Multiparameter Optimization of Pseudomonas aeruginosa Elastase Inhibitors for Systemic Administration
Ahmed S. Abdelsamie, Jelena Konstantinović, Andreas M. Kany, Christian Schütz, Dominik Kolling, Samira Speicher, Andreas Klein, Roya Shafiei, Mélodie Bouté, Katharina Mundry, Yu Mi Park, Brigitta Loretz, Rolf Müller, Jean-Michel Sallenave, Claus-Michael Lehr, Jesko Koehnke

TL;DR
This paper describes the development of optimized inhibitors for a key enzyme in a dangerous bacteria, with improved properties for treating lung infections.
Contribution
The study introduces a multiparameter optimization of LasB inhibitors with systemic administration and lung retention capabilities.
Findings
Optimized inhibitors show improved activity and selectivity with favorable ADMET properties.
Intravenous administration leads to favorable lung retention for the first time in this scaffold.
Physicochemical properties correlate with protein binding and lung activity.
Abstract
Targeting the extracellular protease elastase (LasB) of the high-priority pathogen Pseudomonas aeruginosa is a promising strategy to develop second-generation, narrow-spectrum antibiotics with a novel mode of action. P. aeruginosa is responsible for a variety of infections, particularly of the lung. Herein, we report the structure-based optimization of a previously reported potent and selective phosphonate-based LasB inhibitor scaffold. Having improved the activity while maintaining high selectivity and favorable ADMET properties, we also demonstrate, for the first time within this scaffold, that intravenous administration leads to favorable lung retention. We could rationally align this with in vitro plasma protein binding. We further observed a link between physicochemical properties like logD7.4 and protein binding, including surfactant proteins that can impair compound activity in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1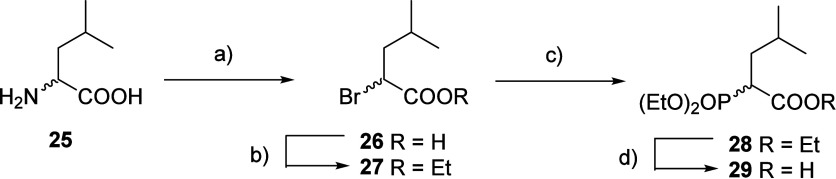 2
2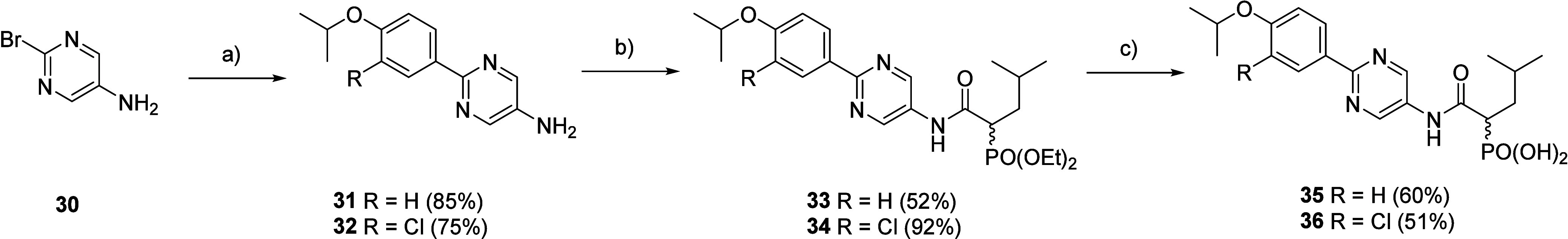 3
3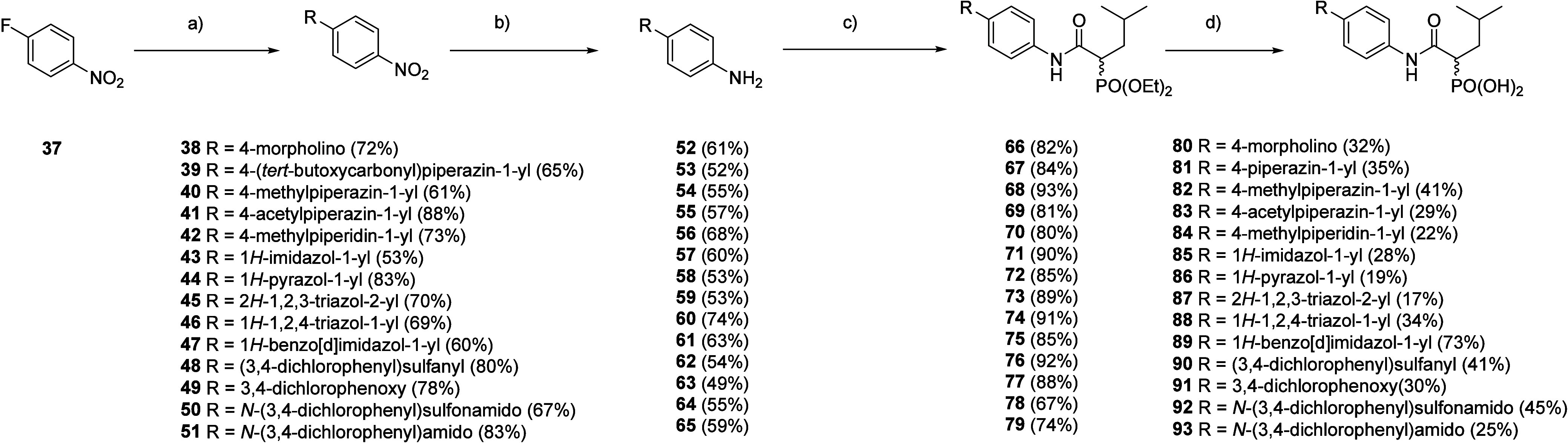 4
4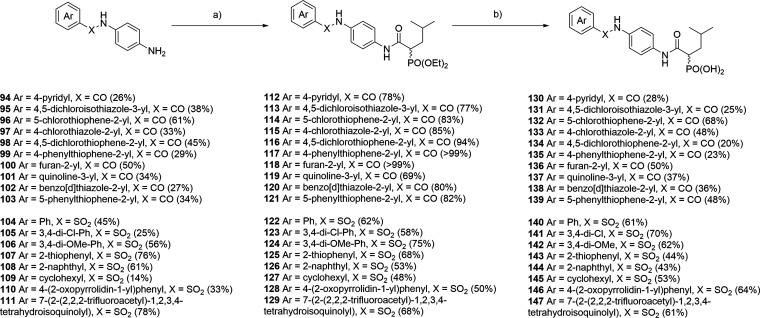 5
5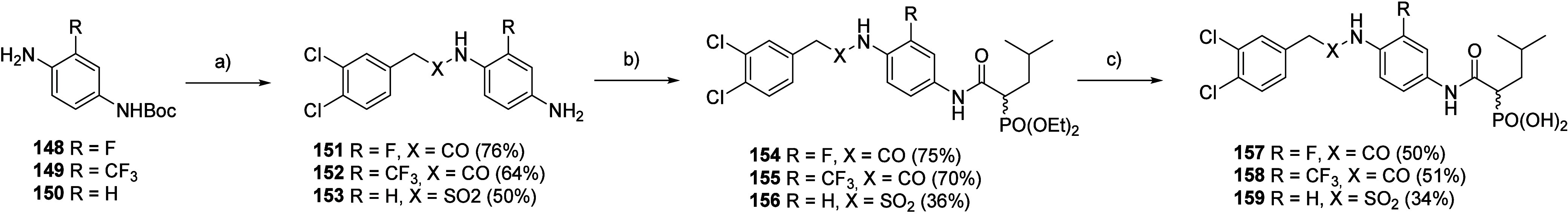 6
6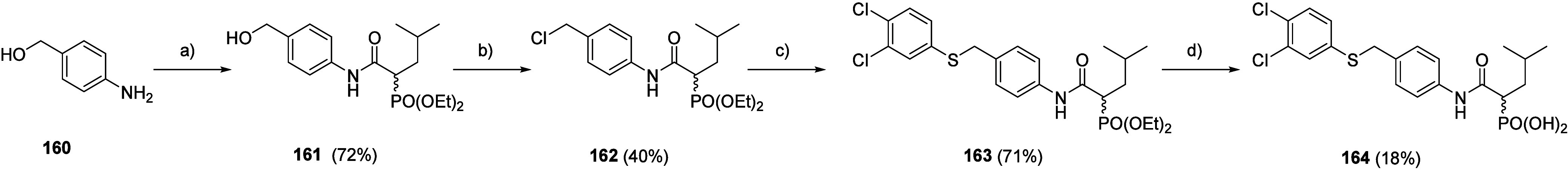 7
7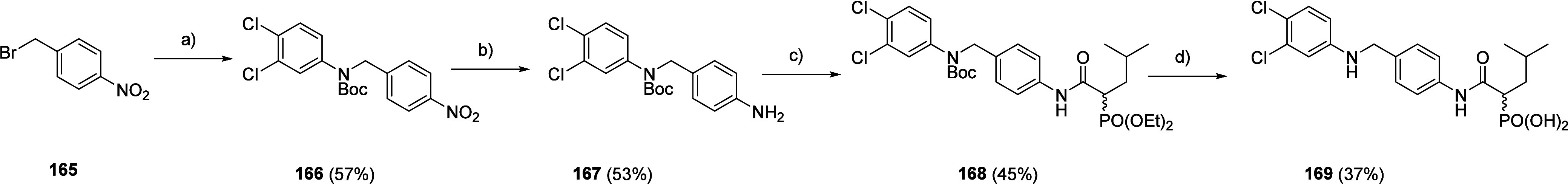 8
8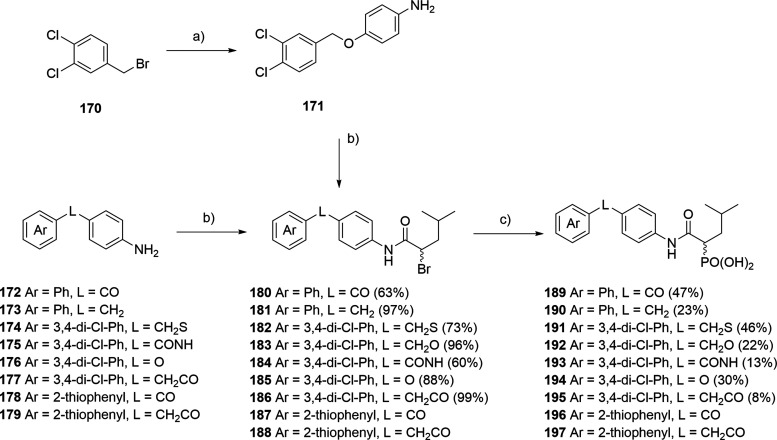 9
9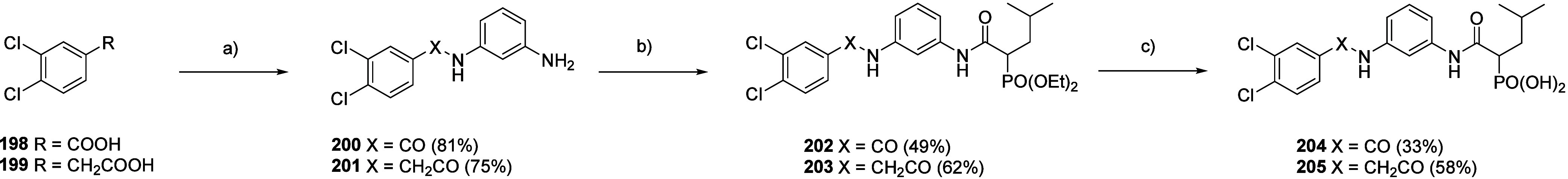 10
10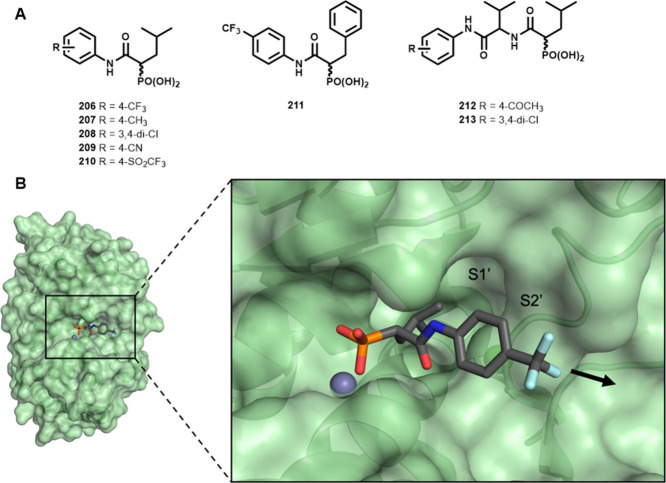 1
1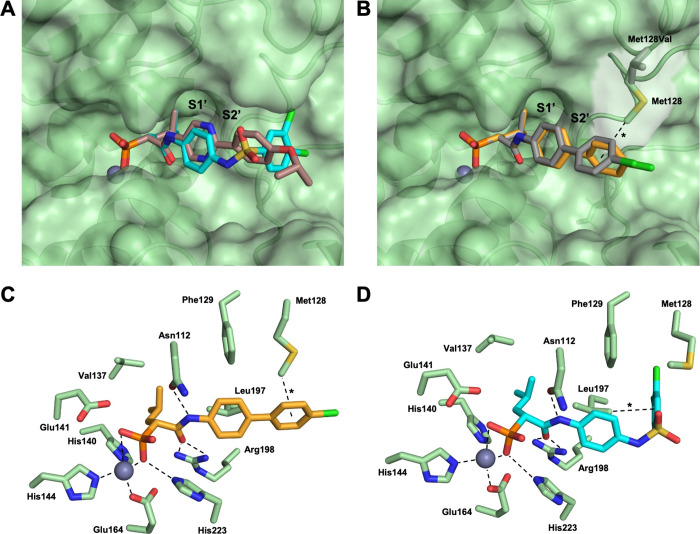 2
2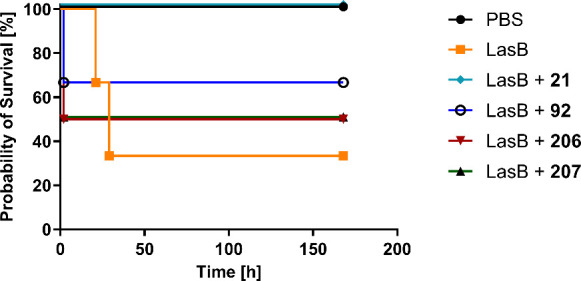 3
3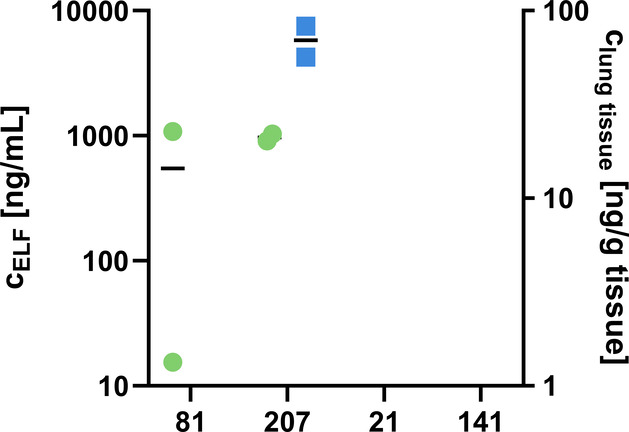 4
4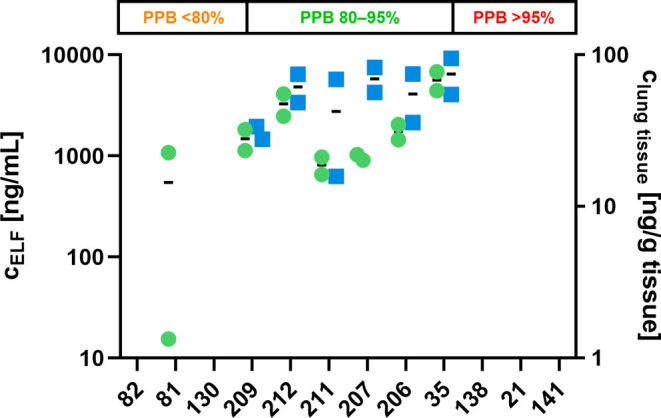 5
5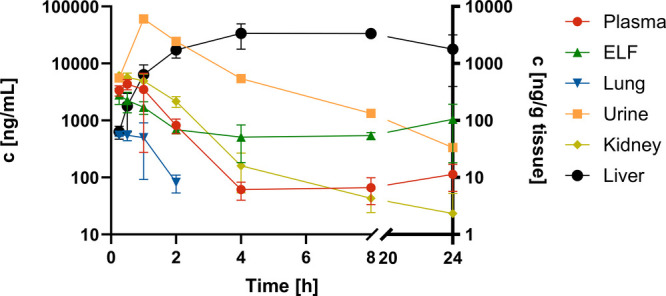 6
6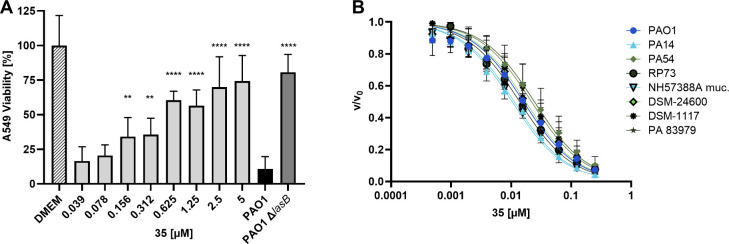 7
7| cpd | FRET IC50 [μM] | FRET IC50 in the presence of 1% surfactant [nM] | fold increase in the presence of surfactant | FRET IC50 in the presence of 0.8 mg/mL DPPC [nM] | surfactant protein binding [%] |
|---|---|---|---|---|---|
|
| 8.5 ± 0.5 | 45 | 5.3 | 10.3 ± 0.7 | 37.2 ± 0.8 |
|
| 9.5 ± 0.4 | 31 ± 13 | 3.2 | n.d. | n.d. |
|
| 15.3 ± 0.8 | 34 ± 11 | 2.2 | n.d. | n.d. |
|
| 174 ± 10.4 | 221 ± 55 | 1.3 | 120 ± 8 | n.d. |
|
| 110 ± 7.8 | 159 ± 52 | 1.4 | n.d. | n.d. |
|
| 68.5 ± 4.0 | 68.7 ± 0.71 | 1.0 | 46.8 ± 1.9 | 0 |
|
| 13.2 ± 0.4 | 153 ± 43 | 12 | 12.5 ± 0.4 | 44.2 ± 2.5 |
| cpd | logD7.4 | S9 Clint [μL/mg/min] mouse | plasma | Calu-3 Papp [10–6 cm/s] | A549 viability inh. [%] | PPB [%] mouse |
|---|---|---|---|---|---|---|
|
| 0.79 | <5.8 | >240 | 0.44 ± 0.35 | <10 | 98.3 ± 0.1 |
|
| 0.32 | <5.8 | >240 | 1.25 ± 0.73 | <10 | 97.2 ± 0.9 |
|
| 0.36 | <5.8 | >240 | 0.49 ± 0.13 | <10 | 95.0 ± 0.6 |
|
| –1.83 | <5.8 | >240 | 0.75 ± 0.28 | <10 | 60.6 ± 8.6 |
|
| –1.36 | <5.8 | >240 | 0.63 ± 0.18 | <10 | 33 ± 13 |
|
| –1.18 | <5.8 | >240 | 1.63 ± 1.02 | <10 | 61.7 ± 5.1 |
|
| 0.32 | <5.8 | >240 | n.d. | <10 | 96.4 ± 0.76 |
|
| 0.36 | <5.8 | >240 | 0.37 ± 0.20 | <10 | 98.3 ± 0.1 |
|
| 0.02 | <5.8 | >240 | 2.76 ± 1.15 | <10 | 93.8 ± 1.0 |
|
| –0.64 | <5.8 | >240 | 0.97 ± 0.21 | <10 | 91.8 ± 2.3 |
|
| 0.06 | <5.8 | >240 | 1.27 ± 0.33 | <10 | 98.9 ± 0.70 |
|
| –0.84 | <5.8 | >240 | n.d. | <10 | 79.6 ± 1.2 |
|
| 0.23 | <5.8 | >240 | n.d. | <10 | 97.5 ± 0.41 |
|
| 0.10 | <5.8 | >240 | n.d. | 10 ± 4 | 89.9 ± 0.77 |
|
| –0.58 | <5.8 | >240 | 0.86 ± 0.39 | <10 | 86.9 ± 2.1 |
|
| 0.41 | <5.8 | >240 | 0.71 ± 0.24 | <10 | 97.6 ± 0.59 |
| PK parameter | 21 | 81 | 207 |
|---|---|---|---|
|
| 2.30 ± 0.4 | 13.3 ± 12.6 | 1.90 ± 1.2 |
| C0 [μg/mL] | 8.55 ± 6.96 | 0.911 ± 0.481 | 2.64 ± 1.03 |
| AUC0‑t [μg/mL h] ELF | 4.68 ± 1.18 | 0.789 ± 0.278 | 4.41 ± 1.65 |
| MRT [h] | 2.8 ± 0.6 | 18.4 ± 18.5 | 2.5 ± 1.4 |
| VZ_obs [L/kg] | 1.19 ± 0.4 | 11.6 ± 1.6 | 0.99 ± 0.1 |
| Clobs [mL/min/kg] lung | 5.89 ± 1.2 | 19.66 ± 20.3 | 7.25 ± 3.9 |
| PK parameter |
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|
|
| 2.20 ± 0.7 | 1.10 ± 0.2 | 0.33 ± 0.0 | n.d. | 1.53 ± 0.2 | 1.54 ± 0.2 | 1.12 ± 0.1 | 3.42 ± 0.1 |
| C0 [μg/mL] | 0.900 ± 0.131 | 0.116 ± 0.060 | 0.194 ± 0.021 | 1.18 ± 0.043 | 8.14 ± 0.0458 | 8.33 ± 4.06 | 5.19 ± 0.86 | 18.96 ± 13.95 |
| AUC0‑t [μg/mL h] | 2.145 ± 0.293 | 0.216 ± 0.0348 | 0.075 ± 0.021 | 0.309 ± 0.037 | 17.5 ± 4.59 | 16.27 ± 1.92 | 7.40 ± 1.48 | 13.16 ± 3.02 |
| MRT [h] | 3.0 ± 1.0 | 1.4 ± 0.4 | 0.42 ± 0.0 | n.d. | 2.17 ± 0.4 | 2.1 ± 0.0 | 1.4 ± 0.3 | 4.1 ± 0.9 |
| VZ_obs [L/kg] | 2.3 ± 0.1 | 14.38 ± 4.8 | 11.52 ± 3.0 | n.d. | 0.23 ± 0.0 | 0.25 ± 0.0 | 0.43 ± 0.1 | 0.54 ± 0.2 |
| Clobs [mL/min/kg] lung | 36.7 ± 3.1 | 217 ± 84 | 168 ± 32 | n.d. | 3.78 ± 0.5 | 3.85 ± 0.4 | 5.95 ± 0.1 | 7.2 ± 0.7 |
| PK parameter |
|
|---|---|
|
| 3.2 ± 0.3 |
|
| 0.33 ± 0.1 |
| AUC0‑t [μg/mL*h] ELF | 19.2 ± 8.5 |
| ELF/plasma ratio | 2.38 |
|
| 0.16 ± 0.05 |
|
| 0.58 ± 0.4 |
| AUC0‑t [μg/g*h] lung | 0.17 ± 0.07 |
- —Alexander von Humboldt-Stiftung10.13039/100005156
- —Combating Antibiotic-Resistant Bacteria Biopharmaceutical Accelerator10.13039/100019590
- —European Research Council10.13039/501100000781
