (m, 2H, H-2, H-3), 3.31 (ddd, 1H, J = 9.8, 3.1, 1.8 Hz, H-5), 3.18 (dd, 1H, J = 10.6, 3.4 Hz, H-3′), 2.10, 2.06, 2.01 (3 × s, 3 × 3H, 3 × Me_Ac_). ^13^C{^1^H} NMR (CDCl_3_, 101 MHz, HSQC, HMBC): δ 170.4, 170.0, 169.2 (3 × CO), 138.3, 138.0 (2 × C q), 128.8, 128.5, 128.33 (3 × 2CH_Ph_), 128.28 (CH_Ph_), 128.0 (2CH_Ph_), 127.9 (CH_Ph_), 103.1 (C-1), 100.3 (C-1′), 81.1 (C-3), 76.3 (C-4), 75.3 (CH_2_ O-3Bn), 74.9 (C-5), 73.9 (CH_2_ O-6Bn), 71.7 (C-5′), 70.3 (C-2′), 67.5 (C-6/4′), 65.8 (C-2), 61.7 (C-3′), 60.9 (C-6′), 57.3 (MeO), 20.9, 20.8, 20.7 (3 × Me Ac). HRMS-ESI (m/z): [M + Na]^+^ calculated for C_33_H_40_N_6_O_12_Na, 735.2596; found 735.2594.
Methyl 4-O-(2,4,6-Tri-O-acetyl-3-azido-3-deoxy-β-d-galactopyranosyl)-2-azido-2-deoxy-β-d-glucopyranoside
(2,3́′-diN3-Lacβ-OMe, S6)
A solution of Na_2_S_2_O_4_ (454 mg, 2.61 mmol) in water (15 mL) was slowly added dropwise to a mixture of S5 (380 mg, 0.53 mmol) and NaBrO_3_ (393 mg, 2.61 mmol) in EtOAc (20 mL) and water (10 mL).? The mixture was stirred vigorously for 6 h. A change of color from colorless to orange-red and back to colorless occurred, indicating the end of the reaction. The mixture was diluted with DCM (100 mL) and washed with water (60 mL). The aqueous phase was extracted four times with DCM (50 mL). The combined organic phases were dried and concentrated. Column chromatography in EtOAc/PE 2:1 provided S6 (255 mg, 90%, see the Supporting Information, Scheme S4, for the structure) as a colorless gel. R _ f _ 0.15 (EtOAc/PE 1:1), +2 (c 0.81, CHCl_3_). ^1^H NMR (CDCl_3_, 400 MHz, H–H COSY): δ 5.41 (dd, 1H, J = 3.4, 1.0 Hz, H-4′), 5.20 (dd, 1H, J = 10.6, 7.9 Hz, H-2′), 4.61 (d, 1H, J = 7.9 Hz, H-1′), 4.28 (d, 1H, J = 1.5 Hz, OH-3), 4.26–4.20 (m, 1H, H-6′a), 4.18 (d, 1H, J = 8.2 Hz, H-1), 4.03–3.97 (m, 2H, H-5′, H-6′b), 3.84 (dd, 1H, J = 12.3, 2.1 Hz, H-6a), 3.68–3.61 (m, 4H, H-3, H-4, H-6b, OH-6), 3.61 (dd, 1H, J = 10.6, 3.4 Hz, H-3′), 3.56 (s, 3H, MeO), 3.35–3.32 (m, 1H, H-5), 3.29–3.24 (m, 1H, H-2), 2.18 (s, 2 × 3H, 2 × Me_Ac_), 2.11 (s, 3H, Me_Ac_). ^13^C{^1^H} NMR (CDCl_3_, 101 MHz, HSQC, HMBC): δ 170.7 (CO_O‑6′Ac_), 170.0 (CO_O‑4′Ac_), 169.6 (CO_O‑2′Ac_), 102.6 (C-1), 102.2 (C-1′), 81.2 (C-4), 74.3 (C-3), 74.0 (C-5), 72.6 (C-5′), 69.8 (C-2′), 67.7 (C-4′), 65.5 (C-2), 62.2 (C-6), 61.8 (C-3′), 60.8 (C-6′), 57.6 (MeO), 20.8, 20.71, 20.68 (3 × Me Ac). HRMS-ESI (m/z): [M + Na]^+^ calcd for C_19_H_28_N_6_O_12_Na, 555.1657; found 555.1659.
