Do Amino-Oxetanes Resemble Amides? A Matched Molecular Pairs Property and Structural Comparison
Hikaru Ishikura, Callum S. Begg, Juan J. Rojas, Luka Blagojevic, Gavin J. Smith, Joyce Luk, Rosemary A. Croft, Charles Romain, Chulho Choi, James A. Bull

TL;DR
Amino-oxetanes behave similarly to amides in many ways, making them promising replacements in drug design.
Contribution
A matched molecular pair study comparing amino-oxetanes and amides reveals their isosteric potential and structural differences.
Findings
Amino-oxetanes show comparable physicochemical properties to amides, including H-bond donor and acceptor capabilities.
Crystal structure analysis reveals conformational differences, with amino-oxetanes resembling sulfonamides more in torsion angles and exit vectors.
Amino-oxetanes offer a design option to modulate drug properties and topology.
Abstract
Oxetanes display properties comparable to ketone carbonyl groups and are increasingly explored as bioisosteres. However, does the comparison hold for the most common carbonyl derivatives: do amino-oxetanes resemble amides? Here, we present a matched molecular pair study of 12 3-aryl-3-amino-oxetane and benzamide matched molecular pairs to assess their viability as isosteres. Across the surveyed physicochemical properties (pH stability, solubility, lipophilicity, clearance, permeability), amino-oxetanes exhibited broadly comparable profiles to their amide counterparts. Amino-oxetanes maintain both the H-bond acceptor and H-bond donor capabilities of analogous amides. These findings support the potential of amino-oxetanes as amide replacements. However, crystal structure analysis highlights the conformational differences and alternative exit vectors available through introduction of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1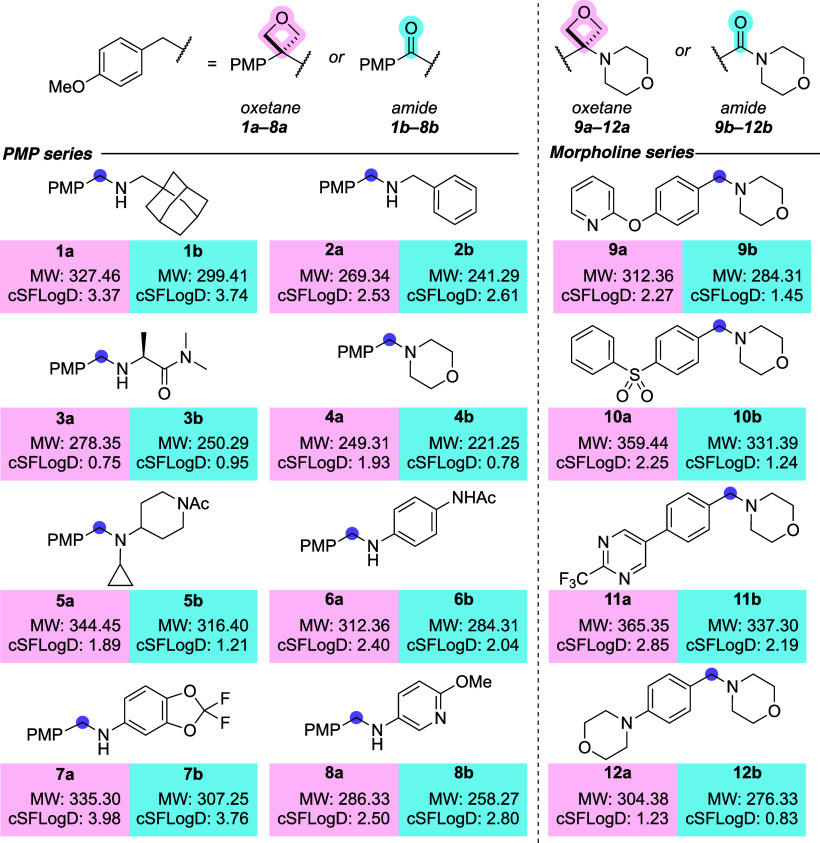 2
2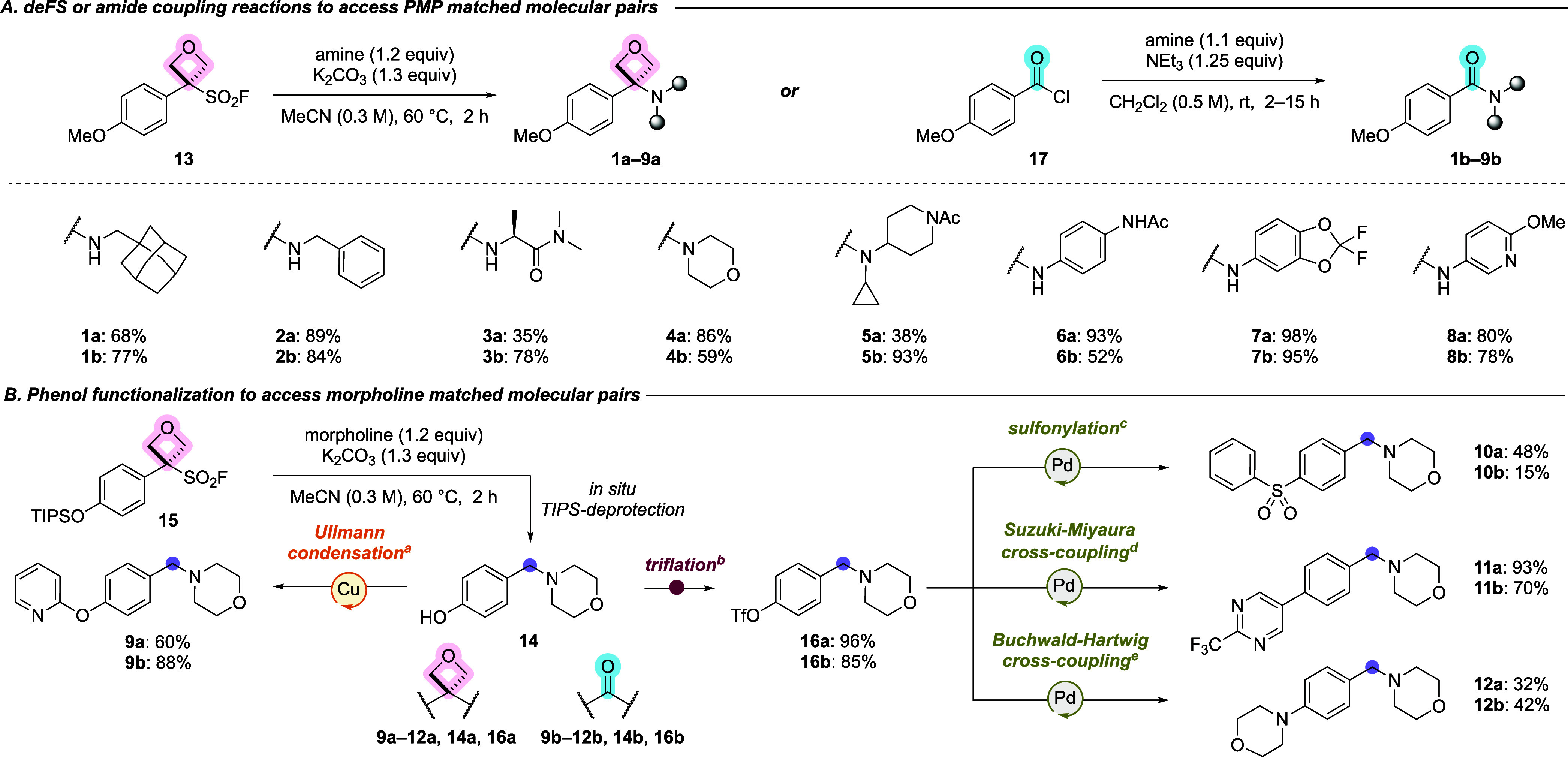 1
1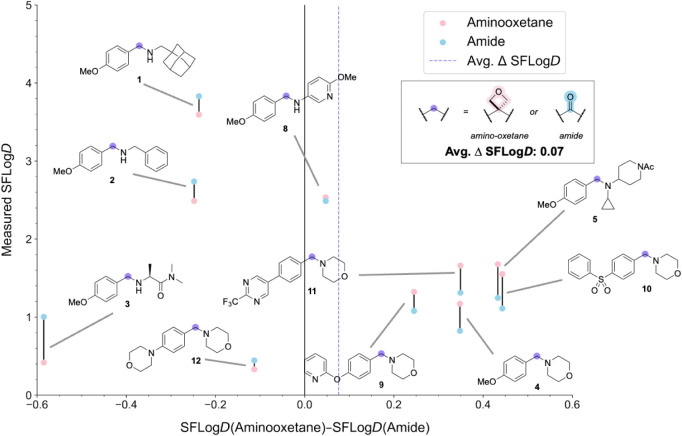 3
3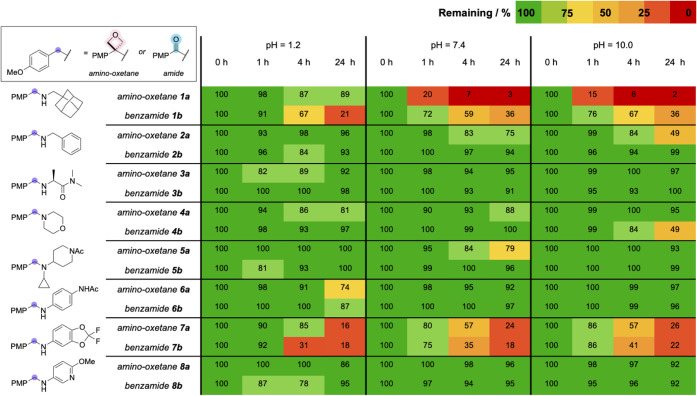 4
4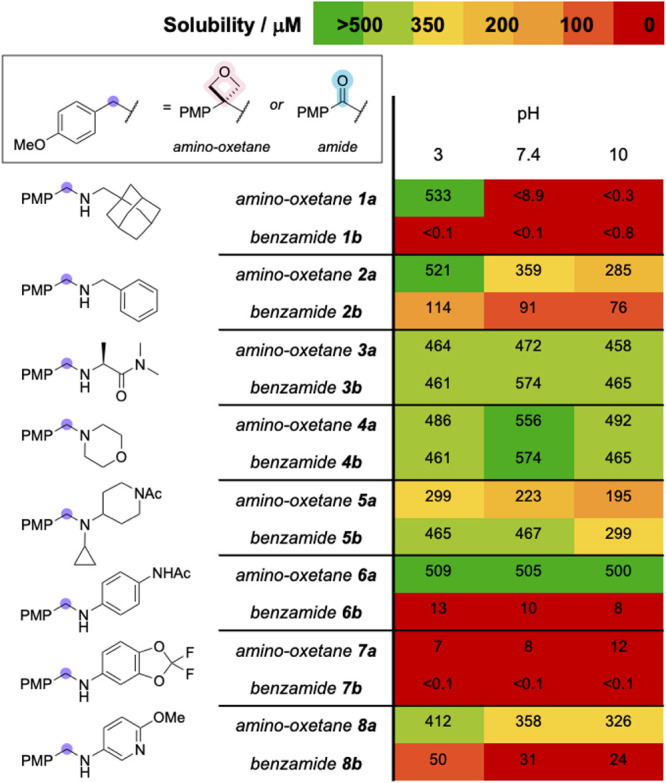 5
5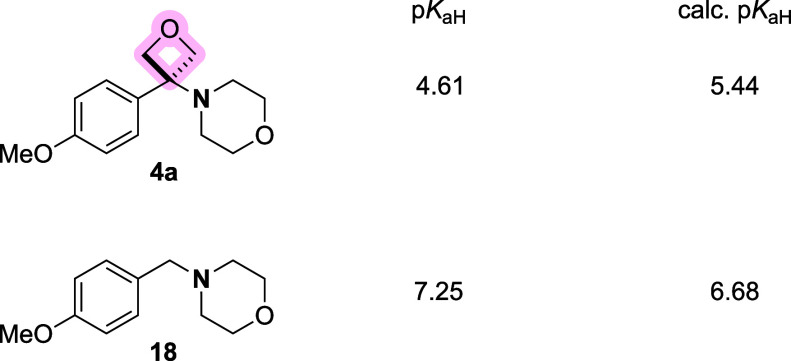 6
6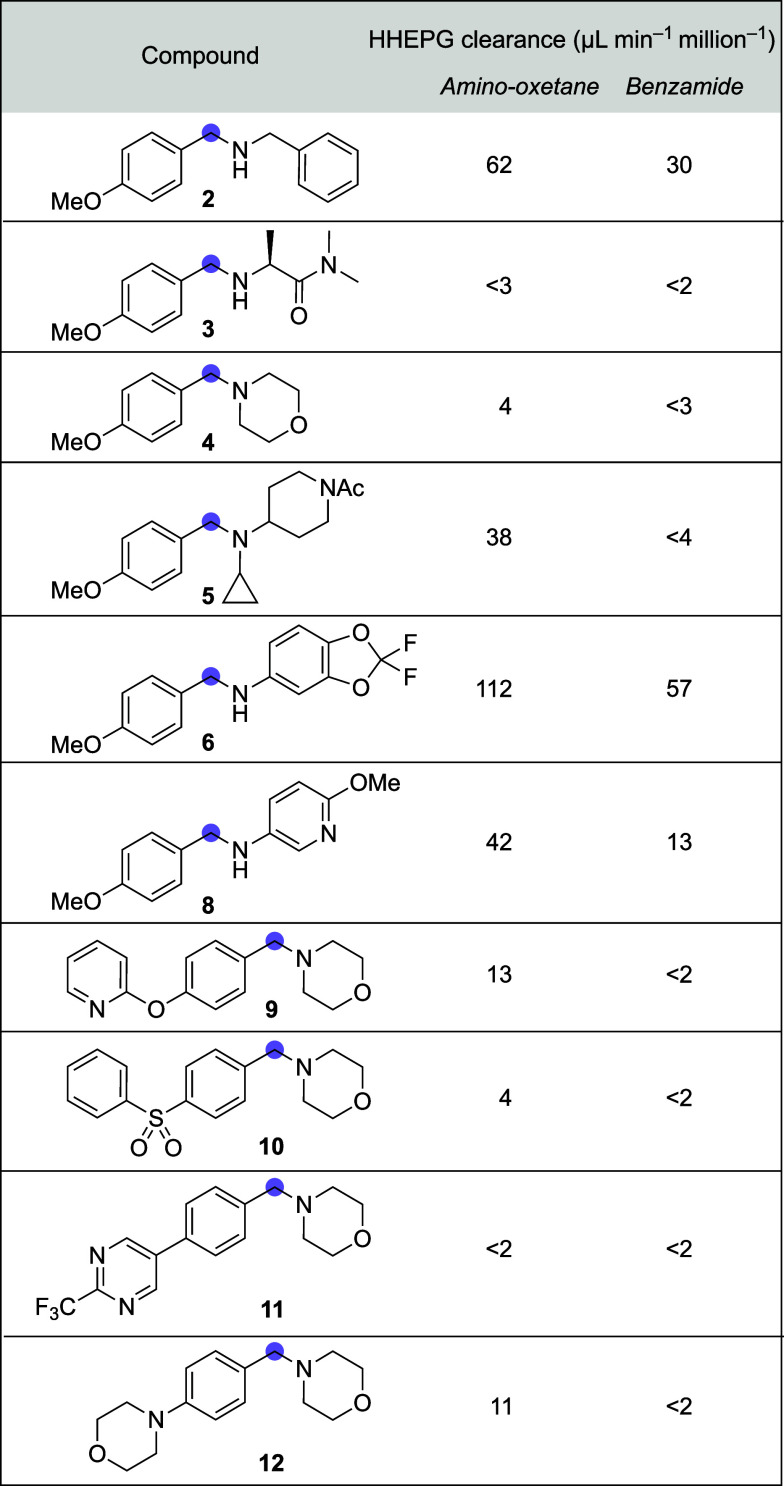 7
7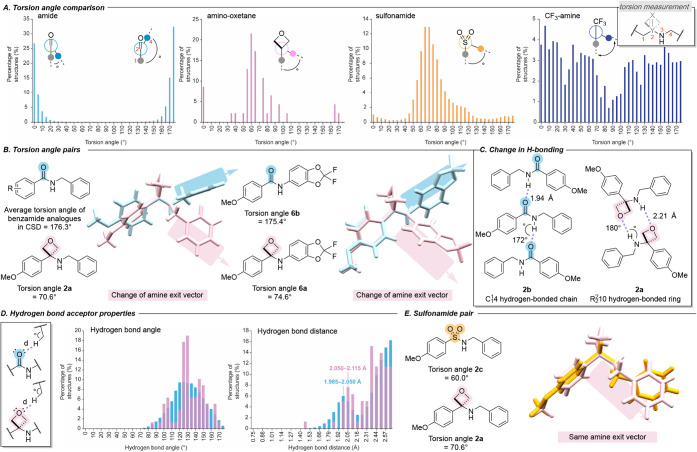 8
8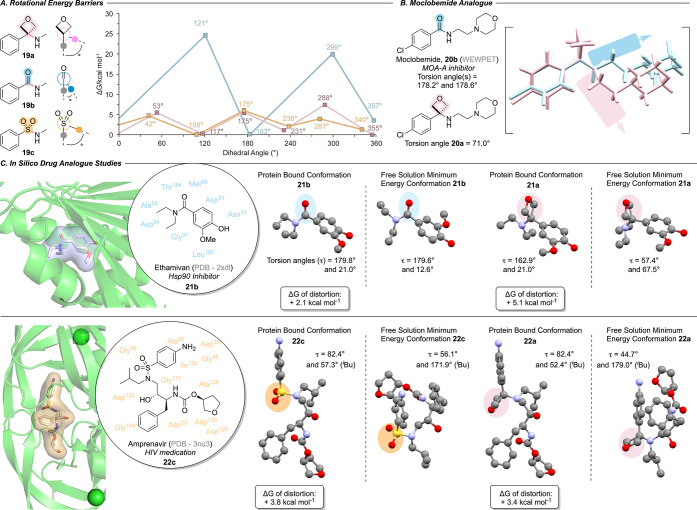 9
9- —Pfizer10.13039/100004319
- —Engineering and Physical Sciences Research Council10.13039/501100000266
- —Royal Society10.13039/501100000288
- —Royal Society10.13039/501100000288
- —Royal Society10.13039/501100000288
- —Imperial College London10.13039/501100000761
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCrystallography and molecular interactions · Chemical Synthesis and Analysis · Crystal structures of chemical compounds
Introduction
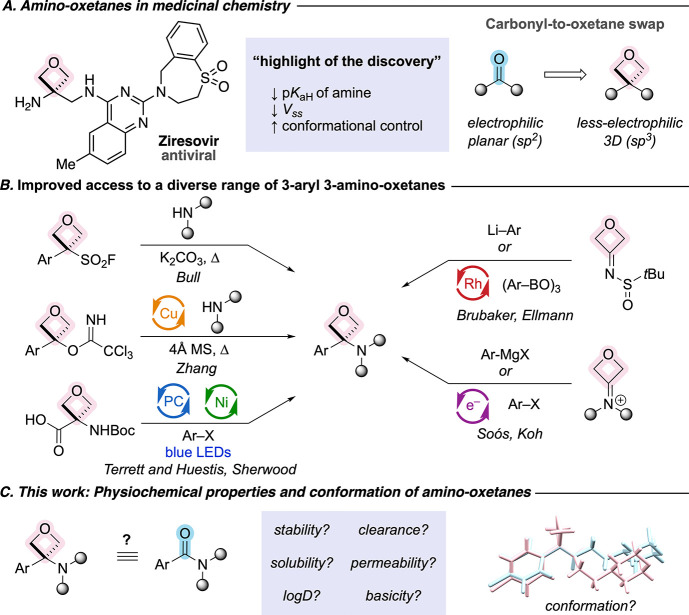
Oxetanes have been the subject of several studies on their potential in medicinal chemistry in recent years. This growing interest has contributed to the advancement of ten oxetane-containing compounds to clinical trials, marking the emergence of the first synthetic oxetane derivatives in the clinic and the first FDA approved fully synthetic oxetane containing drug (rilzabrutinib, approved September 2025). ?−? ? The low molecular weight, high polarity and three-dimensionality of oxetane motifs have offered distinct benefits through modulation of binding and physicochemical properties, as well as the potential to exploit novel chemical and intellectual property space. A prominent example is Ziresovir, where the inclusion of the oxetane was deemed the “highlight of the discovery” as it was able to attenuate amine basicity and thus volume of distribution, while maintaining biological activity through conformational control (FigureA).? Carreira in collaboration with Hoffmann-La Roche pioneered the area through the use of oxetanes as a nonlipophilic alternative to gem-dimethyl groups to block metabolism of methylene units.? Similarly, oxetanes can be considered as surrogates for carbonyl groups, maintaining comparable H-bonding ability, dipole moment, lone pair orientation, and polar surface area while eliminating an electrophilic center with potential issues of metabolic and chemical instability. ?−? ? Ongoing advances in synthetic methodology have further facilitated these applications in drug design and development. ?,? The exploration of oxetanes complements broader medicinal chemistry efforts to optimize key functional groups in drug design through bioisosteric replacements.?
A. Examples of amino-oxetanes in medicinal chemistry. B. Synthetic strategies to access amino-oxetane derivatives. C. This work: comparison of the physicochemical properties and conformation of amino-oxetanes and amides.
In line with these efforts, there remains ongoing demand for alternative chemical motifs that expand accessible chemical space and provide beneficial physicochemical properties. Amides represent the most prevalent functional group in drug molecules. As a subset, benzamides are a commonly employed pharmacophore present in around 120 FDA-approved drug molecules.? In this context, amino-oxetanes have been proposed as possible amide bioisosteres. ?,? Carreira and Shipman independently reported studies on the incorporation of oxetanes in peptidomimetics, where the carbonyl-to-oxetane replacement maintains bioactivity while resulting in improved stability against enzymatic metabolism. ?,? Notably, the conformational effect of the oxetane ring described in these peptide derivatives also facilitated macrocyclization. ?,?