- —Helmholtz-Gemeinschaft10.13039/501100001656
- —Agence Nationale de la Recherche10.13039/501100001665
- —Bundesministerium f?r Bildung und Forschung10.13039/501100002347
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Natural Products and Biosynthesis · Antimicrobial Peptides and Activities · Pneumonia and Respiratory Infections
Introduction
Representing one of the ESKAPE pathogens that is classified as high priority by the World Health Organization, Pseudomonas aeruginosa undoubtedly threatens public health. ?,? The situation is worsened by the rise of resistance against commonly used antibiotics urgently calling for the development of novel treatment options. ?−? ? In this regard, the concept of developing ‘pathoblockers’ targeting bacterial virulence is of particular interest as it offers several advantages: Given that bacteria are not killed but rather impaired in their detrimental effects on the host, selection pressure is reduced. Additionally, the human microbiome is spared as antivirulence targets tend to be species-specific, paving the way for narrow-spectrum antibiotics. ?−? ? While developing a novel pathoblocker against P. aeruginosa , we and others have focused on the development of elastase (LasB) inhibitors. ?−? ? ? ? ? The secreted zinc metalloprotease LasB plays a pivotal role in an infection with P. aeruginosa , facilitating invasion by cleaving components of the host connective tissue and simultaneously favoring immune evasion as several components of the immune system are substrates to LasB. ?−? ? Its extracellular localization constitutes an essential advantage considering the challenges associated with crossing the Gram-negative cell wall.? During recent years, we described the hit discovery and -optimization of a thiol-based LasB inhibitor scaffold that we recently advanced significantly in terms of potency, drug metabolism and pharmacokinetics (DMPK) and drug likeness by replacing the thiol with a phosphonate.? Having access to a rational pipeline of structure-based optimization guided by high-resolution cocrystal structures, in vitro and ex vivo potency assays, in vitro ADMET (absorption, distribution, metabolism, excretion and toxicity) and in vivo PK/PD, we are now optimizing the scaffold according to a specific target-lead and target-product profile (TLP/TPP).
P. aeruginosa is responsible for various diseases, particularly lung infections. This affects immunocompromised patients and people suffering from cystic fibrosis (CF)? or noncystic fibrosis bronchiectasis (NCFB).? Furthermore, hospital-acquired or ventilator-associated pneumonia (HAP/VAP) caused by P. aeruginosa pose a significant threat to patients, e.g., in intensive-care units (ICU). Mortality in ICUs has also been connected to the presence of LasB.? While CF and NCFB patients are capable of inhaling a drug, hospitalized HAP/VAP patients are treated via intravenous (IV) administration.? Apart from these lung infections, a significant proportion of bacterial keratitis, especially contact lens-associated keratitis, where topical treatment is usually applied, is caused by * P. aeruginosa.*
Having demonstrated in vivo proof-of-concept for combination treatment of a LasB inhibitor with a standard-of-care (SOC) antibiotic for both P. aeruginosa lung infection? and Pseudomonas keratitis,? this study aims at exploring the potential of systemic dosing of LasB inhibitors to prepare IV treatment of HAP/VAP patients. We achieved this challenge via multiparameter optimization combining structure-based optimization of the phosphonate scaffold with in vitro activity and ADMET profiling.
Results
Chemistry
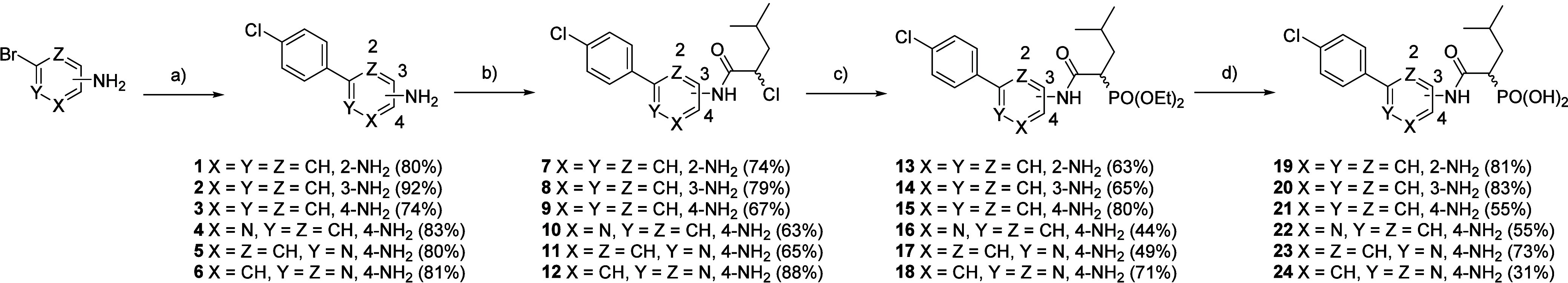
Synthesis of the first derivatives commenced with a Suzuki cross-coupling reaction between corresponding bromoanilines and 4-chlorphenyl boronic acid. Subsequent EDC·HCl-mediated amide coupling with commercially available 2-chloro-4-methylpentanoic acid gave rise to the corresponding phosphonate precursors, which were subjected to an Arbuzov reaction, followed by TMSBr-mediated deprotection (Scheme).
Synthesis of Compounds 19–24
For further synthetic studies, we aimed for a more divergent strategy. Therefore, phosphonate building block 29 was synthesized. Conversion of commercially available racemic leucine into the corresponding α-bromo acid, followed by esterification and subsequent Arbuzov reaction delivered the desired building block after saponification in 35% overall yield (Scheme).? This reaction can be performed on a large scale.
Synthesis of Phosphonate Building Block 29
With this building block in hand, the iso-propoxy-substituted derivatives 35 and 36 were synthesized according to Scheme. Starting with a Suzuki reaction of 2-bromopyrimidin-5-amine and the corresponding boronic acids, the required arylamines were subjected to peptide coupling with building block 29. Subsequent deprotection yielded the desired phosphonates.
Synthesis of Compounds 35 & 36
Synthesis of heterocyclic analogues (Scheme) was achieved via a sequence of S_N_Ar, reduction and subsequent peptide coupling with the aforementioned phosphonate building block (Scheme) and subjection to phosphonate deprotection conditions as mentioned above (see Scheme).
Synthesis of Hetaryl-Substituted Phosphonates 80–93
For the linker installation, we started with synthesis of the desired substituted aniline-derived building blocks, as well as for evaluation of the aromatic residue in the Western part. Compounds 130 – 147 were synthesized from commercially available tert-butyl (4-aminophenyl) carbamate and corresponding acid- or sulfonyl chlorides, followed by Boc-deprotection (see for details). Subsequent coupling to phosphonate building block 29 with EDC·HCl and HOBt gave the desired phosphonates after deprotection using the established TMSBr protocol (Scheme).