Methyl 2-Deoxy-4-O-{3-deoxy-3-[4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl]-β-d-galactopyranosyl}-2-[4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl]-β-d-glucopyranoside
(20)
Copper(II) sulfate pentahydrate (12 mg, 0.05 mmol), ascorbic acid (8 mg, 0.05 mmol), 2-ethynylpyridine (58 μL, 0.57 mmol), and disaccharide S6 (125 mg, 0.23 mmol) reacted according to the general procedure for azide–alkyne cycloaddition. The acetyl-protected intermediate obtained after the aqueous workup and concentration was not chromatographically purified but immediately subjected to deacetylation according to the general procedure. Column chromatography in DCM/MeOH 5:1 afforded 20 (94 mg, 65% over two steps) as a colorless gel, R f 0.35 (DCM/MeOH 5:1), −1 (c 0.78, MeOH). ^1^H NMR (DMSO-d 6, 400 MHz, COSY): δ 8.81 (s, 1H, CH Tria), 8.61 (br s, 2H, CH Py), 8.44 (s, 1H, CH Tria), 8.05–8.03, 7.93–7.87, 7.37–7.32 (3 × m, 3 × 2H, CH Py), 4.98 (d, 1H, J = 8.3 Hz, H-1), 4.89 (dd, 1H, J = 11.0, 3.1 Hz, H-3′), 4.64 (d, 1H, J = 7.5 Hz, H-1′), 4.39 (dd, 1H, J = 10.4, 8.3 Hz, H-2), 4.24 (dd, 1H, J = 10.4, 7.8 Hz, H-3), 4.11 (dd, 1H, J = 11.0, 7.5 Hz, H-2′), 3.88–3.83 (m, 2H, H-6a, H-4′), 3.81 (dd, 1H, J = 6.4, 5.8 Hz, H-5′), 3.76 (dd, 1H, J = 11.9, 4.4 Hz, H-6b), 3.69 (dd, 1H, J = 10.0, 7.8 Hz, H-4), 3.69–3.64 (m, 1H, H-5), 3.51–3.49 (m, 2H, H-6′a, H-6′b), 3.31 (s, 3H, OMe). ^13^C{^1^H} NMR (DMSO-d 6, 101 MHz, HSQC, HMBC): δ 150.2, 150.0 (2 × CH_Py_), 149.63, 149.61 (2 × C q(Py)), 147.0, 146.6 (2 × C q(Tria)), 137.3, 137.2 (2 × CH_Py_), 123.5 (CH_Tria_), 123.0, 122.9 (2 × CH_Py_), 122.5 (CH_Tria_), 119.4, 119.3 (2 × CH_Py_), 103.9 (C-1′), 100.6 (C-1), 80.5 (C-4), 76.3 (C-5′), 75.2 (C-5), 72.4 (C-3), 67.5 (C-4′), 67.3 (C-2′), 65.8 (C-2), 65.6 (C-3′), 60.2 (C-6′), 60.0 (C-6), 56.3 (OMe). HRMS-ESI (m/z): [M + H]^+^ calculated for C_27_H_33_N_8_O_9_, 613.2365; found 613.2361.
2,3′-Bisruthenium Complex 14-Cl
Complex 14-Cl was prepared according to the general complexation procedure starting from dichloro(p-cymene)ruthenium(II) dimer (34 mg, 0.055 mmol) in DCM (1 mL) and disaccharide 20 (34 mg, 0.055 mmol) in a 1:1 mixture of MeOH/DCM (2 mL). The product 14-Cl (62 mg, 91%) precipitated from the MeOH solution by the addition of methyl tert-butyl ether as a yellow solid mixture of four diastereomers. ^1^H NMR (CD_3_OD, 400 MHz, H–H COSY): δ 9.42–9.41 (m, 8 × 1H, Hz, CH Py), 9.16, 9.15, 9.12, 9.11, 9.09, 9.08 (7 × s, 8 × 1H, CH Tria), 8.20–8.10 (m, 8 × 2H, CH Py), 7.69–7.64 (m, 8 × 1H, CH Py), 6.22–6.11, 6.07–6.02, 5.98–5.89, 5.85–5.78 (4 × m, 4 × 8 × 1H, CH _ p‑cym_), 5.23, 5.22 (2 × d, 2 × 1H, J = 8.3 Hz, H-1), 5.20–5.10 (m, 4 × 1H, H-3′), 5.03–5.01 (m, 2 × 1H, H-1), 4.83, 4.82, 4.81, 4.80 (4 × d, 4 × 1H, J = 7.5 Hz, H-1′), 4.60–4.57 (m, 2 × 1H, H-3), 4.60–4.54 (m, 4 × 1H, H-2), 4.46–4.40 (m, 2 × 1H, H-3), 4.41–4.29 (m, 4 × 1H, H-2′), 4.23, 4.21, 4.17, 4.16 (4 × dd, 4 × 1H, J = 3.1, 0.9 Hz, H-4′), 4.06–3.94 (m, 4 × 4H, H-4, H-6a, H-6b, H-5′), 3.84–3.68 (m, 3 × 4H, H-5, H-6′a, H-6′b), 3.50, 3.43 (2 × s, 4 × 3H, OMe), 2.79–2.66 (m, 8 × 1H, CH _ i‑Pr_), 2.26, 2.25, 2.23, 2.22 (4 × s, 8 × 3H, Me _ p‑cym_), 1.19–1.02 (m, 8 × 6H, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 156.8, 156.7 (8 × CH Py), 150.0, 149.9, 149.8, 149.7 (8 × C q(Py)), 147.77, 147.75, 147.7, 147.62, 147.57 (8 × C q(Tria)), 141.4 (8 × CH_Py_), 127.44, 127.41, 127.37, 127.1, 126.2, 126.0 (8 × CH_Py_, 8 × CH_Tria_), 123.6, 123.51, 123.48 (8 × CH_Py_), 106.5, 106.42, 106.38, 106.1 (8 × C q_CH i‑Pr_), 105.40, 105.35, 105.3 (4 × C-1′), 104.2, 103.9, 103.8 (8 × C q_Me), 102.2, 101.9 (4 × C-1), 87.50, 87.45, 87.4, 87.3 (8 × CH p‑cym_), 86.14, 86.05, 86.0, 85.9 (8 × CH_ p‑cym_), 85.6, 85.4, 85.2 (8 × CH_ p‑cym_), 84.7, 84.6, 84.3 (8 × CH_ p‑cym_), 80.9 (4 × C-4), 77.8, 77.7, 77.6 (4 × C-5′), 76.8 (4 × C-5), 74.0, 73.4 (4 × C-3), 70.0, 69.91, 69.85 (4 × C-2), 69.7 (C-3′), 69.33, 69.29, 69.0 (4 × C-2′, 4 × C-4′), 62.1 (4 × C-6′), 61.5, 61.4 (4 × C-6), 57.6, 57.4 (4 × OMe), 32.3, 32.2 (8 × CH_ i‑Pr_), 22.63, 22.57, 21.91, 21.85, 21.8 (8 × Me _ i‑Pr_), 18.9, 18.78, 18.75 (8 × Me _ p‑cym_). HRMS-ESI (m/z): [M – 2Cl]^2+^ calculated for C_47_H_60_Cl_2_N_8_O_9_Ru_2_, 577.0975; found 577.0977.