It is only recently that 3-aryl-3-amino-oxetanes have been synthetically available (FigureB). Early methodologies, including addition into oxetane sulfilimines and dual photoredox/nickel catalyzed arylation, ?,? afforded primary amino-oxetanes, which could be further functionalized.? We reported the defluorosulfonylative coupling (deFS) reaction of oxetane sulfonyl fluorides with a diverse range of functionalized amine nucleophiles to afford 3-aryl-3-amino-oxetanes under mild condition in an amide-like disconnection. ?,? Similarly, Zhang employed Lewis acid activation of oxetanyl trichloroacetimidates to access these targets.? Soós and Koh have developed complementary approaches exploiting oxetanone-derived iminium ions, reacting with Grignard reagents or undergoing electrochemical single-electron reduction to furnish secondary and primary 3-aryl amino-oxetanes, respectively. ?,?
With improved access to these derivatives, we sought to evaluate whether amino-oxetanes function as effective amide mimics. Do the physicochemical properties or conformational features of amino-oxetanes align closely enough to amides that can support their use as isosteres? Alternatively, is a comparison with other amide isosteres more appropriate, or indeed should amino-oxetanes be better considered as a functional group with their own unique properties?
Here we make comparison with benzamides through the preparation of a series of arylamino-oxetane and benzamide matched molecular pairs (MMP) and analysis of their physiochemical properties including lipophilicity, chemical stability, solubility, clearance, and cell permeability (FigureC). We demonstrate that reasonable comparisons can be made with benzamides in terms of several important properties, however, 3-aryl amino-oxetanes adopt a distinct conformation, which differs significantly from that of a benzamide, and is much closer to that of sulfonamide derivatives.
Results & Discussion
Compound Design and Synthesis
We designed a series of amino-oxetane (1a–12a) and benzamide (1b–12b) matched molecular pairs with calculated properties to sit within a range of lead-like and drug-like chemical space.? Two series were established: 1) a p-methoxyphenyl (PMP) series with varying amine components, and 2) a morpholine series with varying aromatic groups (Figure). This allowed for the interrogation of the effect of the carbonyl-to-oxetane replacement across a set of compounds spanning a range of MW, cSFlogD, and electronic features.?
Amino-oxetane (1a–12a) and benzamide (1b–12b) matched molecular pairs. Calculated cSFlogD (pH = 7.4) values predicted using ACD/Laboratories, version 12.1. See SI for full calculated molecular properties.
Amino-oxetanes 1a–8a were synthesized in one-step from the PMP oxetane sulfonyl fluoride 13 and the corresponding amine through a deFS reaction (SchemeA).? Amino-oxetanes 9a–12a were synthesized from a common phenolic amino-oxetane intermediate 14a, which in turn was derived from the deFS reaction of oxetane sulfonyl fluoride 15 with morpholine (SchemeB). As in our previous reports, in situ deprotection of the TIPS-group was observed to reveal the phenolic handle.? From amino-oxetane 14a, Ullmann condensation afforded amino-oxetane 9a. Triflation of the phenol allowed for Suzuki–Miyaura and Buchwald–Hartwig cross-coupling to afford amino-oxetanes 11a and 12a, as well as Pd-catalyzed sulfonylation to give amino-oxetane 10a. All synthetic transformations on amino-oxetane 14a were well tolerated, in-line with their previously established chemical stability.? Benzamides 1b–8b were synthesized by amidation of p-methoxybenzoyl chloride 17 with the corresponding amine (SchemeA). Benzamides 9b–12b were synthesized in a similar manner to amino-oxetanes 9a–12a from commercially available amide 14b through phenol derivatization (SchemeB).
A. Synthesis of PMP-Aminooxetane and Amide Matched Molecular Pairs (1–8). B. Synthesis of Morpholine-Oxetane and Amide Matched Molecular Pairs (9–12). Compounds 1a–12a, 13, 14a, and 15, and 16a Were Previously Reported (see reference )
Properties of a Set of Matched Molecular Pairs
First, we evaluated the lipophilicity (SFlogD, pH = 7.4) of the amino-oxetane and benzamide pairs. The amino-oxetanes exhibited a logD range of 0.33–3.60, with an average value of 1.68 across 10 compounds. In comparison, the benzamide counterparts showed a range of 0.44–3.83 and an average of 1.61. Although previous works on 3,3-disubstituted oxetanes have reported an increase in logD by +0.1 to +0.7 units, ?,? we found on average only a slight increase in logD of +0.07 units for the carbonyl-to-oxetane replacement (Figure).