Synthesis of Compounds 130–147 with Sulfonamide or Amide Linker
Corresponding F- and CF_3_-substituted congeners 157 and 158 were synthesized in a similar fashion as mentioned before, relying on the established synthetic route consisting of amidation, Boc-deprotection, peptide coupling and phosphonate deprotection (Scheme).
Synthesis of Compounds 157–159
With respect to the monofunctional linkage, sulfur-containing compound 164 was synthesized by converting commercially available aminobenzylalcohol 160 into the corresponding phosphonate 161, which was then transformed to benzyl chloride 162. Nucleophilic substitution with 3,4-dichlorobenzenethiol, followed by phosphonate deprotection gave rise to the desired congener (Scheme).
Synthesis of Compound 164
For the respective amino analogue 169, tert-butyl 3,4-dichlorophenylcarbamate was reacted with commercially available benzyl bromide 165 and again our reliable sequence of reduction, peptide coupling and deprotection was applied (Scheme).
Synthesis of Compound 169
In case of the oxygen linker, nucleophilic substitution of 4-(bromomethyl)-1,2-dichlorobenzene with Boc-protected amino phenol afforded intermediate 171, which was then subjected to amide coupling with 2-bromo-4-methylpentanoic acid (Scheme). The same peptide coupling conditions were used with commercially available building blocks. Subsequent Arbuzov reaction, followed by treatment with TMSBr yielded the desired analogues.
Synthesis of Compounds 189–197
Furthermore, we explored the attachment of meta-substituted residues to the phenyl core with compounds 204 and 205. Synthesis commenced via the route already established for para derivatives using peptide coupling sequences (Scheme).
Synthesis of Compounds 204 & 205
Based on the recently obtained cocrystal structure of a first-generation phosphonate-based inhibitor (206) in complex with LasB (PDB code: 8CC4),? we sought to explore a possible growth vector in position 4 of the aromatic ring (Figure). We incorporated different structural features to understand their impact on the on-target activity and on physicochemical properties that may drive in vivo exposure. Our synthetic endeavors started with incorporation of an additional aryl moiety attached directly to the phenyl core via a direct C–C linkage. All of the newly synthesized compounds were tested in a FRET-based inhibition assay, as established by Nishino and Powers.? Compound 21 bearing the additional aryl moiety in 4-position proved to be the most active compound, displaying single-digit nanomolar activity (Table). The corresponding 3-substituted derivative 20 showed activity in the lower double-digit nanomolar range, whereas LasB inhibition dropped for o rtho-substituted compound 19, which was already expected due to likely clashes in the binding pocket. Incorporation of a nitrogen atom in 3-position of the initial phenyl ring led to compound 23 in a similar activity range as the parent compound. When moving to the 2-position (compound 22), activity decreased most likely due to a disadvantageous conformation as a result of the nitrogen lone pair being in close proximity to the carbonyl group. To further investigate the role of the binding angle between the two aryl moieties, we synthesized compound 24, inspired by previous work from Hamed et al. ? However, this led to a slight decrease in activity, comparable to compound 20. Additionally, the chloro-substituent was exchanged for an electron-donating iso-propoxy-substituent (35), which gave a small improvement in LasB inhibition. When combining this motif with a chloro-substituent in 3-position (36), activity slightly decreased again, yet still in a gratifying range. In order to probe a heterocyclic substituent as a decoration in 4-position of the core structure, we started with various five-membered heteroaromatic compounds, this time with direct N–C connection. Addition of imidazole (compound 85) led to increased activity in comparison to parent compound 206. Corresponding pyrazole compound 86, as well as the triazoles 87 and 88 displayed similar activities in between the parent compound (206) and 85. Exchanging the 4-substituent to morpholine (compound 80) resulted in no significant change in LasB inhibition. When we introduced a piperazine (compound 81), however, the IC_50_ value increased to 174 nM. Methylation of the nitrogen atom to mask the hydrogen-bond donor led to a less than 2-fold change in IC_50_ (82). When converted into the corresponding amide 83 with its hydrogen-bond acceptor function, a greater than 5-fold improved activity toward low double-digit nanomolar range was observed. A similar result was obtained when compound 84, the piperidine analogue of 82, was tested, prompting the question which role hydrogen bonding in this area might play in binding.
(A) Structures of phosphonate-based inhibitors published previously. 206–211 correspond to compounds 4b, 4a, 4c, 4k, 4l, and 4y reported in Konstantinovic et al.; 212 and 213 correspond to compounds 30 and 9 in Kiefer et al. (B) Co-crystal structure of LasB in complex with a recently published phosphonate inhibitor 206 (PDB code: 8CC4, adapted from Konstantinovic et al. ). The potential growth vector for further structure-based optimization is indicated by an arrow.
1: Pseudomonas aeruginosa LasB Inhibition in the FRET-Based Inhibition Assay and Chromatographic logD7.4 for Bicyclic Phosphonates
Based on the promising results from our first growth-vector exploration, we aimed to investigate the chemical space around the phenyl core. Our aim was to explore different linker moieties between the aromatic rings also comparing hydrogen bond donor/acceptor functions, rather than connecting them directly (Table), and to modify the Western aromatic part of the molecule (Table). Starting with introduction of bifunctional linkers, amide-connected compounds 93 and 193 were synthesized, both showing good double-digit nanomolar activity, with a preference for N–C linkers. Adding an additional methylene spacer did not affect the inhibitory activity (compound 195). This compound was further modified to study the influence of conformational changes. When introducing a fluorine substituent in 3-position (compound 157), activity was only slightly decreased, yet incorporation of a trifluoromethyl substituent (compound 158) in the same position led to a more pronounced drop in potency. Since installation of the additional substituent to the core structure in 3-position still maintained good activity, this substitution pattern was combined with the corresponding linker units, delivering compounds 204 and 205, showing a significant drop in activity. It is also worth mentioning that the methylene spacer on the amide function in 3-position yielded an almost 2.5-fold more potent LasB inhibitor (205). Bioisosteric replacement of the amide linker with a sulfonamide showed similar trends in structure–activity relationship for compounds 92, 141 and 159, yet with the sulfonamide linkage being more potent. Changing toward monofunctional linker units yielded compounds comparable to sulfonamide-containing inhibitors. Again, hydrogen-bond donor (compound 169) or acceptor (e.g., compounds 91, 169) functions, as well as an added methylene spacer did not significantly impact potency. Incorporation of a carbonyl linkage (compound 189) or a simple methylene linker (compound 190), however, resulted in decreased activity.
2: Pseudomonas aeruginosa LasB Inhibition from the FRET-Based Assay and Chromatographic logD7.4 for Linker-Modified Phosphonates
3: Pseudomonas aeruginosa LasB Inhibition from the FRET-Based Assay and Chromatographic logD7.4 for Phosphonates with Modified Aryl Unit
As mentioned before, we also focused on modification of the additional aryl unit. For our investigations we kept the N–C connection and started with bioisosteric replacement of the phenyl ring with thiophene. In this case, the additional methylene spacer proved to slightly decrease activity (compound 197), whereas a simple amide linkage (compound 196) resembled the activity of compound 93. Furthermore, mono- and dichlorinated derivatives 131 – 134 were synthesized, all with solid double-digit nanomolar activity. Exchanging thiophene for furan gave a similarly potent compound (136). Similar potency was displayed by compounds 137, bearing a quinoline motif, and 138 with a benzothiazole, which inspired us to investigate further growing possibilities. Disconnecting the phenyl moiety from the latter resulted in compounds 135 and 139 with comparable activity, yet slightly in favor of the 2,5-disubstitution pattern regarding thiophene. Next, we focused on further sulfonamide modifications. Omitting the dichloro motif of 141 led to a more than 2-fold drop in activity (compound 140) and replacement with the corresponding dimethoxy pattern decreased activity even further (compound 142). Also, for the sulfonamide derivatives, replacing the Western phenyl moiety with a thiophene (compound 143) led to a similar activity as for the phenyl derivative, while a pyridine derivative showed a higher IC_50_ (130). Even replacement of the aromatic Western part with a cyclohexyl substituent gave an active compound (145). Further enlargement was possible as well, with naphthyl-substituted compound 144 being in a similar activity range. Incorporation of a pyrrolidone substituent, however, led to a drop in potency (compound 146). It is worth noting that all of the above-mentioned compounds do not show any antibacterial activity against PA14 in agreement with our pathoblocker approach (Table S1).
X-ray Crystallography
The extensive optimization of LasB inhibitors was based primarily on the strategy of exploring the growth vector previously identified by X-ray crystallography, resulting in compounds with improved in vitro LasB inhibition. To gain more detailed insights into the binding mode and to confirm the design principle, the most potent biaryl compounds, 21 and 35, and the frontrunner compound with a sulfonamide linker, 141, were cocrystallized with LasB. The LasB–compound complex structures were determined to high resolution. Our optimized compounds share structural motifs with previously published LasB inhibitors, such as the phosphonate for zinc coordination at the active site, as well as the iso-butyl moiety and the amide linker attached to the aryl residue (Figure S1). ?,? Thus, key interactions, such as hydrogen bonds with the side chains of His223, Glu141 and Asn112, hydrophobic interactions with Leu197 in the S2’ pocket, and bidentate hydrogen bonds with Arg198 were conserved in all three cocrystal structures (Figure, Figure S4). In addition to these common interactions, there are major differences in the binding mode of the biaryl compounds in terms of the substitution pattern of the additionally introduced aromatic ring and the linker region. In the cocrystal structures of the two biaryl compounds without linker (21 and 35), an additional CH–π interaction with the conformationally flexible side chain of Met128 was observed (Figure, C, Figure S4, B). While only one distinct conformation of the inhibitor was observed for 21, the electron density observed for 35 could confidently be assigned to an alternative conformation of the compound. As a result, the side chain of Met128 also adopted two distinct conformations, one allowing CH–π interactions with the aromatic moiety, while the other induced a slight rotation of the terminal aromatic ring around the C–C bond (Figure S4). We hypothesize that the CH–π interaction between LasB and 21 was stronger than that between the protein and 35 (no alternative conformation observed). The presence of a pyrimidine substituent within the biphenyl part of 35 enforces a more planar orientation between the two aromatic rings, which constrains conformational flexibility and potentially impairs an effective CH–π interaction. In agreement with this supposition, the in vitro inhibition of LasB was almost twice as strong for 21 (IC_50_ of 8.5 ± 0.5 nM) as that of 35 (IC_50_ of 15.3 ± 0.8 nM).
Crystal structure of LasB (green) in complex with 21, 141, and 35 (PDB code: 9FRY, 9FRZ and 9FS0). (A) Surface representation and superposition of LasB in complex with 141 (carbon atoms: cyan) and 35 (carbon atoms: dark red). (B) Surface representation and superposition of wild-type LasB and the Met128Val mutant in complex with 21. The compound bound to the wild-type structure is represented as sticks with carbon atoms colored in dark yellow. The CH–π interaction between the side chain of LasBMet128 and the aromatic ring of 21 is highlighted by an asterisk. The ring conformation is slightly rotated in the cocrystal structure of LasB Met128Val in complex with 21, for which carbon atoms are colored in gray. (C) Interactions between LasB and 21. The CH–π interaction is indicated by an asterisk. (D) Interactions between LasB and 141. The dichlorine-substituted aromatic ring is engaged in a CH–π interaction with the side chain of LasB Leu197 and highlighted by an asterisk.
In a previously reported sequence analysis, mutations in the lasB gene were identified in clinically relevant isolates of P. aeruginosa , which were characterized biochemically and in terms of activity.? Most mutations had no effect on LasB activity and Met128Val was the only mutant located near the active site (present in 16 out of 2746 analyzed P. aeruginosa LasB sequences, 0.5% prevalence). In light of the favorable CH–π interaction of 21 and 35, we sought to investigate the effects of the Met128Val mutation on the compounds’ activity. The Met128Val mutation has a comparatively minor impact on the in vitro activity of compounds, as LasB inhibition remains within a favorable double-digit nanomolar range. Compound 21 shows an almost 7-fold loss in inhibition (IC_50_ of 58.9 ± 11.7 nM), whereas 35 is less affected with a 2.7-fold reduction (IC_50_ of 42.0 ± 10.4 nM), consistent with an alternative binding conformation not reliant on CH–π interactions. The high-resolution crystal structure of LasB Met128Val in complex with 21 revealed merely a slight rotation of the monochlorine-substituted aromatic ring, similar to the alternative conformation observed in the cocrystal structure with 35 (Figure, A, Figure S4).
To obtain detailed information on the effects of the introduction of a linker between the two aryl moieties, we obtained the high-resolution crystal structure of LasB in complex with 141. Similar to the LasB structures in complex with 21 and 35, the key interactions were retained, while the largest difference in binding was observed for the sulfonamide and the Western part of the compound. Bound to LasB, the sulfonamide linker induces a compound conformation in which the dichloro-substituted ring is oriented at nearly 90° to the aromatic ring engaged in the lipophilic S2’ pocket. This prevents the previously observed interaction with Met128 and enables instead a CH–π interaction of the terminal aromatic ring with the side chain of Leu197 (Figure, D).
Taken together, the binding mode of the different biaryls differs subtly depending on the substitution pattern of the aromatic ring and the linker region, as seen in the overlay (Figure, B). The shared and respective compound-specific interactions underline the structural versatility of the biaryl class in targeting LasB, which also becomes clear with the overall high activity of most of the designed inhibitors. These findings allow for further rational, structure-based design of LasB inhibitors.
LasB Inhibition in the Presence of Pulmonary Surfactant
In vivo, the LasB inhibitors eventually need to be active in the lungs where pulmonary surfactant is present. It has been shown that surfactant can significantly impair the activity of the antibiotic daptomycin.? Hence, we assessed potential effects of surfactant on the in vitro activity of selected LasB inhibitors by adding 1% porcine lung surfactant to the FRET assay. The results show that the activity of several compounds is unaffected by the presence of surfactant (e.g., 81, 82, 130), while some LasB inhibitors show a > 3-fold shift in IC_50_ (e.g., 21, 141, 195, Table).
4: IC50 Values in the FRET-Based LasB Inhibition Assay in the Presence and Absence of 1% Porcine Pulmonary Surfactant (equaling ∼0.8 mg/mL) or 0.8 mg/mL DPPC (Dipalmitoylphosphatidylcholine)
Pulmonary surfactant is composed of phospholipids and surfactant proteins. In order to determine the reason for the reduced activity of some LasB inhibitors, we measured binding to surfactant proteins applying rapid equilibrium dialysis. This setup uses a semipermeable membrane between two chambers, allowing distribution of compounds based on their binding to proteins, and is usually applied to determine plasma protein binding (PPB) as done below. We adapted the setup using assay buffer containing 1% surfactant instead of plasma. Indeed, we observed that 21 and 141, both giving a
4-fold increase in FRET IC_50_, show significant protein binding of ∼ 40%, while 130 not losing activity also does not bind to surfactant proteins. Hence, the impact on activity could be attributed to the binding to surfactant proteins in contrast to what has been reported for daptomycin, where the loss of activity is due to the interaction with phospholipids.? Since the membrane used in the assay has a molecular weight cutoff of 8 kDa, interactions with phospholipids can be excluded, as these would permeate. Confirming these observations, the in vitro activity of compounds 21, 130, 141 and 81 with and without shift in the presence of surfactant is not impaired by 0.8 mg/mL diphosphatidylcholine (DPPC), the main component of pulmonary surfactant (Table).?