1,1′-Sulfanediyl-bis-{3-dideoxy-3-[4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl]-β-d-galactopyranoside}
(21)
Copper(II) sulfate pentahydrate (12 mg, 0.05 mmol), ascorbic acid (8 mg, 0.05 mmol), 2-ethynylpyridine (88 μL, 0.87 mmol), and 1,1′-sulfanediyl-bis(2,4,6-tri-O-acetyl-3-azido-3-deoxy-β-d-galactopyranoside)? (240 mg, 0.36 mmol) reacted according to the general procedure for azide–alkyne cycloaddition. Column chromatography (EtOAc → EtOAc/CH_3_OH 10:1) provided disaccharide S7 (203 mg, 64%, see the Supporting Information, Scheme S5, for the structure) as a white amorphous solid, R _ f _ 0.30 (EtOAc). ^1^H NMR (DMSO-d 6, 400 MHz, COSY): δ 8.61 (ddd, 2H, J = 4.8, 1.8, 1.2 Hz, CH Py), 8.61 (s, 2H, CH Tria), 8.02 (dt, 2H, J = 7.9, 1.2 Hz, CH Py), 7.90 (ddd, 2H, J = 7.9, 7.6, 1.8 Hz, CH Py), 7.36 (ddd, 2H, J = 7.6, 4.8, 1.2 Hz, CH Py), 5.75–5.67 (m, 2 × 2H, H-2, H-3), 5.52 (dd, 2H, J = 2.8, 1.2 Hz, H-4), 5.25 (d, 2H, J = 9.6 Hz, H-1), 4.34 (ddd, 2H, J = 6.7, 5.9, 1.2 Hz, H-5), 4.14 (dd, 2H, J = 11.4, 6.7 Hz, H-6), 4.08 (dd, 2H, J = 11.4, 5.9 Hz, H-6′), 2.05, 2.02, 1.87 (3 × s, 3 × 6H, Me). ^13^C{^1^H} NMR (DMSO-d 6, 101 MHz, HSQC, HMBC): δ 170.0, 169.3, 169.0 (6 × CO), 149.7 (2 × CH_Py_), 149.5 (2 × C q(Py)), 147.3 (2 × C q(Tria)), 137.3, 123.3 (2 × 2CH_Py_), 122.9 (2 × CH_Tria_), 119.6 (2 × CH_Py_), 81.6 (2 × C-1), 74.6 (2 × C-5), 68.8 (2 × C-4), 66.8 (2 × C-2), 61.6 (2 × C-3), 61.4 (2 × C-6), 20.6, 20.3, 20.2 (6 × Me). HRMS-ESI (m/z): [M + Na]^+^ calculated for C_38_H_42_N_8_O_14_SNa, 889.2433; found 889.2437.
A 0.1 M solution of sodium methanolate in methanol was added dropwise to a solution of disaccharide S7 (180 mg, 0.21 mmol) in a mixture of MeOH (10 mL) and DCM (10 mL) until pH = 9. The reaction mixture was stirred overnight at rt. It was neutralized with ion-exchange resin DOVEX 50W(H^+^), filtered, and concentrated to yield 21 (126 mg, 99%, 63% over two steps) as a white amorphous solid, R _ f _ 0.10 (CH_2_Cl_2_/CH_3_OH 7:1). ^1^H NMR (DMSO-d 6, 400 MHz, COSY): δ 8.60 (dt, 2H, J = 4.8, 1.5 Hz, CH Py), 8.47 (s, 2H, CH Tria), 8.04 (dt, 2H, J = 8.0, 1.2 Hz, CH Py), 7.90 (ddd, 2H, J = 8.0, 7.6, 1.5 Hz, CH Py), 7.34 (ddd, 2H, J = 7.6, 4.8, 1.2 Hz, CH Py), 5.53 (d, 2H, J = 6.9 Hz, OH-2), 5.32 (d, 2H, J = 7.4 Hz, OH-4), 4.96 (d, 2H, J = 9.5 Hz, H-1), 4.92 (dd, 2H, J = 10.6, 3.1 Hz, H-3), 4.77 (br s, 2H, OH-6), 4.19 (ddd, 2H, J = 10.6, 9.5, 6.9 Hz, H-2), 3.99 (dd, 2H, J = 7.4, 3.1 Hz, H-4), 3.74 (t, 2H, J = 6.4 Hz, H-5), 3.56–3.54 (m, 2 × 2H, H-6, H-6′). ^13^C{^1^H} NMR (, 101 MHz, HSQC, HMBC): δ 150.2 (2 × C q(Py)), 149.6 (2 × CH_Py_), 146.6 (2 × C q(Tria)), 137.2, 122.8 (2 × 2CH_Py_), 122.5 (2 × CH_Tria_), 119.3 (2 × CH_Py_), 83.5 (2 × C-1), 79.3 (2 × C-5), 67.6 (2 × C-4), 67.0 (2 × C-3), 66.9 (2 × C-2), 60.1 (2 × C-6). HRMS-ESI (m/z): [M + Na]^+^ calculated for C_26_H_30_N_8_O_8_SNa, 637.1800; found 637.1812.