Change in SFLogD of the carbonyl-to-oxetane replacement plotted against the measured SFLogD for selected amino-oxetane (pink, 1a–5a and 8a–12a) and benzamide matched molecular pairs (blue, 1b–5b and 8b–12b). Zero on the x-axis represents no change in SFLogD from the replacement. Positive values on the x-axis represent an increase in SFLogD in the aminooxetane while negative values represent a decrease in SFLogD in the aminooxetane. The purple dotted line represents the average change in SFLogD from the carbonyl-to-oxetane replacement.
The effect on lipophilicity was heavily dependent on the nature of the peripheral substituents, for example, pairs 10a and 10b showed an increase in SFlogD of +0.44, whereas pairs 3a and 3b exhibited a decrease of −0.59 units.
Overall, this indicates that replacing benzamides with amino-oxetanes does not result in an unfavorable systematic increase in lipophilicity. The observed trends in SFlogD were generally well captured by the cSFlogD. For the PMP series, cSFlogD values were typically within 0.2 units of the experimental data. In contrast, for the morpholine series, while the predicted trends remained directionally consistent, deviations of around 1 unit were observed for the amino-oxetanes. Nevertheless, the ability of cSFlogD to reliably capture the general trends supports its utility as a quick and effective tool for predicting lipophilicity changes resulting from carbonyl-to-oxetane substitutions, despite the limited training data available for amino-oxetane structures.
We then subjected the pairs to a pH stability assay. Most of the surveyed pairs demonstrated good stability for 24 h under acidic, neutral, and basic conditions (Figure). Amino-oxetane 1a exhibited reduced stability under neutral and basic conditions, however, this is likely due to solubility limitations in the assay rather than intrinsic chemical instability. Additionally, some degradation was also observed in the matched molecular pair 7, which we speculate may be attributed to hydrolytic cleavage of the benzodioxole ring despite the stabilizing difluoride motif.? These findings indicate that amino-oxetanes possess comparable chemical stability across a range of pH conditions to their benzamide analogues.
Stability of selected pairs (indicated by % of compound remaining) at different time points at different pH (1.2, 7.4, 10.9).
Kinetic solubility of selected amino-oxetanes and benzamides was subsequently assessed across acidic, neutral, and basic conditions (Figure). Although the aqueous solubility varied depending on the peripheral substituents, amino-oxetanes were generally more soluble than their benzamide counterparts, with the exception of amino-oxetane 5a. Specifically, aniline-containing amino-oxetanes 6a and 8a, and benzylamine-containing amino-oxetane 2a exhibited higher solubility across all tested pH values, presumably due to disruption of the conjugated π-system. Amino-oxetanes 3a and 4a displayed solubility comparable to their analogous benzamides 3b and 4b. Despite the overall low solubility of pair 1 under neutral and basic conditions, amino-oxetane 1a showed markedly improved solubility at pH 3, likely due to protonation of the basic amine. Conversely, matched molecular pair 7 remained poorly soluble under all conditions tested. Taken together, these results suggest that amino-oxetanes offer a solubility advantage over benzamides in many cases, particularly under acidic conditions.?
Kinetic solubility of selected pairs.
The influence of basicity is an ongoing rationale for inclusion of the oxetanes in drug candidates, providing the potential to attenuate off-target effects including hERG channel inhibition. In early studies, Carreira demonstrated that the inclusion of an oxetane in the α-, β-, or γ-position decreased neighboring amine basicity.? As comparison against the benzamide analogue is not possible, we instead looked at the head-to-head comparison between amino-oxetane 4a and benzylamine 18 (Figure). Amino-oxetane 4a was considerably less basic (pK aH = 4.61, calculated = 5.44) compared to benzylamine 18 (pK aH = 7.25, calculated = 6.68), clearly demonstrating the potential of oxetanes to attenuate amine basicity. Notably, the calculated pK aH for both evaluated amino-oxetanes were similar to experimentally obtained values (within 1 pK a unit) despite the lack of similar substructures within the database (Table S2 for further data).
Comparison of pK aH data.
Clearance by human hepatocytes (HHEPG) for selected amino-oxetane and benzamide pairs were generally within an acceptable range (<150 μL min^–1^ million^–1^, Figure). Little difference in HHEPG clearance was observed for most pairs with more pronounced changes between pairs in accordance with previous studies.? Only amino-oxetane 7a demonstrated a significantly higher clearance than its benzamide counterpart, although remaining within an acceptable range.
Clearance by human hepatocytes of selected pairs.
All pairs also demonstrated excellent permeability in a Ralph Russ Canine Kidney (RRCK) cell-based permeability assay. No significant difference was observed between the amino-oxetane and benzamide series, with values ranging from 25 to 50 × 10^–6^ cm s^–1^ (see Table S3).
Through analysis of the physicochemical properties, we have demonstrated that the amino-oxetane motif is not a liability and retains similar lipophilicity, chemical and metabolic stability, and permeability in comparison to benzamides. The carbonyl-to-oxetane swap simultaneously confers an increase in aqueous solubility. The introduction of an oxetane also reduces the pK aH of neighboring amines allowing for its use to attenuate amine basicity.
Conformational Comparisons
To investigate the three-dimensional conformations of arylamino-oxetanes, we began with a survey of X-ray motifs commonly regarded as amide isosteres in the Cambridge Structural Database (CSD), focused on amino-oxetanes, sulfonamides, and α-trifluoroethyl amines. Notable differences in torsion angles were revealed: while amides predominantly adopt predictable cis or trans conformations (ca. 0° or 180°), amino-oxetanes favor a gauche conformation, providing an alternative exit vector for the amine substituent (FigureA). Interestingly, the torsion angle distribution of amino-oxetanes more closely resembles sulfonamides than amides, whereas α-trifluoroethyl amines exhibit a more stochastic distribution.
SCXRD analysis of amino-oxetane, benzamide, and sulfonamide structures. A. Torsion angles of commonly proposed amide bioisosteres in the CSD: amides, aminooxetanes, sulfonamides, and trifluoroethylamines. B. Torsion angles of pairs 2 and 6 determined by SCXRD. C. Change in hydrogen bonding conformation of amide and amino-oxetanes based on data available in the CSD. D. Hydrogen bond acceptor properties of amide and amino-oxetanes based on data available in the CSD. E. Torsion angles of sulfonamide and oxetane pair 2 determined by SCXRD.
To enable direct comparisons of the 3-aryl amino-oxetane functionality against benzamides, we recrystallized pairs 2 and 6 for analysis by single-crystal X-ray diffraction (SCXRD). Structural overlays (amide: blue; oxetane: pink; FigureB) highlight the conformational shift induced by introduction of the oxetane. Further analysis of N-benzyl benzamide derivatives bearing varying substituents on the benzamide ring confirmed that electronic effects exert minimal influence on torsion angles, especially in comparison to the influence of the carbonyl-to-oxetane swap (). While oxetanes cannot be conformationally considered direct isosteres of amides, they introduce orthogonal conformational control, including altered exit vectors, enabling the development of alternative electron density surfaces in drug design.
These conformational changes also affect hydrogen bonding morphology within the crystal structures (FigureC). For example, the amide 2b forms a hydrogen-bonded chain (NH···O: 1.94(2) Å), while the oxetane 2a forms an hydrogen-bonded ring (NH···O: 2.21(1) Å). Presumably the change from chain to ring is a consequence of the modified torsion angle between the pair, with the gauche conformation of the oxetane promoting the ring arrangement. These observations confirm that the aminooxetane can act both as an H-bond donor and acceptor, as would be expected for an amide.
Encouraged by these findings, we analyzed hydrogen bond acceptor angles and distances in the CSD. Similar angle and distance distributions were observed for hydrogen bonding to the oxetane oxygen versus the amide carbonyl oxygen (FigureD). There were only limited examples of amino-oxetane NH hydrogen bond donors in the CSD; hence the same analysis could not be performed.
To further compare torsion angles of oxetane and sulfonamide derivatives, we synthesized and recrystallized an analogous sulfonamide and amino-oxetane pair (2a and 2c) and performed SCXRD analysis. The results showed that the torsion angle of the amino-oxetane (70.6°) more closely resembles that of the sulfonamide (60.0°), contrasting with the amide (176.4°, FigureE). By comparison to sulfonamides, amino oxetanes offer lower molecular weight and reduction of the NH acidity commonly associated with secondary sulfonamides. Indeed, secondary sulfonamides can be significantly ionized at physiological pH.? In addition, amino oxetanes exhibit lower TPSA (TPSA = 30.49 and 55.40 Å^2^ for 2a and 2c). Taken together, these features suggest aminooxetanes may provide improvements to CNS penetration of acidic secondary sulfonamide derivatives, while maintaining key properties as described by CNS MPO (see Table S1 for further details).? Moreover, sulfonamides display a slightly longer hydrogen bond and greater hydrogen bond angle, indicating a less pronounced H-bonding acceptor ability, which is distinct from the hydrogen bond properties seen in amides and aminooxetanes (see SI for details). These insights highlight the conformational properties of amino-oxetanes, which align more closely with sulfonamides while preserving the essential hydrogen-bonding interactions seen in amides. This supports their utility in drug design through orthogonal conformational control, access to novel structural opportunities, and as potential mimics for both amides and sulfonamides.
We next examined the barrier to rotation around the C–N bond computationally (Figure, B3LYP/6-311G+dp-GD3BJ; see SI for details). Amino-oxetanes demonstrated a low rotational barrier (<10 kcal mol^–1^, FigureA and see SI for other example aminooxetanes), which is indicative of free rotation in water.?
Computational studies on the molecular structure and rotational bond energies of amino-oxetanes, amides, and sulfonamides. A. Stable minima and transition states of rotation around the C–N bond in amino-oxetane 19a, benzamide 19b, and sulfonamide 19d. Calculations performed at B3LYP/6-311G+dp-GD3BJ, SMD = water level of theory. ΔG quoted in kcal mol–1 compared to the lowest energy minima. B. Comparison of the torsion angles between moclobemide 20b and oxetane analogue 20a by comparison of single crystal X-ray structures (for synthesis of 20a see reference ). C. Computational comparison of conformational energy penalty of ethamivan, amprenavir, and oxetane analogues 21a and 22a to adopt protein bound conformations. Torsion angles (τ) refer to the state of rotation around the C–N bond with respect to the plane through Cq(aromatic)–Cq–N.
Further NBO analysis of the nitrogen hybridization of the lowest energy conformations of aminooxetane 19a indicated a pyramidal-like sp^2^–sp^3^ hybridization (sp^2.2^; vs sp^1.7^ for the benzamide 19b). In contrast, benzamides displayed predictably high barriers to rotation (>20 kcal mol^–1^). Three minima were observed for both amino-oxetane 19a and sulfonamide 19c, indicative of the three staggered conformers, while benzamide 19b preferred a periplanar conformation.
To directly compare the conformation of drug analogues, we synthesized and obtained a crystal structure of oxetanomoclobemide (20a). Comparison with the crystal structure of moclobemide (20b) obtained from the CSD (WEWPET),? again demonstrated the difference in preferred torsion angles around the central C–N bond in the solid state (FigureB).
The lower rotational barrier of aminooxetanes provides the potential to more flexibly accommodate binding sites in proteins of interest. The distortion energies from minimum energy conformation for ligands to adopt their bioactive conformations has previously been calculated to be ≤3 kcal mol^–1^ for around 70% of studied ligand-protein complexes.? To provide a quantitative comparison, we investigated the in silico conformational energy penalty (ΔG of distortion) associated with adopting the bioactive (protein-bound) conformation of benzamide drug ethamivan (21b), and sulfonamide amprenavir (22c) and the oxetane analogues (21a, 22a), with a fixed dihedral angle (FigureC and see SI for details). For the benzamide drug ethamivan 21b (PDB: 2XDL),? as expected minimal distortion in the amide was observed (2.1 kcal mol^–1^). For the corresponding aminooxetane 21a to adopt this “planar” conformation as in the amide would cost 5.1 kcal mol^–1^. Such an increase in binding energy would suggest up to a 100-fold decrease in affinity.?
In contrast, for the sulfonamide drug amprenavir 22c (PDB: 3NU3),? the distortion energy in the binding conformation vs minimal energy conformation for the native sulfonamide was 3.8 kcal mol^–1^. Amino-oxetane analogue 22a showed a very similar value of 3.4 kcal mol^–1^ indicating greater conformational compatibility and potential binding affinity compared to the carbonyl-to-oxetane swap.
Conclusions
In summary, we have analyzed the physicochemical properties and conformation of amino-oxetanes, with direct comparisons to analogous benzamides. We have demonstrated that the amino-oxetanes are chemically stable under a range of pH values. No change in lipophilic profile, metabolic stability, or permeability is observed when compared to benzamide derivatives. In many cases, significant increases in solubility were observed by replacement of the carbonyl with an oxetane. Additionally, amine basicity was significantly reduced by introduction of an oxetane adjacent to an alkyl amine. Finally, we have highlighted the utility of calculated ADME values in predicting property changes from the carbonyl-to-oxetane replacement, even in the context of a limited training set of aminooxetanes. With improved synthetic access to these motifs, we anticipate further refinements in predictive models to support drug discovery campaigns.