Assessment of Selectivity over Human Off-Targets
Since LasB is a zinc metalloprotease, we continued to explore potential off-target effects on mammalian metalloproteases. Particularly, human matrix-metalloproteases are of interest due to their versatile roles in several physiological and also pathological processes.? We further investigated activity against COX-1 and tumor-necrosis factor α-converting enyzme (TACE). Importantly, all new derivatives both with and without linker could maintain the excellent selectivity profile determined previously for the monoaryl phosphonates (Table S1). Hence, growing the molecule deeper into the pocket does not lead to unwanted off-target activities.
Determination of In Vivo Efficacy in Mice Treated
Intranasally with LasB
Based on the previously demonstrated increased lethal effect of WT PAO1 bacteria versus ΔlasB PAO1? and that of purified LasB instilled intranasally into mouse lungs,? we applied the latter model to test a selection of potent inhibitors from this series before investigating drug metabolism and pharmacokinetic (DMPK) properties. As shown in Figure, LasB inhibition led to an increase in survival, with 100% survival observed for the most potent compound tested, 21.
Survival of mice treated with LasB inhibitors. Pure LasB was instilled intranasally into mice together with preincubated LasB inhibitors 21, 92, 206 and 207 at a molar ratio of 1/10. Curves represent groups of 3–4 animals.
DMPK Profiling
In preparation of in vivo PK studies, we determined the in vitro ADMET profile of the most promising biaryls. The results showed generally high kinetic solubility, low logD_7.4_ mostly in the negative range and high metabolic and plasma stability. Additionally, Calu-3 cell permeability was shown to be low (P_app_ < 2 × 10^–6^ cm/s, Table). This profile is highly similar to what we have reported before on monaryls? and dipeptides (Figure).? For representative biaryl 35, we further confirmed these findings across species with high stability in rat and minipig liver fractions and plasma (Table S2). Regarding potential cytotoxicity, the newly designed inhibitors maintained the nontoxic properties on the lung cell line A549. This clean profile was also confirmed against HepG2 and HEK293 cells (Table S1). Furthermore, selected biaryls with linker (141) and without (23, 35), were shown to be nontoxic against zebrafish larvae (Table S3), underlining the safety of the scaffold.
5: In Vitro ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) Profiling of Selected Biaryls
The primary aim of this study was to assess bioavailability of our LasB inhibitors in the lungs after IV administration. As anticipated, the biaryls show good ELF/plasma ratios indicating a good retention in ELF and lung tissue when administered topically via intratracheal instillation (Table S4). Despite their low cell permeability, we were eager to assess lung permeation after systemic administration for this highly potent compound scaffold. Aiming to obtain first information on their lung permeation from the bloodstream, we subjected a small set of compounds to a murine cassette PK study. We combined four structurally diverse phosphonates: biaryl with linker 141, biaryl without linker 21, biaryl without linker 81 that is substituted with a polar piperidine and 207 as one representative from the monoaryl class reported previously,? dosing at 2 mg/kg each.
Looking at the resulting terminal lung levels (Figure), it became apparent that only two of the four compounds applied were detected in lung tissue and epithelial lining fluid (ELF) after 5 h post administration, namely monoaryl 207 and biaryl 81. Looking at the plasma concentration time profiles, three of the four compounds still had considerable levels at 5 h, i.e. 21, 207 and 81, whereas 141 was cleared from plasma already after 15 min post administration (Figure S8, A). Despite similar terminal plasma levels for the three compounds 21, 207 and 81, only two distributed well into ELF and lung tissue suggesting that 21 did not penetrate well despite sufficient plasma concentrations. Considering the in vitro ADMET properties of the compounds tested, it became apparent that the only difference lies in PPB, with the two compounds reaching the lung being characterized by PPB below 98% (Table). In line with the high unbound fraction of 81, this compound shows comparably high clearance (Table). However, 207 as well as 81 showed similarly high parent compound levels in urine, suggesting additional possibly metabolic clearance of 81 compared to 207 (Figure S8, B). Compounds 207 and 21 are similar in terms of plasma t 1/2, volume of distribution and clearance. Since reduced free drug levels resulting from high binding to plasma proteins might not be sufficient to enable significant lung exposure, these results prompted us to investigate in more detail which PPB window would be beneficial for lung exposure for the phosphonate class.
Lung levels after first intravenous cassette showing epithelial lining fluid (ELF) levels in green and lung tissue levels in blue.
6: Pharmacokinetic (PK) Parameters after Intraven Cassette Dosing of 207, 21 and 81 at 2 mg/kg Each
Hence, we determined PPB for a wider range of potent inhibitors (Table). To obtain a better perspective about general trends within the phosphonate class, we included representatives from classes other than the biaryls, namely the initial monoaryls (206–211 ?) and two dipeptides (212, 213 ?). As we observed PPB to correlate with logD_7.4_ (R^2^ = 0.8628, Figure S9), we based our selection of LasB inhibitors to be tested in protein binding assays on the expected PPB as derived from measured logD_7.4_. Taken together, we identified a broad range of protein binding among the phosphonates from 33–99%. Expansion of the initial small correlation of murine PPB with logD_7.4_ with the newly generated data resulted in a similar trend (R^2^ = 0.8385), confirming applicability of our approach to base PPB testing on experimental logD_7.4_ (Figure S9).
As a next step, we subjected a selection of compounds across a PPB range from 33–95% to cassette PK studies at 2 mg/kg dosed intravenously as above. Since we did not detect compound in the lung when PPB was above 98%, we excluded compounds with very high PPB > 98%. Terminal lung levels determined are shown in Figure. Confirming our hypothesis derived from the initial smaller PK study, there was indeed a trend between lung exposure and PPB. We determined favorable lung exposure for the PPB range between 80 and 95% where actually all compounds were detected in lung tissue and ELF. Below 80%, lung levels dropped significantly or were not detectable. It is worth mentioning that corresponding terminal plasma levels of compounds that were not detected in ELF and/or lung tissue were in a similar range as for compounds detected in lung tissue and ELF. Thus, detectability in lung tissue and ELF was not only dependent on the plasma concentration time profile (Figure S10, A). While this might seem to be inconsistent with the proposed general trend, renal excretion supposedly serves as an explanation for this observation as it is known that low PPB leads to faster renal excretion of the free drug.? The significantly higher clearance of 82 and 130 confirms this hypothesis (Table). Moreover, both compounds were detected at high concentrations in urine (Figure S10, B). However, also compounds showing good lung retention and moderate clearance appeared at high concentrations in urine, such as 35. Thus, we assume that additional clearance mechanisms take place for low protein-bound compounds which need to be investigated further. Taken together, the findings suggested a sweet spot of PPB where free drug levels are on the one hand high enough to lead to sufficient permeation into the lung but on the other hand not so low that fast elimination of the free drug impairs lung permeation. Based on the data reported here, we have identified this sweet spot to be between 80 and 95% for the phosphonates under these conditions.
Combined lung levels of additional intravenous cassettes showing epithelial lining fluid (ELF) levels in blue and lung tissue levels in red. Compounds on the x-axis are ranked by PPB from low (82) to high (141).
7: Pharmacokinetic Parameters after intravenous Cassette Dosing of 82, 130, 209, 205, 211, 204, 138 and 35 at 2 mg/kg Each
Based on lung tissue and ELF levels as well as on FRET IC_50_ in the one-digit nanomolar range, not critically impaired by lung surfactant, we selected 35 as the most promising inhibitor from the biaryl class for a focused PK study determining lung levels and tissue distribution after subcutaneous administration of 30 mg/kg (Figure, Table). We did observe that tissue concentrations in kidney and lung were mainly following the plasma concentration profile. Moreover, kidney concentrations were much higher compared to lung tissue, which could be a result of active transport into kidneys albeit no accumulation was observed. Also, urine levels showed high parent compound concentrations following the plasma kinetics. Similarly, ELF concentrations were following plasma kinetics suggesting that compound concentrations in lung tissue and ELF can be correlated with plasma kinetics. This is important as plasma can serve as a surrogate for estimating concentrations in relevant compartments. The measured C max of 3.2 ± 0.3 μg/mL for 35 in ELF equals ∼ 8 μM, which was >500-fold above IC_50_ as it is assumed that ELF levels represent mainly unbound concentrations. Moreover, a favorable ELF/plasma ratio of 2.38 was determined providing further evidence that 35 reaches good concentrations in target compartments. However, 35 also showed high concentrations in liver, reaching a delayed C max compared to plasma concentrations suggesting different transports. Nevertheless, liver concentrations decreased slowly so that dosing schemes would need to be designed to avoid accumulation in liver. Additional studies would be needed to reveal, if transport processes result in higher concentrations of 35 in liver and kidney.
Focused pharmacokinetic (PK) study of 35 at 30 mg/kg SC. Concentrations in plasma, epithelial lining fluid (ELF) and urine are shown as well as tissue levels in lung, kidney and liver.
8: Pharmacokinetic (PK) Parameters of 35 in a Focused PK Study Dosing 30 mg/kg SC
Advanced In Vitro Profiling of Frontrunner 35
We proceeded to determine the capacity of frontrunner 35 to mitigate LasB-associated cytotoxicity, unveiling intriguing results. Our compound exhibited exceptional potency when applied to cells treated with PAO1 culture supernatant (csn), as illustrated in Figure, A. We observed an average cell viability of 81%, when cells encountered the challenge of LasB-deficient ΔlasB PAO1 csn. In contrast, the cell viability dropped to 11% in the presence of PAO1 csn, proving LasB is a significant extracellular virulence factor of P. aeruginosa PAO1, as demonstrated previously in NCI-H292, Calu-3, CFBE epithelial cells? and in macrophages.? In the presence of our compound, a notable enhancement in cell viability was detected, exhibiting an effect that falls between the impact of the wild-type and ΔlasB PAO1. Specifically, the utilization of 5 μM 35 resulted in nearly complete inhibition of LasB activity. These findings underscore the pronounced selectivity of our compound for LasB, demonstrating its proficiency in rescuing cells from the toxic effects of this virulence factor. Moreover, a dose-dependent pattern of LasB inhibition was evident within a low micromolar range, further highlighting the potential of 35 as a promising candidate for in-depth investigations.
(A) Dose-dependent inhibitory impact of 35 against LasB in 10% (v/v) Pseudomonas aeruginosa PAO1 culture supernatant (csn). Each bar reflects the results of three separate experiments, and data are presented with standard deviation (SD). To assess statistical significance, a one-way ANOVA analysis was conducted, followed by Dunnett’s multiple comparisons test, comparing the mean value of each concentration with the mean value of PAO1 without any treatment with the compound. (*** p ≤ 0.0001, *** p ≤ 0.001, ** p ≤ 0.01, * p ≤ 0.05). (B) In vitro activity of 35 against supernatants generated from P. aeruginosa strains PAO1, PA14, PA54, RP73, NH57388A, DSM-24600, DSM-1117, and multiple drug-resistant clinical isolate 83979. IC50 values were determined based on a minimum of three independent experiments, each carried out in duplicate.*
In a next step, we conducted a comprehensive exploration of 35’s potential to inhibit the activity of LasB across additional P. aeruginosa strains, including clinical isolates by implementing the FRET-based LasB inhibition assay. Therefore, we generated supernatants from these strains and adjusted LasB concentrations by diluting them until a comparable proteolytic activity toward the FRET substrate was achieved. Importantly, the IC_50_ values determined does not vary crucially ranging from 10 to 25 nM (Table S5, Figure, B) and matches the in vitro IC_50_ values on the isolated enzyme of 15 nM very well. These results highlight that the inhibitor is indeed active against all tested and potentially additional P. aeruginosa strains with more clinical relevance than the lab strains PAO1 and PA14. This even includes isolates from chronic infections such as RP73 and NH57883A. Notably, the proteolytic activity in these supernatants was significantly lower (Table S5) than in the other strains, which is likely due to the known down-regulation of LasB during chronicization of infections.
Discussion and Conclusions
In order to broaden the scope of potential application of inhibitors targeting LasB from P. aeruginosa , compound properties need to be tailored to the respective indication and patient population. In this study, we aimed at transferring the good retention of LasB inhibitors in the lung after pulmonary administration into favorable lung exposure after IV dosing. In this context, we performed structure-based optimization of our previously reported LasB inhibitors? and improved activity via growing the LasB inhibitor deeper into the active site of the protease. The binding mode of the selected frontrunner compounds 21, 35 and 141 was elucidated using X-ray crystallography, and the successful structure-based compound growth strategy was thereby confirmed. While some changes in the structures led to a significant drop in activity (e.g., substitution in ortho- or meta-position), various changes in the molecule seem to be tolerated well by the LasB binding pocket. Additional beneficial key interactions, typical for the biaryl class – such as CH–π interactions with amino acid side-chain residues - were identified, and the binding mode of 21 to a clinically isolated LasB mutant could be elucidated. Selected inhibitors were further shown to be active against instilled LasB in vivo. We based the further optimization of compound pharmacokinetics on their in vitro ADMET profile. While metabolic stability often turns out to be a parameter that needs to be optimized in drug development,? this is not applicable to the phosphonate-based LasB inhibitors as they seem to be inert toward liver metabolism. This is likely due to their high polarity as confirmed by the low logD_7.4_, rendering them soluble enough for renal excretion of the nonmetabolized compound in vivo. This has been observed before ?,? and could be confirmed in this study. Whereas PPB is generally used to assess free drug levels in order to determine doses for PD studies,? we conclude from the data reported here that it is indeed a parameter that needs to be optimized toward a specific range for this distinct compound class to achieve sufficient exposure in target compartments, such as lung or ELF, after systemic dosing. To rationalize compound selection for PPB measurements from our large set of inhibitors, we employed a correlation with logD_7.4_ as a predictive tool. This reduced the need for protein-binding studies and accelerated the identification of compounds within a suitable logD_7.4_ and with this also PPB range. On top of this rather technical advantage, the in vitro assay-driven preselection of phosphonates for in vivo PK studies assessing lung permeation further reduces the number of animal studies. To what extent this approach is applicable to other compound scaffolds and whether this specific range is transferable to other species or strains of course needs to be determined individually. Here, we could identify a sweet spot of PPB between 80–95% that roughly corresponds to a logD_7.4_ range from –1 to 0.5 or clogP from 1.0 to 1.9. Accordingly, computed logP values can further be used as a parameter guiding straightforward synthetic compound optimization.
In this context, we identified surfactant protein binding to impair the activity of some inhibitors. Notably, 141 and 21, which do bind to surfactant proteins, also show relatively high PPB > 98%, whereas 130 does not bind to surfactant proteins at all and shows only low PPB (61.7%). Thus, the interaction with surfactant can also be linked to compound polarity and optimizing the physicochemical properties of the scaffold turned out advantageous in both regards, resulting in compounds that are more likely to be available in the lung and, once there, less likely to be impaired by binding to surfactant proteins.