Bisruthenium TDG Complex 15-Cl
Complex 15-Cl was prepared according to the general complexation procedure starting from dichloro(p-cymene)ruthenium(II) dimer (39 mg, 0.064 mmol) in DCM (1 mL) and disaccharide 21 (39 mg, 0.063 mmol) in a 1:1 MeOH/DCM mixture (2 mL). The product 15-Cl (52 mg, 67%) precipitated from MeOH solution by the addition of methyl tert-butyl ether as a yellow solid mixture of three diastereomers in approximately a 1:1:0.3 ratio. ^1^H NMR (CD_3_OD, 400 MHz, COSY): δ 9.42–9.41 (m, 6H, CH Py), 9.15, 9.14, 9.13, 9.12 (4 × s, 6H, CH Tria), 8.18–8.09 (m, 12H, CH Py), 7.67–7.63 (m, 6H, CH Py), 6.14–6.12, 6.08–6.03, 5.94–5.90, 5.85–5.81 (4 × m, 4 × 6H, CH _ p‑cym_), 5.23, 5.22, 5.18 (3 × dd, 6H, J = 10.6, 2.9 Hz, H-3), 5.03, 5.02, 5.01 (3 × d, 6H, J = 9.6 Hz, H-1), 4.80–4.70 (m, 6H, H-2), 4.33, 4.32, 4.28, 4.27 (4d, 6H, J = 2.9 Hz, H-4), 3.98–3.93 (m, 6H, H-5), 3.88–3.72 (m, 12H, H-6, H-6′), 2.76 (hept, 6H, J = 6.8 Hz, CH _ i‑Pr_), 2.23 (s, 9H, Me _ p‑cym_), 2.21 (2 × s, 9H, Me _ p‑cym_), 1.17, 1.16, 1.12, 1.07 (4 × d, 36H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 156.8, 156.74, 156.70 (6 × CH Py), 150.0, 149.9 (6 × C q(Py)), 147.73, 147.71, 147.6 (3 × 2C q(Tria)), 141.5, 141.41, 141.35, 127.4, 127.3 (12 × CH_Py_), 126.0, 125.9, 125.72, 125.67 (6 × CH_Tria_), 123.49, 123.46, 123.4, 123.3 (6 × CH_Py_), 106.51, 106.47, 106.45 (6 ×C q_CH i‑Pr_), 103.9, 103.7, 103.6 (3 × 2C q_Me), 87.5, 87.4, 87.3 (3 × 2CH p‑cym_), 86.33, 86.29, 86.2 (3 × 2C-1), 86.02, 86.00, 85.9 (6 × CH_ p‑cym_), 85.6, 85.44, 85.42 (6 × CH_ p‑cym_), 84.8, 84.68, 84.65 (3 × 2CH_ p‑cym_), 81.12, 81.07, 81.05 (3 × 2C-5), 71.03, 71.00, (6 × C-3), 69.58, 69.55, 69.4 (3 × 2C-4), 68.4, 68.19, 68.16 (6 × C-2), 62.53, 62.51 (6 × C-6), 32.32, 32.25 (6 × CH_ i‑Pr_), 22.67, 22.66, 22.6 (3 × 2Me _ i‑Pr_), 21.96, 21.95, 21.8 (6 × Me _ i‑Pr_), 18.80, 18.77, 18.75 (3 × 2Me _ p‑cym_). HRMS-ESI (m/z): [M – 2Cl]^2+^ calculated for C_46_H_58_Cl_2_N_8_O_8_Ru_2_S, 578.0782; found 578.0786.
Bisruthenium TDG Complex 15-BF
Complex 15-BF was prepared according to the general procedure for counterion exchange starting from complex 15-Cl (59 mg, 0.048 mmol) and AgBF_4_ (19 mg, 0.098 mmol). The product 15-BF precipitated by the slow addition of methyl tert-butyl ether as a yellow solid (38 mg, 59%) mixture of three diastereomers in a ratio of approximately 1.5:1:1. ^1^H NMR (CD_3_OD, 400 MHz, COSY): δ 9.43–9.41 (m, 3 × 2H, CH Py), 9.16, 9.14, 9.13, 9.12 (4 × s, 6H, CH Tria), 8.18–8.10 (m, 3 × 4H, CH Py), 7.67–7.64 (m, 3 × 2H, CH Py), 6.14–6.12, 6.08–6.03, 5.94–5.91, 5.86–5.82 (4 × m, 4 × 6H, CH _ p‑cym_), 5.23 (2 × dd, 3H, J = 10.6, 2.9 Hz, H-3), 5.22, 5.18 (2 × dd, 3H, J = 10.6, 2.9 Hz, H-3), 5.03, 5.01, 5.00 (3 × d, 3 × 2H, J = 9.5 Hz, H-1), 4.77, 4.73, 4.72, 4.71 (4 × dd, 6H, J = 10.6, 9.5 Hz, H-2), 4.33, 4.32, 4.28, 4.27 (4 × d, 6H, J = 2.9 Hz, H-4), 3.98–3.93 (m, 6H, H-5), 3.88–3.73 (m, 12H, H-6, H-6′), 2.82–2.70 (m, 6H, CH _ i‑Pr_), 2.24, 2.22. 2.21 (3 × s, 3 × 6H, Me _ p‑cym_), 1.17, 1.16, 1.12, 1.11, 1.07 (5 × d, 3 × 12H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 156.8, 156.74, 156.70 (3 × 2CH Py), 150.0, 149.9 (6C q(Py)), 147.73, 147.71, 147.6 (3 × 2C q(Tria)), 141.5, 141.42, 141.36 (6CH Py), 127.4, 127.3 (6CH Py), 126.02, 125.97, 125.74, 125.69 (6CH_Tria_), 123.54, 123.50, 123.4 (3 × 2CH Py), 106.51, 106.49, 106.47, 106.45 (6C q_CH i‑Pr_), 103.9, 103.70, 103.68, 103.6 (6C q_Me), 87.5, 87.4, 87.3 (3 × 2CH p‑cym_), 86.30, 86.27, 86.2 (6C-1), 86.03, 86.00, 85.9, 85.6, 85.4, 84.8, 84.68, 84.65 (18CH_ p‑cym_), 81.12, 81.07, 81.05 (3 × 2C-5), 71.1, 71.0, 70.8 (3 × 2C-3), 69.6, 69.5, 69.4 (3 × 2C-4), 68.4, 68.20, 68.16 (3 × 2C-2), 62.5 (6C-6), 33.32, 33.26 (6CH_ i‑Pr_), 22.7, 22.6, 22.0, 21.8 (6 × 2Me _ i‑Pr_), 18.80, 18.78, 18.76 (3 × 2Me _ p‑cym_). ^19^F NMR (MeOH-d 4, 376 MHz): δ −155.45 (br s, ^11^BF_4_), −155.50 (br s, ^10^BF_4_). HRMS-ESI (m/z): [M – 2BF_4_]^2+^ calculated for C_46_H_58_Cl_2_N_8_O_8_Ru_2_S, 578.0782; found 578.0784.