Analysis of X-ray crystal structures of 3-aryl-3-amino-oxetanes and benzamides in the CSD has demonstrated the conformational shift induced by the carbonyl-to-oxetane swap. Amino-oxetanes adopt a gauche conformation, providing a distinct amine exit vector in comparison to the planar benzamides, while maintaining H-bonding ability. The torsion angles and exit vectors of the amino-oxetanes are similar to those of sulfonamides, potentially providing a new isosteric replacement.? DFT calculations have also demonstrated amino-oxetanes to be freely rotating in water due to their low barrier of rotation, while benzamides remain conformationally restricted. Computational studies indicated that the distortion energy associated with aminooxetane drug analogues adopting the protein bound conformation of a benzamide drug is higher than that of adopting the conformation of a sulfonamide drug.
We conclude that although amino-oxetanes may not be a direct like-for-like replacement of amides, they maintain similar properties while providing an alternative chemical topology and unlocking the potential to explore new binding sites. We propose amino-oxetanes as interesting motifs in their own right, as well as potential sulfonamide isosteres, and an attractive design option to modulate physicochemical properties without introduction of inherent liabilities.
Experimental Section
General Experimental Considerations
All reactions were run under an inert atmosphere (Ar) with flame-dried glassware, using standard techniques unless otherwise specified. Anhydrous solvents were obtained by filtration through drying columns (CH_2_Cl_2_ and THF) or were purchased from Thermo Scientific Chemicals and used as supplied (MeOH, MeCN, DMSO, DMF, and 1,4-dioxane). Where stated, solvents were degassed by sparging with Ar for 30 min. Water for aqueous solutions and reaction quenches was deionized by electrodeionization using an Arium Advance EDI water purification system. Reactions in sealed tubes were run using Biotage microwave vials (0.5–2 mL, 2–5 mL, 10–20 mL) and aluminum caps with molded butyl/PTFE septa (1,4-dioxane, THF, DMSO, DMF) or simple butyl septa (MeCN). Liquid commercial amines were distilled over KOH pellets before use. Anhydrous K_2_CO_3_ (≥98%, powder, 325 mesh) was purchased from Sigma-Aldrich and flamedried before use. K_3_PO_4_ (≥98%) was purchased from Sigma-Aldrich and ground into a fine powder before use. Cs_2_CO_3_ (99%) was purchased from Sigma-Aldrich and oven-dried before use. All other commercial reagents were used as supplied or purified by standard techniques where necessary.
Manual flash column chromatography was performed using 230–400 mesh silica, with the indicated solvent system according to standard techniques. Automated flash column chromatography was carried out using a Biotage Selekt system with Biotage Sfär HC cartridges (5–25 g) under the indicated conditions. Visualization of the developed chromatogram was performed by UV absorbance (254 nm) and staining with aqueous potassium permanganate solution, aqueous cerium molybdate solution, or *p-*anisaldehyde in ethanol. Infrared spectra (n max, FTIRATR) were recorded in reciprocal centimeters (cm^–1^) using an Agilent Cary 630 FTIR spectrometer. Nuclear magnetic resonance spectra were recorded on 400 or 500 MHz spectrometers. Chemical shifts for ^1^H NMR spectra were recorded in parts per million (ppm) from tetramethylsilane with the residual protonated solvent resonance as the internal standard (CDCl_3_: δ 7.27 ppm, (CD_3_)2_SO: δ 2.50 ppm). Data was reported as follows: chemical shift (multiplicity [s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad], coupling constant (in Hz), integration, and assignment). All multiplet signals were quoted over a chemical shift range. ^13^C NMR spectra were recorded with complete proton decoupling. Chemical shifts were reported in parts per million from tetramethylsilane with the solvent resonance as the internal standard (^13^CDCl_3: δ 77.0 ppm, (CD_3_)2_SO: δ 39.5 ppm). Assignments of ^1^H and ^13^C spectra were based upon the analysis of d and J values. ^19^F NMR spectra were recorded with complete proton decoupling. Chemical shifts are reported in parts per million. ^19^F NMR spectra are indirectly referenced to CFCl_3 automatically by direct measurement of the absolute frequency of the deuterium lock signal by the spectrometer hardware. Melting points were obtained using a Stuart SMP10 digital melting point apparatus and are uncorrected. High-resolution mass spectra (HRMS) were obtained through the Imperial College London mass spectrometry service. HRMS analyses were performed using an electrospray ion source (ESI) or atmospheric pressure chemical ionization (APCI) using an atmospheric solids analysis probe (ASAP). ESI was performed using a Waters LCT Premier (ES-TOF) equipped with an ESI source operated in positive ion mode or a Thermo Scientific Q-Exactive/Dionex Ultimate 3000. APCI was performed using a Thermo Scientific Q-Exactive/Dionex Ultimate 3000 using an ASAP to insert samples into the APCI source operated in positive or negative mode. The sample was introduced at ambient temperature and the temperature increased until the sample vaporized.
p-Methoxyphenyl oxetane sulfonyl fluoride 13 (PMP OSF) and ρ-triisopropylsiloxyphenyl oxetane sulfonyl fluoride 15 (OTIPS OSF),? t-butyl (S)-(1-(dimethylamino)-1-oxopropan-2-yl)carbamate 23,? 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-(trifluoromethyl)pyrimidine 24,? and oxetane triflate 25 were synthesized according to previously reported conditions.? The purity of the synthesized compounds was determined by quantitative ^1^H NMR spectroscopy using 1,3,5-trimethoxybenzene (≥99%) as an internal standard and was at least of 95% purity unless otherwise stated.
General Procedure A: Anhydrous K_2_CO_3_ (1.3 equiv) was added to a reaction tube and flame-dried. Amine (1.2 equiv) and oxetane sulfonyl fluoride (1.0 equiv) were added sequentially and the reaction tube was sealed. Anhydrous acetonitrile (0.3 M) was added, and the reaction mixture was stirred at 60 °C for 2 h. After cooling to rt, the reaction mixture was diluted with EtOAc and filtered through a plug of Celite, eluting with further EtOAc. The solvent was then removed in vacuo. Purification by column chromatography under the stated conditions afforded the amino-oxetane.
General Procedure B: Methoxybenzoyl chloride 17 (68 μL, 0.50 mmol, 1.0 equiv) was added to a solution of amine (0.55 mmol, 1.1 equiv) and triethylamine (87 μL, 0.63 mmol, 1.3 equiv) in CH_2_Cl_2_ (1 mL, 0.5 M). After stirring at rt for 15 h, the reaction mixture was quenched with aq. HCl (1 M, 10 mL) and diluted with CH_2_Cl_2_ (10 mL). The layers were separated, and the aqueous layer was extracted with CH_2_Cl_2_ (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo. Purification by column chromatography under the stated conditions afforded the benzamide.
N-(((3R,5R,7R)-Adamantan-1-yl)Methyl)-3-(4-Methoxyphenyl)Oxetan-3-Amine
(1a)
Prepared according to General Procedure A using K_2_CO_3_, (36.0 mg, 0.26 mmol, 1.3 equiv), 1-adamantanemethylamine (43 μL, 0.24 mmol, 1.2 equiv) and PMP OSF 13 (49.2 mg, 0.2 mmol, 1.0 equiv). Purification by flash column chromatography (10% EtOAc/hexane) afforded amino-oxetane 1a as a colorless oil (45.0 mg, 68%). R_ f _ = 0.18 (10% EtOAc/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 7.29–7.25 (m, 2H, 2 × Ar–CH), 6.95–6.90 (m, 2H, 2 × Ar–CH), 4.94 (dd, J = 6.2, 0.6 Hz, 2H, CHHOCHH), 4.74 (dd, J = 6.2, 0.6 Hz, 2H, CHHOCHH), 3.82 (s, 3H, OCH_3_), 3.16–3.08 (m, 1H, CH), 2.09–2.02 (m, 2H, NCH(CHH)2), 1.96 (br s, 1H, NH), 1.74–1.45 (m, 4H, NCH(CH_2_)2_CH_2 + NCH(CHH)2). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
N-(((3R,5R,7R)-Adamantan-1-yl)Methyl)-4-Methoxybenzamide (1b)
Prepared according to General Procedure B using ((3r,5r,7r)-adamantan-1-yl)methanamine (97 μL, 0.55 mmol, 1.1 equiv). Purification by flash column chromatography (1% MeOH/CH_2_Cl_2_) afforded benzamide 1b as a white solid (115 mg, 77%). R_ f _ = 0.22 (1% MeOH/CH_2_Cl_2_); mp = 195–196 °C; IR (film)/cm^–1^ 3354 (NH), 2892, 2836, 1632 (CO), 1600, 1543, 1505, 1256, 1174, 1036, 838, 767; ^1^H NMR (400 MHz, CDCl_3_) δ 7.77–7.72 (m, 2H, 2 × Ar–CH), 6.96–6.91 (m, 2H, 2 × Ar–CH), 6.12 (s, 1H, NH), 3.85 (s, 3H, OCH_3_), 3.15 (d, J = 6.4 Hz, 2H, CH2N), 2.00 (s, 3H, 3 × CH), 1.76–1.69 (m, 3H, 3 × CHH), 1.68–1.69 (m, 3H, 3 × CHH), 1.56 (d, J = 2.4 Hz, 6H, 3 × CH_2_); ^13^C NMR (101 MHz, CDCl_3_) δ 167.2 (CO), 162.0 (ArC_q_OMe), 128.6 (2 × Ar–CH), 127.3 (Ar–C_q_CO), 113.7 (2 × Ar–CH), 55.4 (OCH_3_), 51.3 (CH_2_N), 40.3 (3 × CH_2_), 36.9 (3 × CH_2_), 34.0 (C_q_), 28.2 (3 × CH); HRMS (TOF-MS-ES^+^) m/z calcd for C_19_H_26_NO_2_ ^+^ [M + H]^+^: 300.1958; found 300.1962.
N-Benzyl-3-(4-Methoxyphenyl)Oxetan-3-Amine
(2a)
Prepared according to General Procedure A using K_2_CO_3_, (36.0 mg, 0.26 mmol, 1.3 equiv), benzylamine (43 μL, 26 μL, 1.2 equiv) and PMP OSF 13 (49.2 mg, 0.2 mmol, 1.0 equiv). Purification by flash column chromatography (40% EtOAc/hexane) afforded amino-oxetane 2a as a white solid (48.0 mg, 89%). R_ f _ = 0.29 (40% EtOAc/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 7.44–7.39 (m, 2H, 2 × Ar–CH), 7.36–7.30 (m, 4H, 4 × Ar–CH), 7.29–7.23 (m, 1H, Ar–CH), 6.99–6.94 (m, 2H, 2 × Ar–CH), 4.97 (d, J = 6.4 Hz, CHHOCHH), 4.77 (d, J = 6.4 Hz, CHHOCHH), 3.85 (s, 3H, OCH_3_), 3.56 (s, 2H, CH_2_Ph), 2.05 (br s, 1H, NH). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
N-Benzyl-4-Methoxybenzamide (2b)
Prepared according to General Procedure B using benzylamine (60 μL, 0.55 mmol, 1.1 equiv). Purification by flash column chromatography (5% Et_2_O/CH_2_Cl_2_) afforded benzamide 2b as a white solid (102 mg, 84%). R_ f _ = 0.32 (5% Et_2_O/CH_2_Cl_2_); mp = 134–136 °C, IR (film)/cm^–1^ 3254 (NH), 1628 (CO), 1609, 1558, 1498, 1323, 1248, 1174, 987, 824, 723, 685; ^1^H NMR (400 MHz, CDCl3) δ: 7.80–7.74 (m, 2H, 2 × Ar–CH), 7.39–7.26 (m, 5H, 5 × Ar–CH), 6.94–6.89 (m, 2H, 2 × Ar–CH), 6.45 (s, 1H, NH), 4.63 (d, J = 5.7 Hz, 2H, CH_2_Ph), 3.85 (s, 3H, OCH_3_); ^13^C NMR (101 MHz, CDCl_3_) δ: 166.8 (CO), 162.2 (Ar–C_q_OMe), 138.4 (Ar–C_q_CH_2_), 128.7 (2 × Ar–CH), 128.7 (2 × Ar–CH), 127.9 (2 × Ar–CH), 127.5 (Ar–CH), 126.6 (Ar–C_q_CO), 113.7 (2 × Ar–CH), 55.4 (OCH_3_), 44.0 (CH_2_). The observed characterization data (^1^H NMR, ^13^C NMR) was consistent with that previously reported.?