The advanced ADMET and IV PK profiling in this study also included representatives from previously published classes. When looking at both tissue and ELF levels, three compounds showed superior behavior: 206, a representative from our previous monoaryl class reported by Konstantinovic et al. has already been shown to be efficacious in a murine lung-infection model after inhalative administration in combination with the SOC levofloxacin.? Second, 212 turned out to be a frontrunner from our dipeptide class reported by Schütz et al. and was shown to be efficacious in combination with meropenem to treat Pseudomonas keratitis. The third superior compound is frontrunner 35 from this study. Its PK were investigated in detail, revealing lung exposure >500-fold above the in vitro IC_50_ value on LasB. The compound was further found to permeate into lung tissue and ELF with a favorable ELF/plasma ratio demonstrating good compound target exposures.
Furthermore, we could also transfer the in vitro activity to other P. aeruginosa strains with no to very little changes in IC_50_ value. This finding is not unexpected as the lasB gene is known to be highly conserved across strains, rendering the LasB inhibitors applicable to various clinical isolates as we have demonstrated previously? and confirmed in this study via X-ray crystallography.
Taken together, our multiparameter approach combining cocrystallization-guided inhibitor optimization with ADMET-DMPK profiling provides a solid platform to advance LasB inhibitors to treatment options in different indications such as HAP/VAP.
Experimental Section
Chemistry
All reagents were used from commercial suppliers without further purification. Procedures were not optimized regarding yield. NMR spectra were recorded on a Bruker AV 500 (500 MHz) spectrometer. Chemical shifts are given in parts per million (ppm) and referenced against the residual proton, ^1^H, or carbon, ^13^C, resonances of the >99% deuterated solvents as internal reference. Coupling constants (J) are given in Hertz (Hz). Data are reported as follows: chemical shift, multiplicity, coupling constants and integration. Liquid chromatography–mass spectrometry was performed on an LC-MS system, consisting of a Dionex UltiMate 3000 pump, autosampler, column compartment and detector (Thermo Fisher Scientific, Dreieich, Germany) and ESI quadrupole MS (MSQ Plus or ISQ EC, Thermo Fisher Scientific, Dreieich, Germany). High-resolution mass was determined by LC-MS/MS using Thermo Scientific Q Exactive Focus Orbitrap LC-MS/MS system. Purity of the final compounds was determined by LC-MS using the area percentage method on the UV trace recorded at a wavelength of 254 nm and found to be >95%.
General Procedure A-1: Amide Coupling Using EDC·HCl Followed
by Boc-Deprotection
Step 1: The acid (1.2–2.0 equiv) was dissolved in DCM. EDC·HCl (1.2–2.0 equiv) was added, followed by the corresponding aniline (1.0 equiv). The resultant mixture was stirred at room temperature (r.t.), until the starting aniline was consumed. The solution obtained was washed with 1 M HCl and sat. aq. NaCl solution. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude product. The crude product obtained was either used in the next step without further purification or purified using column chromatography.
Step 2: Boc-protected aniline (1.0 equiv) obtained in Step 1 was dissolved in DCM. TFA (7.0 equiv) was added, and the mixture was stirred at r.t. overnight. The mixture was evaporated to dryness. Fresh DCM was added, washed with 2 M NaOH and sat. aq. NaCl solution. Dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude product, which was used in the next step without purification.
General Procedure A-2: Amide Coupling Using EDC·HCl and
HOBt
2-(Diethoxyphosphoryl)-4-methylpentanoic acid 214 (1.5 equiv) was dissolved in DCM. EDC·HCl (2.0 equiv), HOBt (2.0 equiv) and DIPEA (2.4 equiv) were added, followed by the corresponding aniline (1.0 equiv). The resultant mixture was stirred at r.t., until the starting aniline was consumed. The solution obtained was washed with 1 M HCl and sat. aq. NaCl solution. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude product. The crude product obtained was either used in the next step without further purification or purified using column chromatography.
General Procedure A-3: Amide Coupling Using TBTU
2-(Diethoxyphosphoryl)-4-methylpentanoic acid 214 (1.2 equiv) was dissolved in DMF or DCM. TBTU (1.5 equiv) and NMM (2.5 equiv) were added, followed by the corresponding aniline (1.0 equiv). The resultant mixture was stirred at r.t. until the starting aniline was consumed. The solution obtained was washed with 1 M NaOH, 1 M HCl and sat. aq. NaCl solution. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude product. The crude product obtained was either used in the next step without further purification or purified using column chromatography.
General Procedure B: Synthesis of Sulfonamides Followed by Boc-Deprotection
Step 1: tert-butyl (4-aminophenyl)carbamate (1.0 equiv) was dissolved in dry DCM and cooled down to 0 °C. Et_3_N (1.2 equiv) was added, followed by the corresponding sulfonyl chloride (1.1 equiv). The ice bath was removed, and the reaction mixture stirred overnight at r.t. Solvents were evaporated, and the crude product obtained was purified using column chromatography.
Step 2: Boc-protected aniline (1.0 equiv) obtained in Step 1 was dissolved in DCM. TFA (7.0 equiv) was added, and the mixture was stirred at r.t. overnight. The mixture was evaporated to dryness. Fresh DCM was added, washed with 2 M NaOH and sat. aq. NaCl solution. Dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude product, which was used in the next step without purification.
Alternatively, Boc-protected aniline (1.0 equiv) obtained in Step 1 was dissolved in DCM/MeOH (1:1). 4 M HCl in dioxane (10.0 equiv) was added, and the mixture was stirred at r.t. overnight. The mixture was evaporated to dryness and used in the next step without purification.
General Procedure C: Synthesis of Phosphonic Acid Derivatives
Step 1: N-Aryl-2-halo-2-alkylacetamide derivative (1.0 equiv) was suspended in triethyl phosphite (10–25 equiv) and heated to 150 °C in a sealed tube for a total of 18 h (or otherwise specified). Most of unreacted triethyl phosphite was evaporated in vacuo, and the resultant oil was purified by column chromatography.
Step 2: To a solution of diethyl phosphonate (1.0 equiv) in dry DCM, bromotrimethylsilane (5.0–7.0 equiv) was added dropwise over a period of 15 min. The reaction mixture was stirred at r.t. overnight (or otherwise specified). If no full conversion was achieved, the excess of bromotrimethylsilane (5.0 equiv) was added the next day. Then, MeOH was added and stirred at r.t. for 30 min to cleave the previously formed TMS ester. Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC.
General Procedure D: Synthesis of Derivatives by Suzuki Coupling
To a mixture of bromo aryl (1 equiv), corresponding boronic acid (1.5 equiv) and potassium carbonate 2 M (1 mL) in a 1,4-dioxane/water mixture (4/1) (2 mL), was added [Pd(dppf)Cl_2_] (0.05 equiv), and the mixture was heated at 150 °C for 20 min under microwave irradiation. The reaction mixture was concentrated in vacuo. The reaction mixtures were diluted with water (5 mL), and the aqueous layer was extracted with DCM (3 × 15 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduced pressure. The product was purified by column chromatography.
General Procedure E: Synthesis of Nitro Derivatives
To a mixture of 1-fluoro-4-nitrobenzene (1.0 equiv) in dry NMP or DMF (10 mL), were added the corresponding aniline, phenol, or thiophenol (1.2 equiv) and potassium carbonate (1.5 equiv). The resulting suspension was stirred at 150 °C for 2–18 h. The reaction mixture was cooled to RT, poured onto ice and filtered. The product was washed with water and dried to give the title compound. The product was used in the next step without further purification.
General Procedure F: Reduction to Afford Amino Derivatives
A mixture of the corresponding nitro derivative (1.0 equiv), Fe (5.0 equiv) and ammonium chloride (0.5 equiv) was dissolved in an ethanol/water (2/1) mixture. The mixture was heated at 100^◦^C for 2 h. Excess ethanol was evaporated under reduced pressure and water (10 mL) was added to the remaining residue, before extraction with ethyl acetate. The organic solvent was then dried over MgSO_4_, filtered and evaporated under reduced pressure. The product was purified by column chromatography.
(1-{[4′-Chloro-(1,1′-biphenyl)-2-yl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (19)
Compound 19 was synthesized according to general procedure C (step 2), using 13 (83 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (water (10 mL) was added). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (59 mg, 81%). ^1^H NMR ((water (10 mL) was added), DMSO) δ 9.27 (s, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.47–7.40 (m, 4H), 7.39–7.34 (m, 1H), 7.30 (dd, J = 7.6, 1.8 Hz, 1H), 7.28–7.24 (m, 1H), 2.94–2.78 (m, 1H), 1.84 (ddd, J = 11.4, 10.7, 3.7 Hz, 1H), 1.38–1.20 (m, 2H), 0.82 (d, J = 6.2 Hz, 3H), 0.78 (d, J =water (10 mL) was added, 3H). ^13^C NMR (126 MHz, DMSO) δ 168.7 (d, J = 5.1 Hz), 138.0, 135.6, 135.2, 132.4, 131.4, 130.5, 128.7, 128.4, 126.8, 126.1, 45.7 (d, J = 127.2 Hz), 36.5 (d, J = 3.9 Hz), 26.6 (d, J = 14.9 Hz), 23.9, 21.7. ^31^P NMR (202 MHz, DMSO) δ 20.68. HRMS (ESI^–^) calculated for C_18_H_20_ClNO_4_P [M-H]^−^ 380.0824, found 380.0822.
(1-{[4′-Chloro-(1,1′-biphenyl)-3-yl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (20)
Compound 20 was synthesized according to general procedure C (step 2), using 14 (83 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (water (10 mL) was added). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (61 mg, 83%). ^1^H NMR (water (10 mL) was added, DMSO) δ ppm: 10.09 (s, 1H), 8.00 (t, J = 1.7 Hz, 1H), 7.66–7.60 (m, 2H), 7.59–7.51 (m, 3H), 7.40 (t, J = 7.9 Hz, 1H), 7.33 (d, J = 7.8 Hz, 1H), 3.01 (ddd, J = 22.4, 11.3, 2.7 Hz, 1H), 2.05–1.91 (m, 1H), 1.58–1.38 (m, 2H), 0.89 (d, J = 2.9 Hz, 3H), 0.87 (d, J = (water (10 mL) was added, 3H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.6 (d, J = 5.4 Hz), 140.6 (s), 139.7 (s), 139.5 (s), 132.9 (s), 129.9 (s), 129.4 (s), 128.8 (s), 121.8 (s), 118.8 (s), 117.6 (s), 46.6 (d, J = 126.7 Hz), 36.2 (d, J = 4.1 Hz), 26.9 (d, J = 14.8 Hz), 22.8 (d, J = 234.2 Hz). ^31^P NMR (water (10 mL) was added, DMSO) δ ppm: 19.81. HRMS (ESI^–^) calculated for C_18_H_20_ClNO_4_P [M–H]^−^ 380.0824, found 380.0822.
(1-{[4′-Chloro-(1,1′-biphenyl)-4-yl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (21)
Compound 21 was synthesized according to general procedure C (step 2), using 15 (83 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (40 mg, 55%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.08 (s, 1H), 7.73–7.70 (m, 2H), 7.68–7.65 (m, 2H), 7.64–7.60 (m, 2H), 7.51–7.47 (m, 2H), 3.01 (ddd, J = 22.4, 11.3, 2.7 Hz, 1H), 2.05–1.87 (m, 1H), 1.57–1.35 (m, 2H), 0.89 (d, J = 2.1 Hz, 3H), 0.87 (d, J = 2.1 Hz, 3H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.5 (d, J = 4.8 Hz), 139.7, 139.0, 133.7, 132.2, 129.3, 128.4, 127.3, 119.8, 46.6 (d, J = 126.8 Hz), 36.2 (d, J = 4.0 Hz), 27.0 (d, J = 14.8 Hz), 23.7, 21.9. ^31^P NMR (202 MHz, DMSO) δ ppm: 19.80. HRMS (ESI^–^) calculated for C_18_H_20_ClNO_4_P [MsH]^−^ 380.0824, found 380.0822.
1-(5-(4-Chlorophenyl)pyridin-2-ylcarbamoyl)-3-methylbutylphosphonic
Acid (22)
Compound 22 was synthesized according to general procedure C (step 2), using 16 (83 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (40 mg, 55%). ^1^H NMR (500 MHz, MeOD) δ ppm: 8.44 (d, J = 2.1 Hz, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.98 (dd, J = 8.7, 2.4 Hz, 1H), 7.54 (d, J = 8.5 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 3.11 (dd, J = 24.0, 12.8 Hz, 1H), 2.12–1.95 (m, 1H), 1.56 (dd, J = 10.8, 4.9 Hz, 2H), 0.88 (d, J = 5.3 Hz, 3H), 0.86 (d, J = 4.3 Hz, 3H). ^13^C NMR (126 MHz, MeOD) δ ppm: 170.1, 150.7, 144.4, 136.9, 135.6, 133.7, 131.5, 128.9, 127.8, 114.2, 35.9 (d, J = 3.3 Hz), 26.8 (d, J = 14.6 Hz), 22.2, 21.3, 20.4. ^31^P NMR (202 MHz, MeOD) δ ppm: 20.28. HRMS (ESI^–^) calculated for C_17_H_19_ClN_2_O_4_P [M-H]^−^ 381.0776, found 381.0776.
1-[6-(4-Chlorophenyl)pyridin-3-ylcarbamoyl]-3-methylbutylphosphonic
Acid (23)
Compound 23 was synthesized according to general procedure C (step 2), using 17 (83 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (53 mg, 73%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.36 (s, 1H), 8.84 (d, J = 2.4 Hz, 1H), 8.17 (dd, J = 8.7, 2.5 Hz, 1H), 8.09–8.03 (m, 2H), 7.94 (d, J = 8.7 Hz, 1H), 7.70–7.40 (m, 2H), 3.04 (ddd, J = 22.4, 11.2, 2.4 Hz, 1H), 2.04–1.93 (m, 1H), 1.57–1.39 (m, 2H), 0.89 (s, 3H), 0.88 (s, 3H). ^13^C NMR (126 MHz, DMSO) δ ppm: 169.1 (d, J = 4.8 Hz), 149.6, 140.7, 137.7, 135.9, 133.7, 129.2, 128.2, 127.1, 120.6, 46.6 (d, J = 126.0 Hz), 36.1 (d, J = 3.9 Hz), 27.0 (d, J = 14.4 Hz), 23.6, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 19.26. HRMS (ESI^–^) calculated for C_17_H_19_ClN_2_O_4_P [M-H]^−^ 381.0776, found 381.0776.