Bisruthenium TDG Complex 15-PF
Complex 15-PF was prepared according to the general procedure for counterion exchange starting from complex 15-Cl (48 mg, 0.039 mmol) and AgPF_6_ (21 mg, 0.083 mmol). The product 15-PF precipitated by slow addition of methyl tert-butyl ether as a yellow solid (46 mg, 81%) mixture of three diastereomers in a ratio of approximately 2:2:1. ^1^H NMR (CD_3_OD, 400 MHz, ^1^H–^1^H COSY): δ 9.41 (d, 3 × 2H, J = 5.7 Hz, CH Py), 9.09, 9.07 (2 × s, 6H, CH Tria), 8.18–8.106 (m, 12H, CH Py), 7.65 (m, 3 × 2H, CH Py), 6.13–6.10 (m, 6H, CH _ p‑cym_), 6.06, 6.03, 5.92, 5.88 (4 × m, 4 × 3H, CH _ p‑cym_), 5.84–5.82 (m, 6H, CH _ p‑cym_), 5.22, 5.21, 5.216 (3 × dd, 3 × 2H, J = 10.7, 2.9 Hz, H-3), 5.01 (d, 2H, J = 9.2 Hz, H-1), 5.00 (2 × d, 4H, J = 9.2 Hz, H-1), 4.77–4.68 (m, 3 × 2H, H-2), 4.32, 4.31, 4.27, 4.26 (4 × d, 6H, J = 2.9 Hz, H-4), 3.96–3.93 (m, 3 × 2H, H-5), 3.88–3.71 (m, 3 × 4H, H-6, H-6′), 2.76 (hept, 3 × 2H, J = 6.8 Hz, CH _ i‑Pr_), 2.23, 2.21, 2.20 (3 × s, 18H, Me _ p‑cym_), 1.16 (d, 18H, J = 6.8 Hz, Me _ i‑Pr_), 1.11, 1.07 (2 × d, 2 × 9H, J = 6.8 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, ^1^H–^13^C HSQC, ^1^H–^13^C HMBC): δ 156.79, 156.76, 156.7 (3 × 2CH Py), 149.98, 149.97, 149.9 (3 × 2C q(Py)), 147.74, 147.72, 147.6 (3 × 2C q(Tria)), 141.5, 141.41, 141.37, 127.40, 127.35 (5 × 2CH Py), 125.87, 125.85, 125.70, 125.66 (6CH_Tria_), 123.48, 123.45, 123.3 (3 × 2CH Py), 106.51 (1C q_CH i-Pr_), 106.50 (4C q_CH i‑Pr_), 106.48 (1C q_CH i‑Pr_), 103.86, 103.85, 103.64 (3 × 2C q_Me), 87.53, 87.47, 87.3 (3 × 2CH p‑cym_), 86.34, 86.3 (6C-1), 86.03, 86.01, 85.9, 85.6, 85.41, 85.38, 84.8, 84.66, 84.65 (18CH_ p‑cym_), 81.12, 81.08 (6C-5), 71.0, 70.8 (6C-3), 69.60, 69.59, 69.4 (3 × 2C-4), 68.44, 68.40, 68.24, 68.20 (3 × 2C-2), 62.58, 62.55 (6C-6), 32.31, 32.25 (6CH_ i‑Pr_), 22.66, 22.65, 22.60, 22.0, 21.9, 21.8 (6 × 2Me _ i‑Pr_), 18.86 (2Me _ p‑cym_), 18.7 (2 × 2Me _ p‑cym_). ^31^P{^1^H} NMR (CD_3_OD, 162 MHz): δ −159.28 (hept, ^1^ J (P–F) = 708.4 Hz). ^19^F NMR (CD_3_OD, 376 MHz): δ −74.57 (d, ^1^ J (F–P) = 708.4 Hz).