N-Benzyl-4-Methoxybenzenesulfonamide (2c)
Et_3_N (0.16 mL, 2.2 mmol, 1.7 equiv) and benzylamine (0.15 mL, 1.4 mmol, 1.05 equiv) were added dropwise to a solution of 4-methoxybenzenesulfonyl chloride (276 mg, 1.3 mmol, 1.0 equiv) in anhydrous CH_2_Cl_2_ (10 mL, 0.1 M) at 0 °C. After stirring at rt for 16 h, the solvent was removed in vacuo. Purification by recrystallization (EtOH) afforded sulfonamide 2c as clear colorless solid blocks (366 mg, 99% yield). R_ f _ = 0.25 (20% EtOAc/pentane); mp = 105–106 °C; IR (film)/cm^–1^ 2880, 2600, 1724, 1597, 1497, 1323, 1258, 1154, 1096, 833, 563; ^1^H NMR (400 MHz, MeOD) δ 7.82–7.71 (m, 2H, 2 × Ar–CH), 7.29–7.16 (m, 5H, 5 × Ar–CH), 7.07–6.98 (m, 2H, 2 × Ar–CH), 4.01 (s, 2H, CH_2_Ph), 3.85 (s, 3H, OCH_3_); ^13^C NMR (101 MHz, MeOD) δ 164.3 (ArC_q_OMe), 138.7 (ArC_q_CH_2_), 133.5 (ArC_q_SO_2_), 130.1 (2 × Ar–CH), 129.4 (2 × Ar–CH), 128.9 (2 × Ar–CH), 128.4 (Ar–CH), 115.2 (2 × Ar–CH), 56.2 (OCH_3_), 47.9 (CH_2_Ph). The observed characterization data (R_ f _, ^1^H, ^13^C) were consistent with that previously reported.?
(S)-2-((3-(4-Methoxyphenyl)Oxetan-3-yl)Amino)-N,N-Dimethylpropanamide (3a)
Trifluoroacetic acid (2.7 mL, 35 mmol, 10 equiv) was added dropwise to a solution of Boc-protected amine 23 (756 mg, 3.5 mmol, 1.0 equiv) in CH_2_Cl_2_ (18 mL, 0.2 M) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min, warmed to 25 °C and stirred for a further 2.5 h. Concentration in vacuo afforded 1-(dimethylamino)-1-oxopropan-2-aminium trifluoroacetate (810 mg) of which a portion was used in the next step without further purification. Anhydrous K_2_CO_3_ (77.4 mg, 0.56 mmol, 2.8 equiv) was added to a reaction tube and flame-dried. (S)-1-(Dimethylamino)-1-oxopropan-2-aminium trifluoroacetate (58.1 mg, 0.25 mmol, 1.25 equiv) and PMP OSF 13 (49.2 mg, 0.20 mmol, 1.0 equiv) were added sequentially and the reaction tube was sealed. Anhydrous acetonitrile (0.66 mL, 0.3 M) was added, and the reaction mixture was stirred at 60 °C for 2 h. After cooling to rt, the reaction mixture was diluted with EtOAc (5 mL) and filtered through a plug of Celite, eluting with further EtOAc (3 × 10 mL). The solvent was then removed in vacuo. Purification by flash chromatography (50% acetone/hexane) afforded amino-oxetane 3a as a colorless oil (19.7 mg, 35%). R_ f _ = 0.20 (50% acetone/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 7.32–7.28 (m, 2H, 2 × Ar–CH), 6.94–6.90 (m, 2H, 2 × Ar–CH), 4.93 (d, J = 6.4 Hz, 1H, CHHOCH_2_), 4.84 (d, J = 6.6 Hz, 1H, CHHOCH_2_), 4.82 (d, J = 6.6 Hz, 1H, CH_2_OCHH), 4.72 (d, J = 6.4 Hz, 1H, CH_2_OCHH), 3.83 (s, 3H, OCH_3_), 3.42 (q, J = 6.9 Hz, 1H, CH), 2.88 (s, 3H, NCH_3_), 2.74 (s, 3H, NCH_3_), 1.75 (br s, 1H, NH), 1.13 (d, J = 6.9 Hz, 3H, CHCH_3_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
N-(1-(Dimethylamino)-1-Oxopropan-2-yl)-4-Methoxybenzamide
(3b)
Trifluoroacetic acid (2.7 mL, 35 mmol, 10 equiv) was added dropwise to a solution of Bocprotected amine 23 (756 mg, 3.5 mmol, 1.0 equiv) in CH_2_Cl_2_ (18 mL, 0.2 M) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min, warmed to 25 °C and stirred for a further 2.5 h. Concentration in vacuo afforded 1-(dimethylamino)-1-oxopropan-2-aminium trifluoroacetate (810 mg) of which a portion was used in the next step without further purification. Methoxybenzoyl chloride (68 μL, 0.50 mmol, 1.0 equiv) was added to a solution of 1-(dimethylamino)-1-oxopropan-2-aminium trifluoroacetate (127 mg, 0.55 mmol, 1.1 equiv) and triethylamine (174 μL, 1.25 mmol, 2.5 equiv) in CH_2_Cl_2_ (1 mL, 0.5 M). After stirring at rt for 14 h, the reaction mixture was quenched with aq. HCl (1 M, 10 mL) and diluted with CH_2_Cl_2_ (10 mL). The layers were separated, and the aqueous layer was extracted with CH_2_Cl_2_ (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo. Purification by flash chromatography (50% acetone/hexane) afforded benzamide 3b as a colorless paste (98.0 mg, 78%). R_ f _ = 0.25 (50% acetone/hexane); IR (film)/cm^–1^ 3317 (NH), 2937, 1625 (CO), 1620 (CO), 1494, 1304, 1248, 1177, 1107, 1028, 846, 767, 730; ^1^H NMR (400 MHz, CDCl_3_) δ 7.82–7.77 (m, 2H, 2 × Ar–CH), 7.24 (d, J = 6.9 Hz, 1H, NH), 6.95–6.90 (m, 2H, 2 × Ar–CH), 5.09 (p, J = 6.9 Hz, 1H, CH), 3.86 (s, 3H, OCH_3_), 3.13 (s, 3H, NCH_3_), 3.02 (s, 3H, NCH_3_), 1.44 (d, J = 6.8 Hz, 3H, CH_3_); ^13^C NMR (101 MHz, CDCl_3_) δ 172.5 (CO), 165.7 (CO), 162.0 (ArC_q_OMe), 128.7 (2 × Ar–CH), 126.1 (Ar–C_q_CO), 113.4 (2 × Ar–CH), 55.2 (OCH_3_), 45.4 (CH), 36.8 (NCH_3_), 35.6 (NCH_3_), 18.5 (CH_3_); HRMS (TOF-MS-ES^+^) m/z calcd for C_13_H_19_N_2_O_3_ ^+^ [M + H]^+^: 251.1390; found 251.1394.
4-(3-(4-Methoxyphenyl)Oxetan-3-yl)Morpholine (4a)
Prepared according to General Procedure A using K_2_CO_3_, (36.0 mg, 0.26 mmol, 1.3 equiv), morpholine (21 μL, 0.24 mmol, 1.2 equiv) and PMP OSF 13 (49.2 mg, 0.2 mmol, 1.0 equiv). Purification by flash column chromatography (80% EtOAc/hexane) afforded amino-oxetane 4a as a white solid (43.0 mg, 86%). R_ f _ = 0.21 (80% EtOAc/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 7.00–6.96 (m, 2H, 2 × Ar–CH), 6.94–6.88 (m, 2H, 2 × Ar–CH), 4.90 (d, J = 5.9 Hz, 2H, CHHOCHH), 4.87 (d, J = 5.9 Hz, 2H, CHHOCHH), 3.83 (s, 3H, OCH_3_), 3.74 (t, J = 4.6 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 2.31 (t, J = 4.6 Hz, 4H, CH_2_NCH_2_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
(4-Methoxyphenyl)(Morpholino)Methanone (4b)
Prepared according to General Procedure B using morpholine (48 μL, 0.55 mmol, 1.1 equiv). Purification by flash column chromatography (60% EtOAc/pentane) afforded benzamide 4b as a colorless paste (65.0 mg, 59%). R_ f _ = 0.20 (60% EtOAc/pentane); IR (film)/cm^–1^ 2959, 2918, 2851, 1625 (CO), 1513, 1420, 1245, 1170, 1110, 1021, 839; ^1^H NMR (400 MHz, CDCl_3_) δ 7.43–7.37 (m, 2H, 2 × Ar–CH), 6.96–6.90 (m, 2H, 2 × Ar–CH), 3.84 (s, 3H, OCH_3_), 3.80–3.51 (br s, 8H, 4 × CH2); ^13^C NMR (101 MHz, CDCl_3_) δ 170.3 (CO), 160.8 (Ar–C_q_OMe), 129.1 (2 × Ar–CH), 127.3 (Ar–C_q_CO), 113.7 (2 × Ar–CH), 66.8 (CH_2_OCH_2_), 55.3 (CH_3_), 47.8 (br, CH_2_N), 43.7 (br, CH_2_N). The observed characterization data (^1^H NMR, ^13^C NMR) was consistent with that previously reported.?
1-(4-(Cyclopropyl(3-(4-Methoxyphenyl)Oxetan-3-yl)Amino)Piperidin-1-yl)Ethan-1-One
(5a)
Prepared according to General Procedure A using K_2_CO_3_, (36.0 mg, 0.26 mmol, 1.3 equiv), 1-(4-(cyclopropylamino)piperidin-1-yl)ethan-1-one (43.7 mg, 0.24 mmol, 1.2 equiv) and PMP OSF 13 (49.2 mg, 0.2 mmol, 1.0 equiv). Purification by flash column chromatography (40% acetone/hexane) afforded amino-oxetane 5a as a white solid (26.4 mg, 38%). R_ f _ = 0.24 (40% acetone/hexane);^1^H NMR (400 MHz, CDCl_3_) δ 7.42–7.29 (m, 2H, 2 × Ar–CH), 7.08–6.83 (m, 2H, 2 × Ar–CH), 5.13–4.99 (m, 2H, CHHOCHH), 4.80 (d, J = 5.7 Hz, 1H, CHHOCH_2_), 4.78 (d, J = 5.7 Hz, 1H, CH_2_OCHH), 4.56 (ddt, J = 13.3, 4.4, 2.4 Hz, 1H, CHHNCH_2_), 3.83 (s, 3H, OCH_3_), 3.70 (ddt, J = 13.6, 4.2, 2.4 Hz, 1H, CH_2_NCHH), 2.86 (td, J = 13.2, 2.1 Hz, 1H, CHHNCH_2_), 2.52 (tt, J = 11.8, 3.6 Hz, 1H, CH), 2.33 (ddt, J = 13.3, 4.4, 2.4 Hz, 1H, CH_2_NCHH), 2.03 (s, 3H, C(O)CH_3_), 1.72–1.65 (m, 1H, CH), 1.61–1.48 (m, 2H, 2 × CHHCH), 1.34 (d, J = 12.6 Hz, 2H, 2 × CHHCH), 0.59–0.47 (m, 4H, 2 × CH_2_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
N-(1-Acetylpiperidin-4-yl)-N-Cyclopropyl-4-Methoxybenzamide (5b)
Prepared according to General Procedure B using 1-(4-(cyclopropylamino)piperidin-1-yl)ethan-1-one (100 mg, 0.55 mmol, 1.1 equiv). Purification by flash column chromatography (5% MeOH/CH_2_Cl_2_) afforded benzamide 5b as a white solid (147 mg, 93%). R_ f _ = 0.15 (5% MeOH/CH_2_Cl_2_); mp = 104–105 °C; IR (film)/cm^–1^ 2959, 1632 (CO), 1602 (CO), 1513, 1420, 1364, 1326, 1241, 1172, 1021, 834, 764, 715; ^1^H NMR (400 MHz, CDCl_3_) δ 7.49–7.44 (m, 2H, 2 × Ar–CH), 6.91–6.86 (m, 2H, 2 × Ar–CH), 4.79 (ddd, J = 6.5, 3.9, 2.1 Hz, 1H, NCHH), 4.42–4.32 (m, 1H, CH), 3.95–3.87 (m, 1H, NCHH), 3.84 (s, 3H, OCH_3_), 3.23–3.12 (m, 1H, NCHH), 2.67–2.54 (m, 2H, NCHH + CH), 2.13 (s, 3H, CH_3_), 1.96 (ddd, J = 24.0, 12.4, 4.2 Hz, 4H, 2 × CH_2_CH), 0.70–0.53 (m, 2H, 2 × CHH), 0.50–0.35 (m, 2H, 2 × CHH); ^13^C NMR (101 MHz, CDCl_3_) δ 172.5 (CO), 168.8 (CO), 160.6 (Ar–C_q_OMe), 129.9 (Ar–C_q_CO), 129.4 (2 × Ar–CH), 113.1 (2 × Ar–CH), 56.4 (CH), 55.3 (OCH_3_), 46.2 (NCH_2_), 41.4 (NCH_2_), 31.1 (CHCH_2_), 30.0 (CHCH_2_), 28.8 (CH), 21.4 (CH_3_), 10.1 (CH_2_), 10.0 (CH_2_); HRMS (TOFMSES^+^) m/z calcd for C_18_H_25_N_2_O_3_ ^+^ [M + H]^+^: 317.1860; found 317.1866.