1-[2-(4-Chlorophenyl)pyrimidin-5-ylcarbamoyl]-3-methylbutylphosphonic
Acid (24)
Compound 24 was synthesized according to general procedure C (step 2), using 18 (83 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (22 mg, 31%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.59 (s, 1H), 9.12 (s, 2H), 8.48–8.08 (m, 2H), 7.69–7.27 (m, 2H), 3.06 (ddd, J = 22.3, 11.0, 2.2 Hz, 1H), 2.01 (ddd, J = 15.3, 10.1, 3.7 Hz, 1H), 1.59–1.32 (m, 2H), 0.88 (d, J = 6.2 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 169.4 (d, J = 4.8 Hz), 157.4, 147.7, 136.3, 135.5, 133.6, 129.4, 129.2, 46.6 (d, J = 125.5 Hz), 36.1 (d, J = 3.9 Hz), 26.9 (d, J = 14.4 Hz), 23.6, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 18.74. HRMS (ESI^–^) calculated for C_16_H_18_ClN_3_O_4_P [M-H]^−^ 382.0729, found 382.0727.
1-[2-(4-Isopropoxyphenyl)pyrimidin-5-ylcarbamoyl-3-methylbutylphosphonic
Acid (35)
Compound 35 was synthesized according to general procedure C (step 2), using 33 (88 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (46 mg, 60%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.46 (s, 1H), 9.05 (s, 2H), 8.24 (d, J = 8.9 Hz, 2H), 7.01 (d, J = 8.9 Hz, 2H), 4.70 (dt, J = 12.1, 6.0 Hz, 1H), 3.04 (ddd, J = 22.4, 11.1, 2.3 Hz, 1H), 2.00 (ddd, J = 15.4, 10.0, 3.7 Hz, 1H), 1.61–1.35 (m, 2H), 1.30 (d, J = 6.0 Hz, 6H), 0.88 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 169.2 (d, J = 4.7 Hz), 159.8, 158.5, 147.8, 132.7, 129.7, 129.4, 115.9, 69.8, 46.6 (d, J = 125.6 Hz), 36.1 (d, J = 3.9 Hz), 26.9 (d, J = 14.5 Hz), 23.6, 22.3, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 18.90. HRMS (ESI^–^) calculated for C_19_H_25_N_3_O_5_P [M-H]^−^ 406.1537, found 406.1533.
1-[2-(3-Chloro-4-isopropoxyphenyl)pyrimidin-5-ylcarbamoyl]-3-methylbutylphosphonic
Acid (36)
Compound 36 was synthesized according to general procedure C (step 2), using 34 (95 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (43 mg, 51%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.54 (s, 1H), 9.07 (s, 2H), 8.29 (d, J = 2.1 Hz, 1H), 8.22 (dd, J = 8.7, 2.1 Hz, 1H), 7.28 (d, J = 8.9 Hz, 1H), 4.78 (dt, J = 12.1, 6.0 Hz, 1H), 3.04 (ddd, J = 22.4, 11.1, 2.3 Hz, 1H), 2.00 (ddd, J = 15.3, 10.2, 3.7 Hz, 1H), 1.56–1.41 (m, 2H), 1.34 (d, J = 6.0 Hz, 6H), 0.88 (d, J = 6.2 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 169.3 (d, J = 5.6 Hz), 157.2, 155.0, 147.7, 133.2, 130.7, 129.1, 127.7, 123.0, 115.6, 71.7, 46.6 (d, J = 125.5 Hz), 36.1 (d, J = 4.1 Hz), 26.9 (d, J = 14.4 Hz), 23.6, 22.2, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 18.79. HRMS (ESI^–^) calculated for C_19_H_24_ClN_3_O_5_P [M–H]^−^ 440.1147, found 440.1144.
1-(4-Morpholinophenylcarbamoyl)-3-methylbutylphosphonic Acid
(80)
Compound 80 was synthesized according to general procedure C (step 2), using 66 (78 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a white solid (21 mg, 32%). ^1^H NMR (500 MHz, DMSO) δ ppm: 9.74 (s, 1H), 7.47 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 9.1 Hz, 2H), 3.79–3.65 (m, 4H), 3.08–2.98 (m, 4H), 2.93 (ddd, J = 22.3, 11.3, 2.8 Hz, 1H), 2.01–1.88 (m, 1H), 1.54–1.35 (m, 2H), 0.87 (d, J = 1.0 Hz, 3H), 0.86 (d, J = 1.0 Hz, 3H). ^13^C NMR (126 MHz, DMSO) δ ppm: 167.6 (d, J = 4.7 Hz), 147.5, 132.4, 120.6, 115.9, 66.6, 49.6, 46.3 (d, J = 127.5 Hz), 36.3 (d, J = 4.1 Hz), 28.0–26.0 (m), 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 20.49. HRMS (ESI^–^) calculated for C_16_H_24_N_2_O_5_P [M-H]^−^ 355.1428, found 355.1426.
1-[4-(Piperazin-1-yl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (81)
Compound 81 was synthesized according to general procedure C (step 2), using 67 (97 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a white solid (23 mg, 35%). ^1^H NMR (500 MHz, D_2_O) δ ppm: 7.33 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.5 Hz, 2H), 3.32 (s, 8H), 2.82 (ddd, J = 13.5, 12.3, 2.2 Hz, 1H), 1.90 (ddd, J = 11.8, 9.3, 6.4 Hz, 1H), 1.47 (dd, J = 26.5, 6.9 Hz, 2H), 0.84 (d, J = 5.7 Hz, 6H). ^13^C NMR (126 MHz, D_2_O) δ ppm: 146.9 (d, J = 5.1 Hz), 131.5, 124.1, 123.4, 118.2, 47.0, 47.4 (d, J = 129.9 Hz), 43.0, 36.4 (d, J = 4.4 Hz), 26.8 (d, J = 14.3 Hz), 22.5, 20.6. ^31^P NMR (202 MHz, D_2_O) δ ppm: 17.60. HRMS (ESI^–^) calculated for C_16_H_25_N_3_O_4_P [M-H]^−^ 354.1588, found 354.1586.
1-(4-Morpholinophenylcarbamoyl)-3-methylbutylphosphonic Acid
(82)
Compound 82 was synthesized according to general procedure C (step 2), using 68 (80 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a white solid (28 mg, 41%). ^1^H NMR (500 MHz, D_2_O) δ ppm: 7.41 (d, J = 8.9 Hz, 2H), 7.07 (d, J = 9.0 Hz, 2H), 3.76 (d, J = 11.5 Hz, 2H), 3.62 (d, J = 9.6 Hz, 2H), 3.21–3.23 (m, 2H), 3.10 (d, J = 11.0 Hz, 2H), 2.94 (s, 3H), 2.93–2.84 (m, 1H), 1.98 (ddd, J = 11.9, 9.3, 6.2 Hz, 1H), 1.63–1.47 (m, 2H), 0.92 (d, J = 6.0 Hz, 6H). ^13^C NMR (126 MHz, D_2_O) δ ppm: 167.7 (d, J = 5.5 Hz), 120.6, 116.8, 50.3, 46.4 (d, J = 127.1 Hz), 36.3 (d, J = 4.0 Hz), 33.9, 30.5, 26.9 (d, J = 15.0 Hz), 23.7, 22.3, 21.9. ^31^P NMR (202 MHz, D_2_O) δ ppm: 17.67. HRMS (ESI^–^) calculated for C_17_H_27_N_3_O_4_P [M-H]^−^ 368.1745, found 368.1740.
1-[4-(4-Acetylpiperazin-1-yl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (83)
Compound 83 was synthesized according to general procedure C (step 2), using 69 (86 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as white solid (22 mg, 29%). ^1^H NMR (500 MHz, DMSO) δ ppm: 9.73 (s, 1H), 7.47 (d, J = 9.0 Hz, 2H), 6.90 (d, J = 9.1 Hz, 2H), 3.57 (dd, J = 10.2, 5.4 Hz, 4H), 3.12–3.05 (m, 2H), 3.03–2.98 (m, 2H), 2.92 (ddd, J = 22.3, 11.3, 2.8 Hz, 1H), 2.04 (s, 3H), 1.95 (tdd, J = 11.8, 7.4, 4.3 Hz, 1H), 1.53–1.32 (m, 2H), 0.87 (d, J = 6.1 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.7, 167.7 (d, J = 5.0 Hz), 147.2, 132.7, 120.6, 116.8 (s), 49.7 (d, J = 59.2 Hz), 46.8, 46.0, 45.8, 41.1, 36.3 (d, J = 4.0 Hz), 26.9 (d, J = 15.0 Hz), 23.7, 21.8, 21.7. ^31^P NMR (202 MHz, DMSO) δ ppm: 20.32. HRMS (ESI^–^) calculated for C_18_H_27_N_3_O_5_P [M-H]^−^ 396.1694, found 396.1687.
1-[4-(4-Methylpiperidin-1-yl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (84)
Compound 84 was synthesized according to general procedure C (step 2), using 70 (80 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as white solid (15 mg, 22%). ^1^H NMR (500 MHz, D_2_O) δ ppm: 7.41 (d, J = 8.9 Hz, 2H), 7.07 (d, J = 9.0 Hz, 2H), 3.76 (d, J = 11.5 Hz, 2H), 3.62 (d, J = 9.6 Hz, 2H), 3.21–3.23 (m, 2H), 3.10 (d, J = 11.0 Hz, 2H), 2.94 (s, 3H), 2.93–2.84 (m, 1H), 1.98 (ddd, J = 11.9, 9.3, 6.2 Hz, 1H), 1.63–1.47 (m, 2H), 0.92 (d, J = 6.0 Hz, 6H). ^13^C NMR (126 MHz, D_2_O) δ ppm: 172.6 (d, J = 4.5 Hz), 146.3, 131.4, 123.4, 118.1, 52.9, 47.4 (d, J = 121.2 Hz), 47.2, 42.8, 36.3 (d, J = 3.9 Hz), 26.8 (d, J = 14.4 Hz), 22.5, 20.6. ^31^P NMR (202 MHz, D_2_O) δ ppm: 17.67. HRMS (ESI^–^) calculated for C_18_H_28_N_2_O_4_P [M-H]^−^ 367.1792, found 367.1791.
1-[4-(1H-Imidazol-1-yl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (85)
Compound 85 was synthesized according to general procedure C (step 2), using 71 (75 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a white solid (18 mg, 28%). ^1^H NMR (500 MHz, D_2_O) δ ppm: 8.85 (s, 1H), 7.66 (s, 1H), 7.52 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.7 Hz, 3H), 2.84 (dd, J = 21.3, 10.9 Hz, 1H), 1.87 (dt, J = 11.6, 7.6 Hz, 1H), 1.44 (dd, J = 11.1, 5.5 Hz, 2H), 0.78 (d, J = 5.9 Hz, 6H). ^13^C NMR (126 MHz, D_2_O) δ ppm: 172.8 (d, J = 5.0 Hz), 138.6, 133.5, 131.2, 122.9, 122.4, 121.1, 120.7, 47.6 (d, J = 120.7 Hz), 36.2 (d, J = 4.6 Hz), 26.8 (d, J = 14.3 Hz), 22.5, 20.6. ^31^P NMR (202 MHz, D_2_O) δ ppm: 17.41. HRMS (ESI^–^) calculated for C_15_H_19_N_3_O_4_P [M-H]^−^ 336.1119, found 336.1117.
1–4-(1H-Pyrazol-1-yl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (86)
Compound 86 was synthesized according to general procedure C (step 2), using 72 (75 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a white solid (12 mg, 19%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.10 (s, 1H), 8.40 (d, J = 2.4 Hz, 1H), 7.77–7.74 (m, 2H), 7.73–7.71 (m, 2H), 7.70 (d, J = 1.6 Hz, 1H), 6.51–6.49 (m, 1H), 3.00 (ddd, J = 22.5, 11.3, 2.7 Hz, 1H), 2.03–1.94 (m, 1H), 1.57–1.38 (m, 2H), 0.88 (d, J = 1.7 Hz, 3H), 0.87 (d, J = 1.8 Hz, 3H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.3 (d, J = 5.0 Hz), 141.0, 138.1, 135.5, 127.8, 120.2, 119.2, 108.0, 46.5 (d, J = 127.0 Hz), 36.2 (d, J = 3.9 Hz), 26.9 (d, J = 14.7 Hz), 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 20.00. HRMS (ESI^–^) calculated for C_15_H_19_N_3_O_4_P [M-H]^−^ 336.1119, found 336.1116.
1-[4-(2H-1,2,3-Triazol-1-yl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (87)
Compound 87 was synthesized according to general procedure C (step 2), using 73 (75 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a white solid (11 mg, 17%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.20 (s, 1H), 8.08 (s, 2H), 7.95 (d, J = 9.0 Hz, 2H), 7.80 (d, J = 9.0 Hz, 2H), 3.02 (ddd, J = 22.5, 11.3, 2.6 Hz, 1H), 1.99 (tdd, J = 11.7, 7.3, 4.2 Hz, 1H), 1.47 (qdd, J = 12.6, 9.7, 4.7 Hz, 2H), 0.88 (d, J = 5.9 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.6 (d, J = 4.8 Hz), 139.4, 136.5, 134.9, 120.2, 119.4, 46.6 (d, J = 126.6 Hz), 36.2 (d, J = 3.9 Hz), 27.0 (d, J = 14.7 Hz), 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 19.65. HRMS (ESI^–^) calculated for C_14_H_18_N_4_O_4_P [M-H]^−^ 337.1071, found 337.1070.
1-[4-(1H-1,2,4-Triazol-1-yl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (88)
Compound 88 was synthesized according to general procedure C (step 2), using 74 (75 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as white solid (22 mg, 34%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.25 (s, 1H), 9.26 (s, 1H), 8.27 (s, 1H), 7.84 (s, 4H), 3.07 (ddd, J = 22.5, 11.2, 2.6 Hz, 1H), 2.05 (tdd, J = 11.7, 7.3, 4.1 Hz, 1H), 1.65–1.40 (m, 2H), 0.94 (d, J = 5.5 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.6 (d, J = 4.7 Hz), 152.7, 142.4, 139.4, 132.3, 120.4, 120.2, 46.6 (d, J = 126.7 Hz), 36.2 (d, J = 4.0 Hz), 27.0 (d, J = 14.7 Hz), 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 19.61. HRMS (ESI^–^) calculated for C_14_H_18_N_4_O_4_P [M-H]^−^ 337.1071, found 337.1070.
(1-{[4-(3,4-Dichlorobenzamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (193)
Compound 193 was synthesized over two steps according to the general procedure C using 184 (209 mg, 0.46 mmol) and triethyl phosphite (2 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 1/1 to 25/75) to give diethyl phosphonate as a white solid (47.6 mg, 20%). The product obtained was then treated with bromotrimethylsilane (80 μL, 0.60 mmol) in DCM (2 mL). TMS ester was cleaved using MeOH (2 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (25 mg, 63%; 13% over two steps). ^1^H NMR (500 MHz, DMSO) δ 10.36 (s, 1H), 9.99 (s, 1H), 8.21 (d, J = 2.0 Hz, 1H), 7.93 (dd, J = 8.4, 2.0 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 9.0 Hz, 2H), 7.59 (d, J = 9.0 Hz, 2H), 2.97 (ddd, J = 22.3, 11.2, 2.3 Hz, 1H), 2.02–1.91 (m, 1H), 1.55–1.35 (m, 2H), 0.87 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8, 162.9, 135.8, 135.3, 134.3, 133.9, 131.3, 130.8, 129.6, 128.1, 120.9, 119.2, 46.0 (d, J = 127.0 Hz), 35.8 (d, J = 3.7 Hz), 26.5 (d, J = 14.8 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.94. HRMS (ESI^–^) calculated for C_19_H_20_Cl_2_N_2_O_5_P^–^ [M–H]^−^ 457.0492, found 457.0497.