^1^H NMR (D_2_O, 400 MHz, COSY): δ 9.40 (dd, 3 × 2H, J = 5.7, 1.4 Hz, CH Py), 9.06, 9.03 (3 × s, 6H, CH Tria), 8.18 (ddd, 3 × 2H, J = 7.7, 7.4, 1.4 Hz, CH Py), 8.09 (dd, 3 × 2H, J = 7.7, 1.5 Hz, CH Py), 7.68 (ddd, 3 × 2H, J = 7.4, 5.7, 1.5 Hz, CH Py), 6.22, 6.19 (2 × m, 6H, CH _ p‑cym_), 6.11, 6.10 (2 × m, 3H, CH _ p‑cym_), 6.07 (m, 3H, CH _ p‑cym_), 5.99, 5.95 (2 × m, 6H, CH _ p‑cym_), 5.87 (2 × m, 3H, CH _ p‑cym_), 5.85 (m, 3H, CH _ p‑cym_), 5.31, 5.28 (2 × dd, 6H, J = 10.7, 3.0 Hz, H-3), 5.24, 5.23, 5.22 (3 × d, 6H, J = 9.8 Hz, H-1), 4.60, 4.59, 4.51, 4.50 (4 × dd, 6H, J = 10.7, 9.8 Hz, H-2), 4.41, 4.40 (2 × d, 6H, J = 3.0 Hz, H-4), 4.14–4.10 (m, 6H, H-5), 3.93–3.87 (m, 6H, H-6), 3.84–3.79 (m, 6H, H-6′), 2.73–2.59 (m, 6H, CH _ i‑Pr_), 2.22, 2.21 (2 × s, 18H, Me _ p‑cym_), 1.09, 1.08, 1.03, 0.99 (4 × d, 36H, J = 6.9 Hz, Me _ i‑Pr_). ^13^C{^1^H} NMR (D_2_O, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 155.8, 155.7 (6CH Py), 148.5 (6C q(Py)), 147.5, 147.4 (6C q(Tria)), 141.2 (6CH Py), 127.3 (6CH Py), 125.7, 125.2 (6CH_Tria_), 123.5 (6CH Py), 105.3, 105.2 (6C q_CH i‑Pr_), 103.9, 103.72, 103.70 (6C q_Me), 86.7, 86.6 (6CH p‑cym_), 85.84, 85.81, 85.6 (6CH_ p‑cym_), 84.83, 84.80 (6C-1), 84.9, 84.64, 84.62 (6CH_ p‑cym_), 83.78, 83.76 (6CH_ p‑cym_), 80.2, 80.1 (6C-5), 69.5, 69.3 (6C-3), 68.6, 68.4 (6C-4), 67.5, 67.4, 67.3 (3 × 2C-2), 61.63, 61.61, 61.60 (3 × 2C-6), 31.4, 31.3, 31.2 (3 × 2CH_ i‑Pr_), 21.92, 21.90, 21.89, 21.4, 21.3 (12Me _ i‑Pr_), 18.6 (6Me _ p‑cym_). ^31^P{^1^H} NMR (D_2_O, 162 MHz): δ −161.97 (hept, ^1^ J (P–F) = 708.9 Hz). ^19^F NMR (D_2_O, 376 MHz): δ −73.32 (d, ^1^ J (F–P) = 708.9 Hz). HRMS-ESI (m/z): [M – 2PF_6_]^2+^ calculated for C_46_H_58_Cl_2_N_8_O_8_Ru_2_S, 578.0781; found 578.0787.
Bisruthenium TDG Complex 16-I
Complex 16-I was prepared according to the general complexation procedure starting from the diiodo(p-cymene)ruthenium(II) dimer (19 mg, 0.019 mmol) in dichloromethane (1 mL) and disaccharide 21 (12 mg, 0.020 mmol) in a mixture of MeOH/DCM 1:1 (1 mL). The product 16-I (25 mg, 80%) precipitated from MeOH solution by the addition of methyl tert-butyl ether as a red solid mixture of three diastereomers in a ratio of approximately 4:4:1. ^1^H NMR (CD_3_OD, 400 MHz, COSY): δ 9.35–9.32 (m, 3 × 2H, CH Py), 9.19 (s, 1H, CH Tria), 9.17, 9.15 (2 × s, 2 × 2H, CH Tria), 9.13 (s, 1H, CH Tria), 8.25–8.08 (m, 3 × 4H, CH Py), 7.60–7.58 (m, 3 × 2H, CH Py), 6.09–5.98, 5.94–5.84 (2 × m, 2 × 12H, CH _ p‑cym_), 5.23, 5.20 (3 × dd, 3 × 2H, J = 10.7, 2.9 Hz, H-3), 5.02 (d, 3H, J = 9.6 Hz, H-1), 4.97, 4.96 (2 × d, 3H, J = 9.6 Hz, H-1), 4.81–4.69 (m, 3 × 2H, H-2), 4.38, 4.36, 4.26, 4.25 (4 × d, 6H, J = 2.9 Hz, H-4), 3.99–3.92 (m, 3 × 2H, H-5), 3.84–3.71 (m, 3 × 4H, H-6, H-6′), 3.06–2.90 (m, 3 × 2H, CH _ i‑Pr_), 2.39, 2.33. 2.32 (3 × s, 3 × 6H, Me _ p‑cym_), 1.22–1.09 (m, 3 × 12H, Me _ i‑Pr_). ^13^C{^1^H} NMR (CD_3_OD, 101 MHz, HSQC, HMBC): δ 158.1, 158.0, 157.9 (3 × 2CH Py), 150.1, 150.0, 149.9, 149.8 (6C q(Py)), 147.58, 147.56, 147.43. 147.41 (6C q(Tria)), 141.0, 140.94, 140.91, 140.8 (6CH Py), 127.00, 126.98, 126.96, 126.9 (6CH Py), 125.8, 125.6 (2 × 3CH_Tria_), 123.7, 123.3, 123.2 (3 × 2CH Py), 108.5, 108.4, 108.0, 107.9 (6C q_CH i‑Pr_), 102.6 (3 × 2C q_Me), 87.4, 86.74, 86.71, 86.69, 86.6, 86.4, 85.77, 85.75, 85.7, 85.6 (24CH p‑cym_, 6C-1), 81.2, 81.1, 81.0 (3 × 2C-5), 70.7, 70.6 (6C-3), 69.4, 69.3 (6C-4), 68.6, 68.1, 68.0 (3 × 2C-2), 62.6, 62.61, 62.57 (3 × 2C-6), 33.1, 33.0, 32.9 (3 × 2CH_ i‑Pr_), 23.2, 23.1, 22.9, 22.1, 22.0, 21.9 (6 × 2Me _ i‑Pr_), 20.1, 19.9 (6Me _ p‑cym_). HRMS-ESI (m/z): [M – 2I]^2+^ calculated for C_46_H_58_I_2_N_8_O_8_Ru_2_S 670.0143; found 670.0135.