N-(4-((3-(4-Methoxyphenyl)Oxetan-3-yl)Amino)Phenyl)Acetamide
(6a)
Prepared according to General Procedure A using K_2_CO_3_, (36.0 mg, 0.26 mmol, 1.3 equiv), N-(4-aminophenyl) acetamide (36 μL, 0.24 mmol, 1.2 equiv) and PMP OSF 13 (49.2 mg, 0.2 mmol, 1.0 equiv). Purification by flash column chromatography (60% acetone/hexane) afforded aminooxetane 6a as a white solid (58.0 mg, 93%). R_ f _ = 0.30 (60% acetone/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 7.57–7.53 (m, 2H, 2 × Ar–CH), 7.19–7.15 (m, 2H, 2 × Ar–CH), 7.13 (br s, 1H, AcNH), 6.94–6.87 (m, 2H, 2 × Ar–CH), 6.26–6.22 (m, 2H, 2 × Ar–CH), 4.95 (d, J = 6.3 Hz, 2H, CHHOCHH), 4.85 (d, J = 6.3 Hz, 2H, CHHOCHH), 4.61 (br s, 1H, NH), 3.81 (s, 3H, OCH_3_), 2.10 (s, 3H, CH_3_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
N-(4-Acetamidophenyl)-4-Methoxybenzamide (6b)
Prepared according to General Procedure B using N-(4-aminophenyl)acetamide (82.6 mg, 0.55 mmol, 1.1 equiv). Purification by flash column chromatography (45% acetone/hexane) afforded benzamide 6b as a white solid (74.0 mg, 72%). R_ f _ = 0.24 (45% acetone/hexane); mp = 265–267 °C; IR (film)/cm^–1^ 3280 (NH), 1677 (CO), 1647 (CO), 1602, 1543, 1517, 1401, 1312, 1252, 1170, 1032, 827, 760, 670; ^1^H NMR (400 MHz, (CD_3_)2_SO) δ 10.03 (s, 1H, NH), 9.90 (s, 1H, NH), 8.00–7.91 (m, 2H, 2 × Ar–CH), 7.71–7.62 (m, 2H, 2 × Ar–CH), 7.58–7.49 (m, 2H, 2 × Ar–CH), 7.10–7.01 (m, 2H, 2 × Ar–CH), 3.83 (s, 3H, OCH_3), 2.03 (s, 3H, CH_3_); ^13^C NMR (101 MHz, (CD_3_)2_SO) δ 168.0 (CO), 164.6 (CO), 161.8 (ArC_q_OMe), 135.1 (Ar–C_q_NH), 134.5 (Ar–C_q_NH), 129.5 (2 × Ar–CH), 127.0 (Ar–C_q_CO), 120.8 (2 × Ar–CH), 119.2 (2 × Ar–CH), 113.6 (2 × Ar–CH), 55.4 (OCH_3), 23.9 (CH_3_); HRMS (TOF-MS-ES^+^) m/z calcd for C_16_H_17_N_2_O_3_ ^+^ [M + H]^+^: 285.1234; found 285.1238.
2,2-Difluoro-N-(3-(4-Methoxyphenyl)Oxetan-3-yl)Benzo[d][1,3]dioxol-5-Amine (7a)
Prepared according to General Procedure A using K_2_CO_3_, (36.0 mg, 0.26 mmol, 1.3 equiv), 2,2-difluorobenzo[d][1,3]dioxol-5-amine-methane (30 μL, 0.24 mmol, 1.2 equiv) and PMP OSF 13 (49.2 mg, 0.2 mmol, 1.0 equiv). Purification by flash column chromatography (40% Et_2_O/hexane) afforded aminooxetane 7a as a white solid (66.0 mg, 98%). R_ f _ = 0.18 (40% Et_2_O/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 7.58–7.51 (m, 2H, 2 × Ar–CH), 6.97–6.91 (m, 2H, 2 × Ar–CH), 6.78 (d, J = 8.6 Hz, 1H, Ar–CH), 6.04 (d, J = 2.3 Hz, 1H, Ar–CH), 5.94 (dd, J = 8.6, 2.4 Hz, 1H, Ar–CH), 4.96 (d, J = 6.4 Hz, 2H, CHHOCHH), 4.83 (d, J = 6.4 Hz, 2H, CHHOCHH), 4.63 (br s, 1H, NH), 3.83 (s, 3H, OCH_3_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
N-(2,2-Difluorobenzo[d][1,3]dioxol-5-yl)-4-Methoxybenzamide
(7b)
Prepared according to General Procedure B using 2,2-difluorobenzo[d][1,3]dioxol-5-amine (68 μL, 0.55 mmol, 1.1 equiv). Purification by flash column chromatography (45% Et_2_O/hexane) afforded benzamide 7b as a white solid (145 mg, 90%). R_ f _ = 0.22 (45% Et_2_O/hexane); mp = 187–191 °C; IR (film)/cm^–1^ 3358 (NH), 1647 (CO), 1602, 1498, 1442, 1241, 1162, 1025, 810, 725; ^1^H NMR (400 MHz, (CD_3_)2_SO) δ 7.88–7.81 (m, 2H, 2 × ArCH), 7.77–7.70 (m, 2H, Ar–CH + NH), 7.13–6.94 (m, 4H, 4 × ArCH), 3.89 (s, 3H, OCH_3); ^13^C NMR (101 MHz, (CD_3_)2_SO) δ 165.0 (CO), 162.1 (Ar–C_q_OMe), 142.5 (Ar–C_q_O), 138.4 (Ar–C_q_O), 136.1 (Ar–C_q_NH), 131.3 (t, J = 252.4 Hz, C_q_F_2) 129.6 (2 × Ar–CH), 126.5 (Ar–C_q_CO), 115.8 (Ar–CH), 113.7 (2 × Ar–CH), 109.9 (Ar–CH), 103.0 (Ar–CH), 55.5 (OCH_3_); ^19^F NMR (377 MHz, (CD_3_)2_SO) δ −49.12; HRMS (TOF-MS-ES^+^) m/z calcd for C_15_H_12_NO_4 ^+^ [M + H]^+^: 308.0729; found 308.0733.
6-Methoxy-N-(3-(4-Methoxyphenyl)Oxetan-3-yl)Pyridin-3-Amine
(8a)
Prepared according to General Procedure A using K_2_CO_3_, (36.0 mg, 0.26 mmol, 1.3 equiv), 6-methoxypyridin-3-amine (29.8 mg, 0.24 mmol, 1.2 equiv) and PMP OSF 13 (49.2 mg, 0.2 mmol, 1.0 equiv). Purification by flash column chromatography (20% Et_2_O/CH_2_Cl_2_) afforded aminooxetane 8a as a pink solid (49.4 mg, 86%). R_ f _ = 0.28 (20% Et_2_O/CH_2_Cl_2_); mp = 74–76 °C, IR (film)/cm^–1^ 3280 (NH), 2948, 1610, 1490, 1375, 1300, 1185, 1028, 976, 816; ^1^H NMR (400 MHz, CDCl_3_) δ 7.57–7.50 (m, 2H, 2 × Ar–CH), 7.19 (d, J = 2.6 Hz, 1H, Ar–CH), 6.97–6.88 (m, 2H, 2 × Ar–CH), 6.74 (dd, J = 8.8, 3.0 Hz, 1H, Ar–CH), 6.56 (dd, J = 8.8, 0.4 Hz, 1H, Ar–CH), 4.98 (d, J = 6.4 Hz, 2H, CHHOCHH), 4.83 (d, J = 6.4 Hz, 2H, CHHOCHH), 4.32 (br s, 1H, NH), 3.82 (s, 6H, 2 × OCH_3_) ^13^C NMR (101 MHz, CDCl_3_) δ 159.0 (Ar–C_q_OMe), 157.7 (ArC_q_OMe), 135.4 (ArC_q_C_q_), 133.2 (Ar–CH), 131.8 (Ar–C_q_NH), 127.1 (2 × Ar–CH), 126.7 (Ar–CH), 114.2 (2 × Ar–CH), 110.9 (Ar–CH), 83.9 (CH_2_OCH_2_), 60.4 (C_q_), 55.3 (OCH_3_), 53.2 (OCH_3_); HRMS (TOF-MS-ES^+^) m/z calcd for C_16_H_19_N_2_O_3_ ^+^ [M + H]^+^: 287.1396; found 287.1402.
4-Methoxy-N-(6-Methoxypyridin-3-yl)Benzamide (8b)
Methoxybenzoyl chloride 17 (74 μL, 0.60 mmol, 1.0 equiv) was added to a solution of 6-methoxypyridin-3-amine (74.8 mg, 0.60 mmol, 1.0 equiv) and triethylamine (95 μL, 0.68 mmol, 1.03 equiv) in CH_2_Cl_2_ (1.1 mL, 0.55 M). After stirring at rt for 13 h, the reaction mixture was quenched with aq. HCl (1 M, 10 mL) and diluted with CH_2_Cl_2_ (10 mL). The layers were separated, and the aqueous layer was extracted with CH_2_Cl_2_ (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo. Purification by flash column chromatography (40% EtOAc/hexane) afforded benzamide 8b as a pink solid (110 mg, 78%). R_ f _ = 0.24 (40% EtOAc/hexane); mp = 180–181 °C; IR (film)/cm^–1^ 3313 (NH), 2940, 1643 (CO), 1606, 1490, 1379, 1278, 1244, 1025, 898, 820, 764, 663;^1^H NMR (400 MHz, CDCl_3_) δ 8.25 (d, J = 2.6 Hz, 1H, Ar–CH), 8.04 (dd, J = 8.9, 2.7 Hz, 1H, Ar–CH), 7.93–7.80 (m, 2H, 2 × Ar–CH), 7.63 (br s, 1H, NH), 7.07–6.91 (m, 2H, 2 × Ar–CH), 6.79 (d, J = 8.9 Hz, 1H, Ar–CH), 3.95 (s, 3H, OCH_3_), 3.89 (s, 3H, OCH_3_); ^13^C NMR (101 MHz, (CD_3_)2_SO) δ 164.8 (CO), 162.0 (Ar–C_q_OMe), 159.7 (Ar–C_q_OMe), 138.8 (Ar–CH), 132.6 (Ar–CH), 130.2 (Ar–C_q_NH), 129.5 (2 × Ar–CH), 126.4 (Ar–C_q_CO), 113.6 (2 × Ar–CH), 109.9 (Ar–CH), 55.4 (OCH_3), 53.2 (OCH_3_). The observed characterization data (^1^H NMR, ^13^C NMR) was consistent with that previously reported.?
4-(3-(4-(Pyridin-2-yloxy)Phenyl)Oxetan-3-yl)Morpholine (9a)
Amino-oxetane 14a (47 mg, 0.2 mmol, 1.0 equiv), picolinic acid (4.9 mg, 40 μmol, 20 mol %), CuI (3.8 mg, 20 μmol, 10 mol %), and K_3_PO_4_ (93 mg, 0.44 mmol, 2.2 equiv) were added to a reaction vial. The reaction vial was sealed then evacuated and backfilled with Ar three times. 2Iodopyridine (28 μL, 0.26 mmol, 1.3 equiv) and anhydrous, degassed DMSO (0.44 mL, 0.45 M) were added sequentially by syringe to the reaction vial. The reaction mixture was heated to 110 °C and stirred for 24 h. After cooling to 25 °C, water (15 mL) followed by EtOAc (15 mL) were added. The layers were separated, and the aqueous layer was extracted with EtOAc (2 ´ 15 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo. Purification by flash column chromatography (80–100% EtOAc/hexane) afforded amino-oxetane 9a as a pale-yellow solid (39 mg, 60%). R_ f _ = 0.13 (80% EtOAc/hexane);^1^H NMR (400 MHz, CDCl_3_) δ 8.22 (dd, J = 5.1, 2.0 Hz, 1H, Ar–CH), 7.71 (td, J = 7.7, 2.0 Hz, 1H, Ar–CH), 7.17 (d, J = 8.5 Hz, 2H, 2 × Ar–CH), 7.13–7.06 (m, 2H, 2 × Ar–CH), 7.02 (dd, J = 7.2, 5.0 Hz, 1H, Ar–CH), 6.94 (d, J = 8.3 Hz, 1H, Ar–CH), 4.93 (d, J = 6.0 Hz, 2H, CHHOCHH), 4.89 (d, J = 6.0 Hz, 2H, CHHOCHH), 3.75 (t, J = 4.6 Hz, 4H, 2 × OCH_2_CH_2_), 2.36 (t, J = 4.6 Hz, 4H, 2 × NCH_2_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
Morpholino(4-(Pyridin-2-yloxy)Phenyl)Methanone (9b)
Benzamide 14b (41 mg, 0.2 mmol, 1.0 equiv), picolinic acid (4.9 mg, 40 μmol, 20 mol %), CuI (3.8 mg, 20 μmol, 10 mol %), and K_3_PO_4_ (93 mg, 0.44 mmol, 2.2 equiv) were added to a reaction vial. The reaction vial was sealed then evacuated and backfilled with Ar three times. 2-Iodopyridine (28 μL, 0.26 mmol, 1.3 equiv) and anhydrous, degassed DMSO (0.44 mL, 0.45 M) were added sequentially by syringe to the reaction vial. The reaction mixture was heated to 110 °C and stirred for 24 h. After cooling to 25 °C, water (15 mL) followed by EtOAc (15 mL) were added. The layers were separated, and the aqueous layer was extracted with EtOAc (2 ´ 15 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo. Purification by flash column chromatography (60–80% EtOAc/hexane) afforded benzamide 9b as a white solid (50.0 mg, 88%). R_ f _ = 0.32 (100% EtOAc); mp = 118–120 °C; IR (film)/cm^–1^ 2963, 2919, 2853, 1629 (CO), 1590, 1506, 1463, 1426, 1260, 1242, 1112, 1013, 884, 842, 777, 547; ^1^H NMR (400 MHz, CDCl_3_) δ 8.22 (dd, J = 5.1, 2.0 Hz, 1H, Ar–CH), 7.73 (ddd, J = 8.6, 7.2, 2.0 Hz, 1H, Ar–CH), 7.50–7.44 (m, 2H, 2 × ArCH), 7.21–7.16 (m, 2H, 2 × Ar–CH), 7.08–7.02 (m, 1H, Ar–CH), 6.96 (d, J = 8.2 Hz, 1H, Ar–CH), 3.71 (br, 8H, 2 × NCH_2_CH_2_); ^13^C NMR (101 MHz, CDCl_3_) δ 169.9 (CO), 163.0 (Ar–C_q_O), 155.7 (Ar–C_q_O), 147.7 (Ar–CH), 139.7 (Ar–CH), 131.1 (Ar–C_q_C_q_), 129.1 (2 × Ar–CH), 120.8 (2 × Ar–CH), 119.1 (Ar–CH), 112.2 (Ar–CH), 66.9 (2 × NCH_2_CH_2_O); HRMS (TOF-MS-ES^+^) m/z calculated for C_16_H_17_N_2_O_3_ [M + H]: 285.1239; found 285.1246.