(4-Methyl-1-oxo-1-{[4-(thiophene-2-carboxamido)phenyl]amino}pentan-2-yl)phosphonic
Acid (196)
Compound 196 was synthesized over two steps according to the general procedure C, using 187 (147 mg, 0.37 mmol) and triethyl phosphite (2 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 1/1 to EtOAc) to give diethyl phosphonate as white solid (137.2 mg, 82%). The product obtained (135 mg, 0.30 mmol) was then treated with bromotrimethylsilane (275 μL, 2.10 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (36.5 mg, 31%; 25% over two steps). ^1^H NMR (500 MHz, DMSO) δ 10.19 (s, 1H), 9.95 (s, 1H), 8.00 (dd, J = 3.8, 0.9 Hz, 1H), 7.84 (dd, J = 5.0, 0.9 Hz, 1H), 7.65–7.53 (m, 4H), 7.21 (dd, J = 4.9, 3.8 Hz, 1H), 2.97 (ddd, J = 22.4, 11.3, 2.5 Hz, 1H), 2.02–1.92 (m, 1H), 1.54–1.33 (m, 2H), 0.87 (d, J = 6.1 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7 (d, J = 4.3 Hz), 159.7, 140.2, 135.5, 133.8, 131.8, 128.9, 128.2, 120.9, 119.3, 46.0 (d, J = 127.1 Hz), 35.8, 26.5 (d, J = 15.0 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 20.02. HRMS (ESI^–^) calculated for C_17_H_20_N_2_O_5_PS^–^ [M-H]^−^ 395.0836, found 395.0840.
(1-{[3-(3,4-Dichlorobenzamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (204)
Compound 204 was synthesized according to the general procedure C (Step 2), using 202 (88 mg, 0.17 mmol) and bromotrimethylsilane (112 μL, 0,85 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a colorless solid (26 mg, 33%). ^1^H NMR (500 MHz, DMSO) δ10.39 (s, 1H), 10.01 (s, 1H), 8.22 (d, J = 2.1 Hz, 1H), 8.12 (t, J = 2.1 Hz, 1H), 7.94 (dd, J = 8.4, 2.1 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.39 (ddd, J = 14.5, 7.6 Hz, 1.9, 2H), 7.25 (t, J = 8.1 Hz, 1H), 3.02 (ddd, J = 22.5, 11.3, 2.8 Hz, 1H), 1.97 (tq, J = 11.9, 7.0, 5.4 Hz, 1H), 1.46 (dddd, J = 32.3, 16.5, 7.7, 4.6 Hz, 2H), 0.87 (dd, J = 6.4, 1.8 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.9 (d, J = 6.7 Hz), 163.1, 139.7, 138.9, 135.2, 134.4, 131.3, 130.8, 129.7, 128.7, 128.1, 115.3, 115.0, 111.5, 46.0 (d, J = 126.7 Hz), 35.8 (d, J = 4.4 Hz), 26.5 (d, J = 15.0 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ19.95. HRMS (ESI^–^) calculated for C_19_H_20_Cl_2_N_2_O_5_P^–^ [M-H]^−^ 457.0492, found 457.0487.
(1-{[4-(Isonicotinamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (130)
Compound 130 was synthesized according to the general procedure C (Step 2), using 112 (102 mg, 0.23 mmol) and bromotrimethylsilane (150 μL, 1.14 mmol) in DCM (4 mL). The TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a yellow solid (25.5 mg, 28%). ^1^H NMR (500 MHz, DMSO) δ 10.45 (s, 1H), 9.96 (s, 1H), 8.77 (dd, J = 4.5, 1.6 Hz, 2H), 7.85 (dd, J = 4.5, 1.6 Hz, 2H), 7.67 (d, J = 9.0 Hz, 2H), 7.64–7.54 (m, 2H), 2.97 (ddd, J = 22.4, 11.3, 2.8 Hz, 1H), 1.97 (tdd, J = 11.7, 7.3, 4.2 Hz, 1H), 1.55–1.37 (m, 2H), 0.91–0.85 (m, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7 (d, J = 4.6 Hz), 163.6, 150.3, 142.0, 135.9, 133.7, 121.6, 120.9, 119.3, 46.0 (d, J = 126.9 Hz), 35.8 (d, J = 3.7 Hz), 26.5 (d, J = 14.7 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.92. HRMS (ESI^–^) calculated for C_18_H_21_N_3_O_5_P^–^ [M-H]^−^ 390.1224, found 390.1218.
(1-{[4-(4,5-Dichloroisothiazole-3-carboxamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (131)
Compound 131 was synthesized according to the general procedure C (Step 2), using 113 (168 mg, 0.32 mmol) and bromotrimethylsilane (211 μL, 0.85 mmol) in DCM (6 mL). The TMS ester was cleaved using MeOH (6 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a colorless solid (38 mg, 25%). ^1^H NMR (500 MHz, DMSO) δ 10.71 (s, 1H), 9.99 (s, 1H), 7.69–7.64 (m, 2H), 7.62–7.58 (m, 2H), 2.97 (ddd, J = 22.4, 11.3, 2.9 Hz, 1H), 2.02–1.91 (m, 1H), 1.55–1.37 (m, 2H), 0.87 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8 (d, J = 6.6 Hz), 158.6, 157.8, 149.6, 136.1, 133.1, 122.8, 120.6, 119.4, 46.1 (d, J = 127.0 Hz), 35.8 (d, J = 2.5 Hz), 26.5 (d, J = 14.7 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.85. HRMS (ESI^–^) calculated for C_16_H_17_Cl_2_N_3_O_5_PS^–^ [M-H]^−^ 464.0009, found 464.0000.
(1-{[4-(5-Chlorothiophene-2-carboxamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (132)
Compound 132 was synthesized according to the general procedure C (Step 2), using 114 (107 mg, 0.22 mmol) and bromotrimethylsilane (145 μL, 1.1 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a colorless solid (66 mg, 68%). ^1^H NMR (500 MHz, DMSO) δ 10.25 (s, 1H), 9.96 (s, 1H), 7.89 (d, J = 4.1 Hz, 1H), 7.59 (s, 4H), 7.26 (d, J = 4.1 Hz, 1H), 2.97 (ddd, J = 22.3, 11.3, 2.9 Hz, 1H), 2.02–1.89 (m, 1H), 1.46 (dddd, J = 32.9, 16.5, 7.3, 4.2 Hz, 2H), 0.87 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7 (d, J = 4.6 Hz), 158.5, 139.3, 135.7, 133.7, 133.4, 128.9, 128.3, 120.9, 119.3, 46.0 (d, J = 127.0 Hz), 35.8 (d, J = 4.0 Hz), 26.5 (d, J = 14.9 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ19.91. HRMS (ESI^–^) calculated for C_17_H_19_ClN_2_O_5_PS^–^ [M-H]^−^ 429.0446, found 429.0443.
(1-{[4-(5-Chlorothiophene-2-carboxamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (133)
Compound 133 was synthesized according to the general procedure C (Step 2), using 115 (106 mg, 0.25 mmol) and bromotrimethylsilane (165 μL, 1.25 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a colorless solid (52 mg, 48%). ^1^H NMR (500 MHz, DMSO) δ 10.24 (d, J = 3.2 Hz, 1H), 9.97 (s, 1H), 8.41 (d, J = 17.4 Hz, 1H), 7.71 (d, J = 8.7 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H), 2.97 (dd, J = 22.3, 11.0 Hz, 1H), 1.97 (s, 1H), 1.57–1.39 (m, 2H), 0.87 (d, J = 6.2 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8 (d, J = 2.7 Hz), 157.8, 157.7, 150.9, 149.7, 148.1, 136.5, 135.8, 133.4, 129.6, 128.1, 121.0, 119.1, 46.1 (d, J = 126.7 Hz), 35.8, 26.5 (d, J = 14.3 Hz), 23.2, 21.4 (rotamers detected). ^31^P NMR (202 MHz, DMSO) δδ 19.95. HRMS (ESI^–^) calculated for C_16_H_18_ClN_3_O_5_PS^–^ [M-H]^−^ 430.0399, found 430.0391.
(1-{[4-(4,5-Dichlorothiophene-2-carboxamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (134)
Compound 134 was synthesized according to the general procedure C (Step 2), using 116 (105 mg, 0.20 mmol) and bromotrimethylsilane (132 μL, 1.00 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a colorless solid (19 mg, 20%). ^1^H NMR (500 MHz, DMSO) δδ 10.33 (s, 1H), 10.00 (s, 1H), 8.07 (s, 1H), 7.59 (s, 4H), 2.98 (dt, J = 20.5, 6.9, 1H), 1.96 (s, 1H), 1.59–1.38 (m, 2H), 0.87 (d, J = 6.3, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8 (d, J = 6.6 Hz), 157.6, 137.5, 136.0, 133.1, 128.9, 128.0, 123.4, 120.7, 119.3, 46.1 (d, J = 126.8 Hz), 35.8, 26.5 (d, J = 14.5 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δδ 19.86. HRMS (ESI^–^) calculated for C_17_H_18_Cl_2_N_2_O_5_PS^–^ [M-H]^−^ 463.0057, found 463.0042.
(4-Methyl-1-oxo-1-{[4-(4-phenylthiophene-2-carboxamido)phenyl]amino}pentan-2-yl)phosphonic
Acid (135)
Compound 135 was synthesized according to the general procedure C (Step 2), using 117 (99 mg, 0.19 mmol) and bromotrimethylsilane (125 μL, 0.95 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a colorless solid (21 mg, 23%). ^1^H NMR (500 MHz, DMSO) δδ 10.25 (s, 1H), 10.03 (s, 1H), 8.47 (d, J = 1.5 Hz, 1H), 8.17 (d, J = 1.4 Hz, 1H), 7.77–7.72 (m, 2H), 7.63 (q, J = 8.9 Hz, 4H), 7.47 (t, J = 7.7 Hz, 2H), 7.35 (t, J = 7.3 Hz, 1H), 2.98 (dd, J = 22.6, 11.2 Hz, 1H), 1.97 (s, 1H), 1.56–1.36 (m, 2H), 0.87 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.9, 159.5, 141.9, 140.9, 135.7, 134.7, 133.8, 129.2, 127.7, 127.5, 126.4, 126.0, 120.7, 119.3, 46.1 (d, J = 127.2 Hz), 35.8 (d, J = 2.2 Hz), 26.5 (d, J = 14.5 Hz), 23.4, 21.5. ^31^P NMR (202 MHz, DMSO) δδ 19.91. HRMS (ESI^–^) calculated for C_23_H_24_N_2_O_5_PS^–^ [M-H]^−^ 471.1149, found 471.1154.
(1-{[4-(Furan-2-carboxamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (136)
Compound 136 was synthesized according to the general procedure C (Step 2), using 118 (109 mg, 0.24 mmol) and bromotrimethylsilane (158 μL, 1.20 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a pale yellow solid (46 mg, 50%). ^1^H NMR (500 MHz, DMSO) δ 10.13 (s, 1H), 9.99 (s, 1H), 7.92 (d, J = 1.6 Hz, 1H), 7.64 (d, J = 9.0 Hz, 2H), 7.56 (d, J = 9.1 Hz, 2H), 7.30 (d, J = 3.4 Hz, 1H), 6.69 (dd, J = 3.5, 1.7 Hz, 1H), 2.97 (ddd, J = 22.3, 11.4, 2.8 Hz, 1H), 1.95 (dq, J = 11.7, 6.9, 4.7 Hz, 1H), 1.55–1.35 (m, 2H), 0.86 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8 (d, J = 5.9 Hz), 156.0, 147.7, 145.7, 135.6, 133.6, 120.8, 119.2, 114.5, 112.2, 46.1 (d, J = 127.4 Hz), 35.9, 26.5 (d, J = 14.7 Hz), 23.3, 21.5. ^31^P NMR (202 MHz, DMSO) δ 19.93. HRMS (ESI^–^) calculated for C_17_H_20_N_2_O_6_P^–^ [M-H]^−^ 379.1064, found 390.1071.
(4-Methyl-1-oxo-1-{[4-(quinoline-3-carboxamido)phenyl]amino}pentan-2-yl)phosphonic
Acid (137)
Compound 137 was synthesized according to the general procedure C (Step 2), using 119 (103 mg, 0.23 mmol) and bromotrimethylsilane (152 μL, 1.15 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a pale yellow solid (38 mg, 37%). ^1^H NMR (500 MHz, DMSO) δ 10.56 (s, 1H), 9.98 (s, 1H), 9.35 (d, J = 2.3 Hz, 1H), 8.95 (d, J = 2.2 Hz, 1H), 8.21–8.07 (m, 2H), 7.90 (ddd, J = 8.4, 6.7, 1.5 Hz, 1H), 7.78–7.70 (m, 3H), 7.66–7.58 (m, 2H), 2.99 (ddd, J = 22.4, 11.4, 2.9 Hz, 1H), 1.98 (tdd, J = 12.0, 7.1, 4.2 Hz, 1H), 1.47 (dddd, J = 32.5, 16.5, 7.3, 4.2 Hz, 2H), 0.88 (dd, J = 6.5, 1.6 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7, 163.8, 149.1, 148.5, 135.9, 135.7, 134.1, 131.4, 129.2, 128.8, 127.7, 127.5, 126.5, 120.7, 119.3, 46.0 (d, J = 126.9 Hz), 35.8 (d, J = 3.8 Hz), 26.5 (d, J = 14.5 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.99. HRMS (ESI^–^) calculated for C_22_H_23_N_3_O_5_P^–^ [M-H]^−^ 440.1381, found 440.1381.
(1-{[4-(Benzo[d]thiazole-2-carboxamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (138)
Compound 138 was synthesized according to the general procedure C (Step 2), using 120 (102 mg, 0.23 mmol) and bromotrimethylsilane (152 μL, 1.15 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a pale yellow solid (36 mg, 36%). ^1^H NMR (500 MHz, DMSO) δ 11.04 (s, 1H), 10.00 (s, 1H), 8.28–8.25 (m, 1H), 8.21 (d, J = 8.1 Hz, 1H), 7.84–7.79 (m, 2H), 7.69–7.64 (m, 1H), 7.64–7.59 (m, 3H), 2.98 (ddd, J = 22.6, 11.3, 2.9 Hz, 1H), 1.97 (tdd, J = 11.8, 7.1, 4.2 Hz, 1H), 1.46 (ddtd, J = 35.5, 12.7, 9.6, 4.7 Hz, 2H), 0.87 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8 (d, J = 5.0 Hz), 164.9, 157.8, 152.7, 136.4, 136.2, 132.9, 127.3, 127.1, 124.1, 123.1, 121.1, 119.2, 46.1 (d, J = 126.7 Hz), 35.8 (d, J = 4.2 Hz), 26.5 (d, J = 14.6 Hz), 23.2, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.90. HRMS (ESI^–^) calculated for C_20_H_21_N_3_O_5_PS^–^ [M-H]^−^ 446.0945, found 446.0947.