Bisruthenium TDG Complex 17-Cl
Complex 17-Cl was prepared according to the general complexation procedure starting from dichloro(benzene)ruthenium(II) dimer (23 mg, 0.046 mmol) in dichloromethane (1 mL) and disaccharide 21 (28 mg, 0.046 mmol) in a 1:1 MeOH/DCM mixture (2 mL). The product 17-Cl (42 mg, 83%) precipitated from the MeOH solution by the addition of methyl tert-butyl ether as a yellow solid mixture of three diastereomers. ^1^H NMR (DMSO-d 6, 400 MHz, COSY): δ 9.59–9.57 (m, 3 × 2H, CH Py), 9.42 (s, 2H, CH Tria), 9.40 (2 × s, 2 × 2H, CH Tria), 8.30–8.22 (m, 3 × 4H, CH Py), 7.69–7.66 (m, 3 × 2H, CH Py), 6.19, 6.18 (2 × s, 3 × 12H, CH _ PhH ), 5.80–5.77 (m, 3 × 2H, OH-2), 5.72–5.65 (m, 3 × 2H, OH-4), 5.23, 5.20 (2 × dd, 3 × 2H, J = 10.6, 2.9 Hz, H-3), 5.04, 5.03 (2 × d, 3 × 2H, J = 9.5 Hz, H-1), 4.92, 4.85 (2 × t, 3 × 2H, J = 6.0 Hz, OH-6), 4.30–4.20 (m, 8H, H-2, H-4), 4.12–4.06 (m, 2 × 2H, H-4), 3.82–3.79 (m, 3 × 2H, H-5), 3.61–3.57 (m, 3 × 4H, H-6, H-6′). ^13^C{^1^H} NMR (DMSO-d 6, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 156.0 (3 × 2CH Py), 148.28, 148.25 (6C q(Py)), 145.7, 145.5 (6C q(Tria)), 141.3, 125.7 (2 × 6CH Py), 125.5, 125.4 (6CH_Tria), 122.1, 122.0 (6CH Py), 86.0 (3 × 12CH_ PhH _), 83.3, 83.21, 83.16, 83.1 (6C-1), 79.0, 78.8 (6C-5), 69.1, 69.0 (6C-3), 67.1 (6C-4), 66.8, 66.6 (6C-2), 59.9, 59.6 (6C-6). HRMS-ESI (m/z): [M – 2Cl]^2+^ calculated for C_38_H_42_Cl_2_N_8_O_8_Ru_2_S, 522.0154; found 522.0158.
Bisruthenium TDG Complex 18-Cl
Complex 18-Cl was prepared according to the general complexation procedure starting from dichloro(hexamethylbenzene)ruthenium(II) dimer (33 mg, 0.049 mmol) in dichloromethane (1 mL) and disaccharide 21 (30 mg, 0.049 mmol) in a mixture of MeOH/DCM 1:1 (2 mL). The product 18-Cl (51 mg, 81%) precipitated from the MeOH solution by the addition of methyl tert-butyl ether as a yellow solid mixture of three diastereomers. ^1^H NMR (DMSO-d 6, 400 MHz, COSY): δ 9.43, 9.42, 9.41 (3 × s, 3 × 2H, CH Tria), 8.91–8.89 (m, 3 × 2H, CH Py), 8.30–8.18 (m, 3 × 4H, CH Py), 7.71–7.67 (m, 3 × 2H, CH Py), 5.80–5.72 (m, 3 × 4H, OH-2, OH-4), 5.25 (dd, 3 × 2H, J = 10.6, 3.2 Hz, H-3), 5.08, 5.04 (3 × d, 3 × 2H, J = 9.5 Hz, H-1), 4.99, 4.96, 4.88, 4.85 (4 × t, 6H, J = 6.1 Hz, OH-6), 4.26–4.09 (m, 3 × 4H, H-2, H-4), 3.84, 3.79 (2 × t, 6H, J = 6.7 Hz, H-5), 3.60–3.58 (m, 3 × 4H, H-6, H-6′), 2.11, 2.10, 2.02 (3 × s, 3 × 36H, Me). ^13^C{^1^H} NMR (DMSO-d 6, 101 MHz, HSQC, HMBC, HSQC TOCSY): δ 153.1 (3 × 2CH Py), 148.04, 147.95, 147.94 (3 × 2C q(Py)), 145.5, 145.2 (6C q(Tria)), 139.7 (3 × 2CH Py), 126.0 (3 × 2CH Py), 125.2 (3 × 2CH_Tria_), 121.6, 121.5 (6CH Py), 96.1, 94.8 (3 × 12C _q_Me), 83.0, 82.92, 82.85 (3 × 2C-1), 79.1, 78.8 (6C-5), 69.0 (6C-3), 67.5 (6C-2), 67.2, 66.8, 66.5 (3 × 2C-4), 59.8, 59.6 (6C-6), 16.6, 15.4, 15.28, 15.26 (36Me). HRMS-ESI (m/z): [M – 2Cl]^2+^ calculated for C_50_H_66_Cl_2_N_8_O_8_Ru_2_S 606.1096; found 606.1098.
Bioanalytical Methods
Detailed experimental procedures for the determination of the affinity to galectins by fluorescence polarization and intrinsic tryptophan fluorescence are described in Supporting Information, Section D. The experimental procedures for the determination of cell viability are described in Supporting Information, Section G.
Production
of Galectins
Preparation and purification of hGal-1, hGal-3-CRD, and monobiotinylated protein constructs of hGal-1 and full-length hGal-3 (hGal-1-AVI, hGal-3-AVI) are described in detail in the Supporting Information, Section C.
Biolayer Interferometry
The kinetic analysis of the best-performing ruthenium complex-based inhibitors targeting galectin constructs (hGal-1, hGal-3) was performed by using biolayer interferometry. The galectin constructs carried a 15-amino acid AVI-tag at the N-terminus, selectively monobiotinylated during heterologous production in E. coli. Experiments were conducted under controlled conditions (25 ± 0.1 °C, 1000 rpm) using an Octet Red96e instrument (FortéBio, Fremont, CA, USA). Optimized galectin concentrations of 10 μg/mL (hGal-1) or 50 μg/mL (hGal-3) were used across all of the experiments. Biotinylated galectin constructs were diluted to their respective optimal concentrations in PBS containing 0.05% Tween-20 and immobilized (300 s) on streptavidin-coated biosensors (Octet SA Biosensors, Sartorius, Göttingen, Germany) via biotin–streptavidin coupling. The interactions between the immobilized constructs and serially diluted inhibitors (62.5–500 μM for hGal-3; 4.9–400 nM for hGal-1) were monitored over a total of 1050 s, including an association phase of 450 s and a dissociation phase of 600 s. Immobilization did not affect galectin activity. BLI data were analyzed using Octet Analysis Studio (Sartorius, Göttingen, Germany). Background signals from nonspecific interactions (<10% of the total response) and sensor drift were corrected using a double-reference subtraction method. Kinetic data were evaluated using a Langmuir 1:1 kinetic model, and all data sets were further validated through steady-state analysis as an additional method of kinetic data evaluation.