4-(3-(4-(Phenylsulfonyl)Phenyl)Oxetan-3-yl)Morpholine (10a)
Phenyllithium (1.82 M, 0.11 mL, 0.2 mmol, 2.0 equiv) was added dropwise to a solution of DABSO (24 mg, 0.1 mmol, 1.0 equiv) in degassed 1,4-dioxane (0.77 mL, 0.13 M). The reaction was stirred at 25 °C for 2 h. Oxetane triflate 16a (49.0 mg, 0.13 mmol, 1.3 equiv), Pd(OAc)2 (3.0 mg, 0.013 mmol, 10 mol %), Cs_2_CO_3_ (65.0 mg, 0.2 mmol, 2.0 equiv), and Xantphos (7.7 mg, 0.013 mmol, 10 mol %) were added to a separate vial. The vial was evacuated and backfilled with nitrogen three times. The 1,4-dioxane suspension was then added via syringe. After stirring at 110 °C for 16 h, the reaction was cooled to rt and CH_2_Cl_2_ (10 mL) was added. The resulting suspension was filtered through Celite and concentrated in vacuo. Purification by flash column chromatography (50% EtOAc/CH_2_Cl_2_) afforded amino-oxetane 10a as a colorless oil (21.0 mg, 58%). Rf = 0.24 (50% EtOAc/CH_2_Cl_2_);^1^H NMR (400 MHz, CDCl_3_) δ 8.01–7.94 (m, 4H, 4 × Ar–CH), 7.64–7.58 (m, 1H, Ar–CH), 7.58–7.51 (m, 2H, 2 × Ar–CH), 7.25–7.18 (m, 2H, 2 × Ar–CH), 4.90 (d, J = 6.4 Hz, 2H, CHHOCHH), 4.83 (d, J = 6.4 Hz, 2H, CHHOCHH), 3.72 (t, J = 4.5 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 2.29 (t, J = 4.5 Hz, 4H, CH_2_NCH_2_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
Morpholino(4-(Phenylsulfonyl)Phenyl)Methanone (10b)
Amide triflate 16b (68.0 mg, 0.2 mmol, 1.0 equiv), Pd(OAc)2 (4.5 mg, 20 μmol, 10 mol %), Xantphos (12.0 mg, 20 μmol, 10 mol %), Cs_2_CO_3_ (98.0 mg, 0.3 mmol, 1.5 equiv), and sodium benzenesulfinate salt (24.0 mg, 0.2 mmol, 1.0 equiv) were added to a reaction vial. The reaction vial was sealed then evacuated and backfilled with Ar three times. Degassed 1,4-dioxane (1.12 mL, 0.18 M) was added by syringe and the reaction mixture was stirred at 110 °C for 16 h. After cooling to rt, the reaction mixture was diluted with CH_2_Cl_2_ (10 mL) and filtered through a plug of Celite, eluting through with more CH_2_Cl_2_ (3 × 10 mL). The solvent was then removed in vacuo. Purification by automated flash column chromatography (25–100% EtOAc/hexane) afforded benzamide 10b as a white solid (11.0 mg, 15%). Rf = 0.43 (100% EtOAc); mp = 124–126 °C; IR (film)/cm^–1^ 3061, 2965, 2919, 2855, 1631 (CO), 1431, 1318, 1278, 1258, 1150, 1109, 1069, 1014, 917, 841, 725, 689, 598, 572, 543; ^1^H NMR (400 MHz, CDCl_3_) δ 8.03–7.98 (m, 2H, 2 × Ar–CH), 7.98–7.90 (m, 2H, 2 × Ar–CH), 7.66–7.49 (m, 5H, 5 × ArCH), 3.91–3.23 (m, 8H, 2 × NCH_2_CH_2_); ^13^C NMR (101 MHz, CDCl_3_) δ 168.5 (CO), 142.9 (Ar–C_q_C_q_), 140.9 (Ar–C_q_SO_2_), 140.1 (Ar–C_q_SO_2_), 133.6 (Ar–CH), 129.5 (2 × Ar–CH), 128.2 (2 × Ar–CH), 127.9 (2 × Ar–CH), 127.8 (2 × Ar–CH), 66.8 (2 × OCH_2_), 48.0 (NCH_2_), 42.5 (NCH_2_); HRMS (TOF-MS-ES^+^) m/z calculated for C_19_H_21_N_2_O_4_S^+^ [M + CH_3_CNH]^+^: 373.1222; found 373.1215.
4-(3-(4-(2-(Trifluoromethyl)Pyrimidin-5-yl)Phenyl)Oxetan-3-yl)Morpholine
(11a)
Amino-oxetane triflate 16a (36.0 mg, 0.1 mmol, 1.0 equiv), Pd(PPh_3_)4 (3.5 mg, 3 μmol, 3 mol %), Cs_2_CO_3_ (65.0 mg, 0.2 mmol, 2.0 equiv), and pyrimidine pinacol ester 24 (41.0 mg, 0.15 mmol, 1.5 equiv) were added to a reaction vial. The reaction vial was sealed then evacuated and backfilled with Ar three times. A degassed solution of 1,4-dioxane:water (7:3, 0.5 mL, 0.2 M) was added by syringe and the reaction mixture was stirred at 110 °C for 20 h. After cooling to 25 °C, the reaction mixture was diluted with Et_2_O (10 mL) and filtered through a plug of Celite, eluting through with more Et_2_O (3 × 10 mL). The solvent was then removed in vacuo. Purification by flash column chromatography (60–80% EtOAc/hexane) afforded amino-oxetane 11a as a white solid (35.0 mg, 93%). R_ f _ = 0.15 (80% EtOAc/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 9.11 (s, 2H, 2 × ArCH), 7.66 (d, J = 8.0 Hz, 2H, 2 × ArCH), 7.29 (d, J = 8.0 Hz, 2H, 2 × Ar–CH), 4.96 (d, J = 6.2 Hz, 2H, CHHOCHH), 4.94 (d, J = 6.2 Hz, 2H, CHHOCHH), 3.77 (t, J = 4.6 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 2.38 (d, J = 4.6 Hz, 4H, CH_2_NCH_2_); ^19^F NMR (377 MHz, CDCl_3_) δ −70.1. The observed characterization data (R_ f _, ^1^H NMR, ^19^F NMR) were consistent with that previously reported.?
Morpholino(4-(2-(Trifluoromethyl)Pyrimidin-5-yl)Phenyl)Methanone
(11b)
Benzamide triflate 16b (68.0 mg, 0.2 mmol, 1.0 equiv), Pd(OAc)2 (2.2 mg, 10 μmol, 5 mol %), SPhos (8.4 mg, 20 μmol, 10 mol %), K_3_PO_4_ (85.0 mg, 0.4 mmol, 2.0 equiv), and pyrimidine pinacol ester 24 (82.0 mg, 0.3 mmol, 1.5 equiv) were added to a reaction vial. The reaction vial was sealed then evacuated and backfilled with Ar three times. Degassed 1,4-dioxane:water (4:1, 2 mL, 0.1 M) was added by syringe and the reaction mixture was stirred at 65 °C for 24 h. After cooling to rt, the reaction mixture was diluted with Et_2_O (10 mL) and filtered through a plug of Celite, eluting through with more Et_2_O (3 × 10 mL). The solvent was then removed in vacuo. Purification by flash column chromatography (60–80% EtOAc/hexane) afforded benzamide 11b as a white solid (48.0 mg, 70%). R_ f _ = 0.24 (80% EtOAc/hexane); mp = 168–170 °C; IR (film)/cm^–1^ 2967, 2921, 2857, 1628 (CO), 1550, 1459, 1431, 1352, 1278, 1192, 1145, 1114, 1009, 842, 650, 566; ^1^H NMR (400 MHz, CDCl_3_) δ 9.10 (s, 2H, 2 × Ar–CH), 7.69 (d, J = 8.2 Hz, 2H, 2 × Ar–CH), 7.62 (d, J = 8.2 Hz, 2H, 2 × Ar–CH), 3.89–3.43 (m, 8H, 2 × NCH_2_CH_2_); ^13^C NMR (101 MHz, CDCl_3_) δ 169.2 (CO), 155.9 (q, J = 36.7 Hz, Ar–C_q_CF_3_), 155.8 (2 × Ar–CH), 136.9 (Ar–C_q_C_q_), 135.3 (Ar–C_q_Ar–C_q_), 134.4 (ArC_q_Ar–C_q_), 128.5 (2 × Ar–CH), 127.6 (2 × Ar–CH), 119.7 (q, J = 274.9 Hz, CF_3_), 66.9 (2 × OCH_2_), 48.2 (NCH_2_), 42.6 (NCH_2_); ^19^F NMR (377 MHz, CDCl_3_) δ −70.1; HRMS (TOF-MS-ES^+^) m/z calculated for C_16_H_15_N_3_O_2_F_3_ ^+^ [M + H]^+^: 338.1116; found 338.1108.
4-(4-(3-Morpholinooxetan-3-yl)Phenyl)Morpholine (12a)
Amino-oxetane triflate 16a (73.0 mg, 0.2 mmol, 1.0 equiv), Pd(OAc)2 (2.2 mg, 10 μmol, 5 mol %), JohnPhos (6.0 mg, 20 μmol, 10 mol %), and K_3_PO_4_ (59.0 mg, 0.3 mmol, 1.5 equiv) were added to a reaction vial. The reaction vial was sealed then evacuated and backfilled with Ar three times. Morpholine (29 μL, 0.24 mmol, 1.2 equiv) followed by anhydrous, degassed THF (0.4 mL, 0.5 M) were added by syringe and the reaction mixture was stirred at 65 °C for 24 h. After cooling to rt, the reaction mixture was diluted with Et_2_O (10 mL) and filtered through a plug of Celite, eluting through with more Et_2_O (3 × 10 mL). The solvent was then removed in vacuo. Purification by flash column chromatography (80% EtOAc/hexane) afforded amino-oxetane 12a as a pale-yellow solid (19.0 mg, 32%). R_ f _ = 0.20 (80% EtOAc/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 6.96 (d, J = 8.5 Hz, 2H, 2 × Ar–CH), 6.91 (d, J = 8.5 Hz, 2H, 2 × Ar–CH), 4.89 (d, J = 6.0 Hz, 2H, CHHOCHH), 4.86 (d, J = 6.0 Hz, 2H, CHHOCHH), 3.88 (t, J = 4.8 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 3.73 (t, J = 4.6 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 3.18 (t, J = 4.8 Hz, 4H, CH_2_NCH_2_), 2.31 (t, J = 4.6 Hz, 4H, CH_2_NCH_2_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
4-Morpholinonebenzene-4-Yl-Morpholine (12b)
Benzamide triflate 16b (73.0 mg, 0.2 mmol, 1.0 equiv), Pd(OAc)2 (2.2 mg, 10 μmol, 5 mol %), JohnPhos (6.0 mg, 20 μmol, 10 mol %), and K_3_PO_4_ (59.0 mg, 0.3 mmol, 1.5 equiv) were added to a reaction vial. The reaction vial was sealed then evacuated and backfilled with Ar three times. Morpholine (29 μL, 0.24 mmol, 1.2 equiv) followed by anhydrous, degassed THF (0.4 mL, 0.5 M) were added by syringe and the reaction mixture was stirred at 65 °C for 24 h. After cooling to rt, the reaction mixture was diluted with Et_2_O (10 mL) and filtered through a plug of Celite, eluting through with more Et_2_O (3 × 10 mL). The solvent was then removed in vacuo. Purification by flash column chromatography (80% EtOAc/hexane) afforded benzamide 12b as a white solid (23.0 mg, 42%). R_ f _ = 0.20 (100% EtOAc); mp = 122–124 °C; IR (film)/cm^–1^ 2960, 2915, 2893, 2852, 1627 (CO), 1608, 1517, 1451, 1426, 1301, 1276, 1262, 1232, 1196, 1115, 1067, 1025, 928, 834, 763, 701, 643;^1^H NMR (400 MHz, CDCl_3_) δ 7.36 (d, J = 8.7 Hz, 2H, 2 × Ar–CH), 6.88 (d, J = 8.7 Hz, 2H, 2 × Ar–CH), 3.85 (t, J = 4.8 Hz, 4H, 2 × OCH_2_), 3.79 – 3.53 (m, 8H, 2 × NCH_2_CH_2_), 3.21 (t, J = 4.8 Hz, 4H, 2 × NCH_2_); ^13^C NMR (101 MHz, CDCl_3_) δ 170.6 (CO), 152.4 (Ar–C_q_N), 129.1 (2 × Ar–CH), 125.6 (ArC_q_C_q_), 114.4 (2 × Ar–CH), 67.0 (2 × OCH_2_), 66.7 (2 × OCH_2_), 48.4 (4 × NCH_2_). The observed characterization data (mp, IR, ^1^H NMR, ^13^C NMR) was consistent with that previously reported.?
4-(3-Morpholinooxetan-3-yl)Phenol (14a)
Prepared according to General Procedure A using K_2_CO_3_, (223 mg, 1.4 mmol), morpholine (0.11 mL, 1.3 mmol) and OTIPS OSF 15 (440 mg, 1.1 mmol). Purification by flash column chromatography (80–100% EtOAc/hexane) afforded aminooxetane 14a as a white solid (263 mg, 98%). R_ f _ = 0.20 (80% EtOAc/hexane); ^1^H NMR (400 MHz, CDCl_3_) δ 6.93 (d, J = 8.1 Hz, 2H, 2 ´ Ar–CH), 6.84 (d, J = 8.1 Hz, 2H, 2 ´ Ar–CH), 5.09 (s, 1H, OH), 4.90 (d, J = 9.1 Hz, 2H, CHHOCHH), 4.89 (d, J = 9.1 Hz, 2H, CHHOCHH), 3.75 (t, J = 4.6 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 2.33 (s, 4H, CH_2_NCH_2_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
4-(3-Morpholinooxetan-3-yl)Phenyl Trifluoromethanesulfonate
(16a)
Triflic anhydride (106 μL, 0.63 mmol, 1.1 equiv) was added dropwise to a solution of pyridine (91 μL, 1.15 mmol, 2.0 equiv) and amino-oxetane 14a (135 mg, 0.57 mmol, 1.0 equiv) in CH_2_Cl_2_ (1.15 mL, 0.5 M). After stirring at rt for 3 h, the reaction was quenched with sat. NaHCO_3_ solution (10 mL). The layers were separated, and the aqueous layer was extracted with CH_2_Cl_2_ (3 ´ 10 mL). The combined organic layers were dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo. Purification by flash column chromatography (80% EtOAc/hexane) afforded amino-oxetane triflate 16a as red gum (141 mg, 96%). R_ f _ = 0.37 (80% EtOAc/hexane);^1^H NMR (400 MHz, CDCl_3_) δ 7.32 (d, J = 8.7 Hz, 2H, 2 × Ar–CH), 7.19 (d, J = 8.7 Hz, 2H, 2 × Ar–CH), 4.92 (d, J = 6.2 Hz, 2H, CHHOCHH), 4.87 (d, J = 6.2 Hz, 2H, CHHOCHH), 3.75 (t, J = 4.6 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 2.32 (s, 4H, CH_2_NCH_2_). ^19^F NMR (377 MHz, CDCl_3_) δ −72.7. The observed characterization data (R_ f _, ^1^H NMR, ^19^F NMR) were consistent with that previously reported.?
4-(Morpholine-4-Carbonyl)Phenyl Trifluoromethanesulfonate (16b)
Triflic anhydride (0.17 mL, 1.0 mmol, 1.0 equiv) was added dropwise to a solution of triethylamine (0.17 mL, 1.2 mmol, 1.2 equiv) and benzamide 14b (207 mg, 1.0 mmol, 1.0 equiv) in CH_2_Cl_2_ (2.5 mL, 0.25 M). After stirring at rt for 3 h, the reaction was quenched with water (30 mL). The layers were separated, and the aqueous layer was extracted with CH_2_Cl_2_ (3 ´ 15 mL). The combined organic layers were washed with sat. aq. NaHCO_3_ (30 mL), dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo. Purification by flash column chromatography (40–60% EtOAc/hexane) afforded benzamide triflate 16b as an offwhite solid (257 mg, 85%). R_ f _ = 0.41 (100% EtOAc); mp = 70–72 °C; IR (film)/cm^–1^ 2968, 2922, 2857, 1633 (CO), 1598, 1499, 1420, 1278, 1251, 1204, 1135, 1111, 1069, 1014, 880, 846, 758, 605, 548, 517; ^1^H NMR (400 MHz, CDCl_3_) δ 7.53 (d, J = 8.2 Hz, 2H, 2 × Ar–CH), 7.35 (d, J = 8.2 Hz, 2H, 2 × Ar–CH), 3.88–3.38 (m, 8H, 2 × NCH_2_CH_2_); ^13^C NMR (101 MHz, CDCl_3_) δ 168.5 (CO), 150.2 (Ar–C_q_OTf), 135.6 (Ar–C_q_C_q_), 129.4 (2 × Ar–CH), 121.8 (2 × Ar–CH), 118.7 (q, J = 321.2 Hz, C_q_F_3_), 66.8 (2 × OCH_2_), 48.2 (NCH_2_), 42.7 (NCH_2_); ^19^F NMR (377 MHz, CDCl_3_) δ −72.7. The observed characterization data (^1^H NMR, ^13^C NMR, ^19^F NMR) were consistent with that previously reported.?
4-(4-Methoxybenzyl)Morpholine (18)
Sodium triacetoxyborohydride (51.0 mg, 0.24 mmol, 1.2 equiv) was added to a solution of p-anisaldehyde (23 μL, 0.2 mmol, 1.0 equiv) and morpholine (21 μL, 0.24 mmol, 1.2 equiv) in anhydrous CH_2_Cl_2_ (0.67 mL, 0.3 M). After stirring at rt for 20 h, the reaction mixture was quenched with aq. NaOH (1M, 10 mL). The phases were separated, and the aqueous layer was extracted with CH_2_Cl_2_ (3 ´ 10 mL). The combined organic layers were concentrated in vacuo. The residue was acidified by additions of aq. HCl (1 M, 7 mL) and washed with Et_2_O (2 ´ 10 mL). The organic layers were discarded, and the aqueous layer was basified by addition of aq. NaOH (1M, 14 mL). The aqueous layer was extracted with CH_2_Cl_2_ (3 ´ 10 mL). The combined organic layers were dried over anhydrous Na_2_SO_4_, filtered, and concentrated in vacuo to afford benzylamine 18 as a clear colorless oil (21.0 mg, 50%). R_ f _ = 0.20 (50% EtOAc/hexane); IR (film)/cm^–1^ 2954, 2929, 2852, 2803, 2761, 1612, 1512, 1456, 1286, 1245, 1176, 1116, 1034, 1006, 914, 866, 829, 795, 568;^1^H NMR (400 MHz, CDCl_3_) δ 7.26 (d, J = 8.6 Hz, 2H, 2 ´ Ar–CH), 6.87 (d, J = 8.6 Hz, 2H, 2 ´ Ar–CH), 3.81 (s, 3H, OCH_3_), 3.73 (t, J = 4.7 Hz, 4H, CH_2_OCH_2_), 3.48 (s, 2H, Ar–C_q_CH_2_), 2.46 (t, J = 4.7 Hz, 4H, CH_2_NCH_2_); ^13^C NMR (101 MHz, CDCl_3_) δ 158.9 (Ar–C_q_OMe), 130.5 (2 ´ Ar–CH), 129.3 (Ar–C_q_CH_2_), 113.7 (2 × Ar–CH ), 66.9 (CH_2_OCH_2_), 62.8 (CH_2_), 55.3 (OCH_3_), 53.4 (CH_2_NCH_2_). The observed characterization data (IR, ^1^H, ^13^C) were consistent with that previously reported.?
3-(4-Chlorophenyl)-N-(2-Morpholinoethyl)Oxetan-3-Amine
(20a)
Oxetane triflate 25 (41.0 mg, 0.1 mmol, 1.0 equiv), KCl (14.9 mg, 0.2 mmol, 2.0 equiv), and KF (2.9 mg, 0.05 mmol, 0.5 equiv) were added to a reaction vial (1). The reaction vial (1) was sealed then evacuated and backfilled with Ar three times. Pd_2_(dba)3 (1.4 mg, 1.5 μmol, 1.5 mol %) and *t-*BuBrettPhos (2.2 mg, 4.5 μmol, 4.5 mol %) were added to a separate reaction vial (2). The reaction vial (2) was sealed then evacuated and backfilled with Ar three times. Anhydrous, degassed 1,4-dioxane (0.1 mL) was added to reaction vial (2) and the reaction mixture was stirred at 120 °C for 5 min. After cooling to 25 °C, the contents of reaction vial (2) were transferred to reaction vial (1) by syringe, and the reaction mixture was diluted with further 1,4-dioxane (0.3 mL). The reaction mixture was then heated to 130 °C and stirred for 20 h. After cooling to 25 °C, the reaction mixture was filtered through a plug of Celite and eluted with Et_2_O (10 mL). The solvent was then removed in vacuo. Purification by flash column chromatography (0–10% MeOH/CH_2_Cl_2_) afforded amino-oxetane 20a as a white crystalline solid (9.7 mg, 32%). R_ f _ = 0.6 (30% MeOH/CH_2_Cl_2_); ^1^H NMR (400 MHz, CDCl_3_) δ 7.41–7.32 (m, 4H, 4 × Ar–CH), 4.92 (d, J = 6.4 Hz, 2H, CHHOCHH), 4.74 (d, J = 6.4 Hz, 2H, CHHOHH), 3.72 (t, J = 4.7 Hz, 4H, CH_2_CH_2_OCH_2_CH_2_), 2.46 (m, 4H, CH_2_CH_2_N), 2.40 (m, 4H, CH_2_NCH_2_). The observed characterization data (R_ f _, ^1^H NMR) was consistent with that previously reported.?
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1b Symes, O. L. ; Ishikura, H. ; Bull, J. A. Oxetanes and Thietanes. In Burger’s Medicinal Chemistry and Drug Discovery; Wiley, 2003; pp. 1–43. 10.1002/0471266949.bmc 308. · doi ↗
- 2c World Health Organization model list of essential medicines: 21st list 2019; World Health Organization, 2019. https://apps.who.int/iris/handle/10665/325771.
- 3U.S. Food and Drug Administration. FDA Approves Drug to Treat Adults with Persistent or Chronic Immune Thrombocytopenia; U.S. Food and Drug Administration, 2025. https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-drug-treat-adults-persistent-or-chronic-immune-thrombocytopenia.
- 4a Zheng X.Liang C.Wang L.Wang B.Liu Y.Feng S.Wu J. Z.Gao L.Feng L.Chen L.Guo T.Shen H. C.Yun H.Discovery of Benzoazepinequinoline (BAQ) Derivatives as Novel, Potent, Orally Bioavailable Respiratory Syncytial Virus Fusion Inhibitors J. Med. Chem.20186122102281024110.1021/acs.jmedchem.8b 0139430339388 · doi ↗ · pubmed ↗
- 5a Wuitschik G.Rogers-Evans M.Müller K.Fischer H.Wagner B.Schuler F.Polonchuk L.Carreira E. M.Oxetanes as Promising Modules in Drug Discovery Angew. Chem., Int. Ed.200645467736773910.1002/anie.20060234317013952 · doi ↗ · pubmed ↗
- 6Dubois M. A. J.Croft R. A.Ding Y.Choi C.Owen D. R.Bull J. A.Mousseau J. J.Investigating 3,3-Diaryloxetanes as Potential Bioisosteres through Matched Molecular Pair Analysis RSC Med. Chem.202112122045205210.1039/D 1MD 00248 A 35024613 PMC 8672821 · doi ↗ · pubmed ↗
- 7a Lassalas P.Oukoloff K.Makani V.James M.Tran V.Yao Y.Huang L.Vijayendran K.Monti L.Trojanowski J. Q.Lee V. M.-Y.Kozlowski M. C.Smith A. B.Brunden K. R.Ballatore C.Evaluation of Oxetan-3-Ol, Thietan-3-Ol, and Derivatives Thereof as Bioisosteres of the Carboxylic Acid Functional Group ACS Med. Chem. Lett.20178886486810.1021/acsmedchemlett.7b 0021228835803 PMC 5554911 · doi ↗ · pubmed ↗
- 8a Shearer J.Castro J. L.Lawson A. D. G.Mac Coss M.Taylor R. D.Rings in Clinical Trials and Drugs: Present and Future J. Med. Chem.202265138699871210.1021/acs.jmedchem.2c 0047335730680 PMC 9289879 · doi ↗ · pubmed ↗