Inhibition of Galectin
Binding to Cancer Cells
The cell binding inhibition assay was performed similarly to the previous studies with other galectins. ?,? Ruthenium complex-based inhibitors or lactose (used as a positive control) were serially diluted in a PBS/BSA buffer (50 mM NaH_2_PO_4_, 150 mM NaCl, and 1% w/v bovine serum albumin, pH 7.5). These were then premixed with Gal-1-AVI (final concentration 5 μg/mL, BSA calibrated) on ice for 30 min. The mixtures were combined with a suspension of MDA-MB-231 breast cancer cells (7.5 × 10^4^ cells in a 100 μL final volume) in PBS/BSA buffer. The resulting mixture was incubated in U-bottom 96-well tissue culture plates (TPP Techno Plastic Products, CH) for 60 min on ice. After incubation, the cells were washed with PBS/BSA buffer and centrifuged (200 × g, 3 min, 4 °C). The surface-bound Gal-1-AVI was labeled with a streptavidin–phycoerythrin conjugate (1:249 dilution; 50 μL; BioLegend, USA) for 45 min on ice. Following another wash with PBS/BSA, the cells were resuspended in PBS/BSA buffer and analyzed by the flow cytometer ZE5 Cell Analyzer (Bio-Rad, CA, USA) using Hoechst 33258 stain (1 mg/mL). Quadruplicate results were obtained. During data analysis with FlowJo software (Tree Star, Ashland, OR, USA), cell aggregates and dead cells were excluded through appropriate gating. The half-maximal inhibitory concentrations (IC 50) were calculated from the nonlinear fitting of the mean fluorescence intensities of phycoerythrin-stained cells against glycomimetic concentrations with values reported along with standard deviations (SD). Under these assay conditions, lactose (positive control) had an IC 50 value of 4.3 ± 0.5 μM.
Jurkat Preaparesis Assay
Jurkat cells (1 × 10^6^/mL) were cultured for 30 min at 37 °C in the presence of 20 μM recombinant hGal-1. For preaparesis analysis, cells were subsequently stained with Annexin V–Alexa Fluor (Invitrogen, cat. no. R37174) according to the manufacturer’s instructions, and signals of 10 000 cells were measured (FacsVerse flow cytometer (BD Biosciences)) and analyzed (BD FACSuiteTM Software). The effect of hGal-1 inhibitors was determined by incubating hGal-1 with equimolar amounts of inhibitors for 1 h at 4 °C, before addition to the cell culture. All experiments were performed in three to five biological replicates, each in three parallels. Results are expressed as the percentage of Annexin V positive cells out of the total cell population. Inhibition of hGal-1-induced preaparesis in Jurkat cells by complexes 16-I and 17-Cl is displayed in the Supporting Information, Figure S20. Representative flow cytometry histograms of Jurkat cells stained with Annexin V–Alexa Fluor 488 including negative controls are shown in the Supporting Information, Figures S21–S23.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hevey R.Ling C.-C.Recent advances toward the development of inhibitors to attenuate tumor metastasis via the interruption of lectin–ligand interactions Adv. Carbohydr. Chem. Biochem.20136912520710.1016/B 978-0-12-408093-5.00005-824274369 · doi ↗ · pubmed ↗
- 2Ernst B.Magnani J. L.From carbohydrate leads to glycomimetic drugs Nat. Rev. Drug Discovery 2009866167710.1038/nrd 285219629075 PMC 7097102 · doi ↗ · pubmed ↗
- 3Aretz J.Wamhoff E.-C.Hanske J.Heymann D.Rademacher C.Computational and Experimental Prediction of Human C-Type Lectin Receptor Druggability Front. Immunol.20145510.3389/fimmu.2014.0032325071783 PMC 4090677 · doi ↗ · pubmed ↗
- 4Shanina E.Kuhaudomlarp S.Siebs E.Fuchsberger F. F.Denis M.da Silva Figueiredo Celestino Gomes P.Clausen M. H.Seeberger P. H.Rognan D.Titz A.Imberty A.Rademacher C.Targeting undruggable carbohydrate recognition sites through focused fragment library design Commun. Chem.202256410.1038/s 42004-022-00679-336697615 PMC 9814205 · doi ↗ · pubmed ↗
- 5Smith B. A. H.Bertozzi C. R.The clinical impact of glycobiology: Targeting selectins, Siglecs and mammalian glycans Nat. Rev. Drug Discovery 20212021724310.1038/s 41573-020-00093-133462432 PMC 7812346 · doi ↗ · pubmed ↗
- 6Vijayan M.Chandra N.Lectins. Curr. Opin Struct. Biol.1999970771410.1016/S 0959-440X(99)00034-210607664 · doi ↗ · pubmed ↗
- 7Leusmann S.Menova P.Shanin E.Titz A.Rademacher C.Glycomimetics for the inhibition and modulation of lectins Chem. Soc. Rev.2023523663374010.1039/D 2CS 00954 D 37232696 PMC 10243309 · doi ↗ · pubmed ↗
- 8Damalanka V. C.Maddirala A. R.Janetka J. W.Novel approaches to glycomimetic design: Development of small molecular weight lectin antagonists Expert Opin. Drug. Discovery 20211651353610.1080/17460441.2021.185772133337918 · doi ↗ · pubmed ↗