(4-Methyl-1-oxo-1-{[4-(5-phenylthiophene-2-carboxamido)phenyl]amino}pentan-2-yl)phosphonic
Acid (139)
Compound 139 was synthesized according to the general procedure C (Step 2), using 121 (98 mg, 0.21 mmol) and bromotrimethylsilane (139 μL, 1.05 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 0.5/9.5 to 10/0). The product was obtained as a pale yellow solid (48 mg, 48%). ^1^H NMR (500 MHz, DMSO) δ 10.21 (s, 1H), 9.94 (s, 1H), 8.01 (d, J = 4.0 Hz, 1H), 7.75 (dd, J = 7.3, 1.7 Hz, 2H), 7.66–7.58 (m, 5H), 7.49–7.44 (m, 2H), 7.41–7.36 (m, 1H), 2.98 (ddd, J = 22.5, 11.3, 2.9 Hz, 1H), 1.98 (tq, J = 12.1, 5.7, 4.8 Hz, 1H), 1.55–1.37 (m, 2H), 0.87 (dd, J = 6.4, 1.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.6 (d, J = 4.8 Hz), 159.4, 148.2, 139.1, 135.5, 133.8, 133.0, 130.0, 129.3, 128.7, 125.7, 124.5, 120.8, 119.3, 46.0 (d, J = 127.0 Hz), 35.8 (d, J = 4.1 Hz), 26.5 (d, J = 14.6 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 20.01. HRMS (ESI^–^) calculated for C_23_H_24_N_2_O_5_PS^–^ [M-H]^−^ 471.1149, found 471.1134.
(4-Methyl-1-oxo-1-{[4-(phenylsulfonamido)phenyl]amino}pentan-2-yl)phosphonic
Acid (140)
Compound 140 was synthesized according to the general procedure C (Step 2), using 122 (156 mg, 0.32 mmol) and bromotrimethylsilane (215 μL, 1.62 mmol) in DCM (4 mL). The TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (83 mg, 61%). ^1^H NMR (500 MHz, DMSO) δ 10.07 (s, 1H), 9.90 (s, 1H), 7.70 (dd, J = 5.3, 3.4 Hz, 2H), 7.58 (ddd, J = 6.4, 3.7, 1.2 Hz, 1H), 7.56–7.49 (m, 2H), 7.44 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 2.98–2.84 (m, 1H), 1.92 (s, 1H), 1.48–1.27 (m, 2H), 0.83 (dd, J = 6.2, 2.2 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7, 139.4, 136.3, 132.9, 132.4, 129.3, 126.7, 121.7, 119.7, 46.0 (d, J = 126.6 Hz), 35.7, 26.4 (d, J = 14.0 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.87. HRMS (ESI^–^) calculated for C_18_H_22_N_2_O_6_PS^–^ [M-H]^−^ 425.0942, found 425.0931.
[1-({4-[(3,4-Dichlorophenyl)sulfonamido]phenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (141)
Compound 141 was synthesized according to the general procedure C (Step 2), using 123 (80 mg, 0.14 mmol) and bromotrimethylsilane (100 μL, 0.72 mmol) in DCM (4 mL). The TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (48.4 mg, 70%). ^1^H NMR (500 MHz, DMSO) δ 10.22 (s, 1H), 9.92 (s, 1H), 7.89 (d, J = 2.1 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.60 (dd, J = 8.4, 2.1 Hz, 1H), 7.53–7.45 (m, 2H), 7.04–6.94 (m, 2H), 2.92 (ddd, J = 22.4, 11.3, 2.6 Hz, 1H), 1.99–1.86 (m, 1H), 1.52–1.33 (m, 2H), 0.83 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8 (d, J = 4.9 Hz), 139.7, 136.9, 136.0, 132.2, 131.8, 131.5, 128.4, 126.9, 122.3, 119.8, 46.0 (d, J = 127.0 Hz), 35.7 (d, J = 3.7 Hz), 26.4 (d, J = 14.7 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.76. HRMS (ESI^–^) calculated for C_18_H_20_Cl_2_N_2_O_6_PS^–^ [M-H]^−^ 493.0162, found 493.0156.
[1-({4-[(3,4-Dimethoxyphenyl)sulfonamido]phenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (142)
Compound 142 was synthesized according to the general procedure C (Step 2), using 124 (195 mg, 0.36 mmol) and bromotrimethylsilane (240 μL, 1.80 mmol) in DCM (6 mL). The TMS ester was cleaved using MeOH (6 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (109 mg, 62%). ^1^H NMR (500 MHz, DMSO) δ 9.86 (s, 1H), 9.85 (s, 1H), 7.44 (d, J = 8.6 Hz, 2H), 7.26–7.20 (m, 2H), 7.03 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 8.6 Hz, 2H), 3.78 (s, 3H), 3.73 (s, 3H), 2.98–2.83 (m, 1H), 1.92 (s, 1H), 1.50–1.30 (m, 2H), 0.83 (d, J = 6.0 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7 (d, J = 5.0 Hz), 152.1, 148.5, 136.2, 132.7, 130.8, 121.7, 120.5, 119.6, 111.0, 109.4, 55.7, 55.7, 46.7–45.3 (m), 35.7, 26.4 (d, J = 12.3 Hz), 23.2, 21.3. ^31^P NMR (202 MHz, DMSO) δ 19.89. HRMS (ESI^–^) calculated for C_20_H_26_N_2_O_8_PS^–^ [M-H]^−^ 485.1153, found 485.1139.
(4-Methyl-1-oxo-1-{[4-(thiophene-2-sulfonamido)phenyl]amino}pentan-2-yl)phosphonic
Acid (143)
Compound 143 was synthesized according to the general procedure C (Step 2), using 125 (175 mg, 0.36 mmol) and bromotrimethylsilane (470 μL, 3.58 mmol) in DCM (5 mL). The TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (68.7 mg, 44%). ^1^H NMR (500 MHz, DMSO) δ 10.19 (s, 1H), 9.95 (s, 1H), 7.86 (dd, J = 5.0, 1.3 Hz, 1H), 7.49 (d, J = 8.9 Hz, 2H), 7.46 (dd, J = 3.7, 1.3 Hz, 1H), 7.10 (dd, J = 4.9, 3.8 Hz, 1H), 7.02 (d, J = 8.9 Hz, 2H), 2.93 (dd, J = 21.7, 10.6 Hz, 1H), 1.97–1.88 (m, J = 15.0, 7.5 Hz, 1H), 1.51–1.30 (m, 2H), 0.93–0.76 (m, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.8, 139.8, 136.7, 133.3, 132.4, 132.1, 127.7, 122.0, 119.7, 46.0 (d, J = 126.7 Hz), 35.8, 26.5 (d, J = 14.7 Hz), 23.4, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.82. HRMS (ESI^–^) calculated for C_16_H_20_N_2_O_6_PS_2_ ^–^ [M-H]^−^ 431.0506, found 431.0506.
(4-Methyl-1-{[4-(naphthalene-2-sulfonamido)phenyl]amino}-1-oxopentan-2-yl)phosphonic
Acid (144)
Compound 144 was synthesized according to the general procedure C (Step 2), using 126 (110.6 mg, 0.21 mmol) and bromotrimethylsilane (275 μL, 2.08 mmol) in DCM (4 mL). The TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (43.2 mg, 43%). ^1^H NMR (500 MHz, DMSO) δ 10.18 (s, 1H), 9.87 (s, 1H), 8.37 (d, J = 1.3 Hz, 1H), 8.09 (dd, J = 19.3, 8.4 Hz, 2H), 7.99 (d, J = 8.1 Hz, 1H), 7.72 (dd, J = 8.7, 1.8 Hz, 1H), 7.70–7.65 (m, 1H), 7.65–7.60 (m, 1H), 7.41 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 2.88 (dd, J = 21.9, 10.8 Hz, 1H), 1.90 (s, 1H), 1.47–1.30 (m, 2H), 0.80 (dd, J = 6.1, 3.0 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7 (d, J = 2.9 Hz), 136.5, 136.3, 134.3, 132.3, 131.6, 129.4, 129.3, 129.0, 128.0, 127.9, 127.7, 122.2, 121.7, 119.7, 46.0 (d, J = 125.0 Hz), 35.7, 26.4 (d, J = 14.6 Hz), 23.3, 21.3. ^31^P NMR (202 MHz, DMSO) δ 19.81. HRMS (ESI^–^) calculated for C_22_H_24_N_2_O_6_PS^–^ [M-H]^−^ 475.1098, found 475.1082.
(1-{[4-(Cyclohexanesulfonamido)phenyl]amino}-4-methyl-1-oxopentan-2-yl)phosphonic
Acid (145)
Compound 145 was synthesized according to the general procedure C (Step 2), using 127 (123.3 mg, 0.25 mmol) and bromotrimethylsilane (230 μL, 1.77 mmol) in DCM (4 mL). TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (57.3 mg, 53%). ^1^H NMR (500 MHz, DMSO) δ 9.95 (s, 1H), 9.61 (s, 1H), 7.53 (d, J = 8.9 Hz, 2H), 7.13 (d, J = 8.9 Hz, 2H), 3.00–2.83 (m, 2H), 2.03–1.90 (m, 3H), 1.73 (d, J = 13.0 Hz, 2H), 1.60–1.30 (m, 5H), 1.24–1.03 (m, 3H), 0.85 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 168.1, 136.2, 133.8, 120.9, 120.3, 58.9, 46.4 (d, J = 127.4 Hz), 36.2, 26.9 (d, J = 14.7 Hz), 26.4, 25.2, 24.8, 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ 19.96. HRMS (ESI^+^) calculated for C_18_H_30_N_2_O_6_PS^+^ [M + H]^+^ 433.1557, found 433.1542.
{4-Methyl-1-oxo-1-[(4-{[4-(2-oxopyrrolidin-1-yl)phenyl]sulfonamido}phenyl)amino]pentan-2-yl}phosphonic
Acid (146)
Compound 146 was synthesized according to the general procedure C (Step 2), using 128 (120 mg, 0.21 mmol) and bromotrimethylsilane (200 μL, 1.48 mmol) in DCM (4 mL). The TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (68.3 mg, 64%). ^1^H NMR (500 MHz, DMSO) δ 9.98 (s, 1H), 9.86 (s, 1H), 7.84–7.75 (m, 2H), 7.71–7.64 (m, 2H), 7.43 (d, J = 8.9 Hz, 2H), 6.98 (d, J = 8.9 Hz, 2H), 3.81 (t, J = 7.0 Hz, 2H), 2.91 (ddd, J = 22.4, 11.3, 2.6 Hz, 1H), 2.48–2.52 (m, 2H), 2.09–1.99 (m, 2H), 1.91 (dtd, J = 11.6, 7.7, 3.7 Hz, 1H), 1.49–1.32 (m, 2H), 0.83 (dd, J = 6.4, 1.8 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 174.7, 167.6 (d, J = 4.8 Hz), 143.0, 136.2, 133.5, 132.5, 127.7, 121.4, 119.6, 118.6, 47.9, 45.9 (d, J = 126.7 Hz), 35.7, 32.4, 26.4 (d, J = 14.7 Hz), 23.2, 21.3, 17.3. ^31^P NMR (202 MHz, DMSO) δ 19.82. HRMS (ESI^–^) calculated for C_22_H_27_N_3_O_7_PS^–^ [M-H]^−^ 508.1313, found 508.1316.
[1-({4-[2-(3,4-Dichlorophenyl)acetamido]phenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (195)
Compound 195 was synthesized over two steps according to the general procedure C using 186 (190 mg, 0.45 mmol) and triethyl phosphite (3 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 1/1 to EtOAc/MeOH = 1/1) to give diethyl phosphonate as a white solid (70.6 mg, 30%). The product obtained (67.2 mg, 0.13 mmol) was then treated with bromotrimethylsilane (120 μL, 0.89 mmol) in DCM (5 mL). TMS ester was cleaved using MeOH (5 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (15.6 mg, 25%; 7.5% over two steps). ^1^H NMR (500 MHz, DMSO) δ 10.14 (s, 1H), 9.91 (s, 1H), 7.62–7.56 (m, 2H), 7.50 (dd, J = 21.8, 9.1 Hz, 4H), 7.31 (dd, J = 8.3, 2.0 Hz, 1H), 3.66 (s, 2H), 2.94 (ddd, J = 22.3, 11.3, 2.5 Hz, 1H), 2.00–1.89 (m, 1H), 1.44 (ddt, J = 29.9, 19.8, 8.3 Hz, 2H), 0.85 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.9, 167.6 (d, J = 3.9 Hz), 137.2, 135.2, 134.3, 131.4, 130.7, 130.4, 129.8, 129.3, 119.5, 119.4, 46.0 (d, J = 126.9 Hz), 41.9, 35.8 (d, J = 3.6 Hz), 26.5 (d, J = 14.9 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 20.01. HRMS (ESI^–^) calculated for C_20_H_22_Cl_2_N_2_O_5_P^–^ [M-H]^−^ 471.0649, found 471.0653.
[4-Methyl-1-oxo-1-({4-[2-(thiophen-2-yl)acetamido]phenyl}amino)pentan-2-yl]phosphonic
Acid (197)
Compound 197 was synthesized over two steps according to the general procedure C using 188 (166 mg, 0.46 mmol) and triethyl phosphite (2 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 1/1 to EtOAc) to give diethyl phosphonate as a white solid (80.7 mg, 38%). The product obtained (77.5 mg, 0.17 mmol) was then treated with bromotrimethylsilane (150 μL, 1.16 mmol) in DCM (3 mL). The TMS ester was cleaved using MeOH (3 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (25.4 mg, 36%; 14% over two steps). ^1^H NMR (500 MHz, DMSO) δ 10.15 (s, 1H), 9.91 (s, 1H), 7.51 (q, J = 9.1 Hz, 4H), 7.43–7.33 (m, 1H), 6.97 (d, J = 3.6 Hz, 2H), 3.84 (s, 2H), 2.94 (dd, J = 21.4, 10.4 Hz, 1H), 1.99–1.89 (m, 1H), 1.52–1.34 (m, 2H), 0.85 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7, 162.0 (d, J = 29.5 Hz), 137.3, 135.2, 134.3, 126.7, 126.4, 125.1, 119.5, 119.4, 46.0 (d, J = 127.1 Hz), 37.5, 35.8 (d, J = 3.0 Hz), 26.5 (d, J = 14.8 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 20.03. HRMS (ESI^–^) calculated for C_18_H_22_N_2_O_5_PS^–^ [M-H]^−^ 409.0993, found 409.0993.
[1-({3-[2-(3,4-Dichlorophenyl)acetamido]phenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (205)
Compound 205 was synthesized over two steps according to the general procedure C using 203 (190 mg, 0.45 mmol) and triethyl phosphite (2 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 7/3 to 3/7) to give diethyl phosphonate as transparent oil (157.3 mg, 74%). The product obtained (155 mg, 0.29 mmol) was then treated with bromotrimethylsilane (200 μL, 1.46 mmol) in DCM (6 mL). The TMS ester was cleaved using MeOH (6 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (81 mg, 58%; 43% over two steps). ^1^H NMR (500 MHz, DMSO) δ 10.18 (s, 1H), 9.93 (s, 1H), 7.94 (s, 1H), 7.59 (t, J = 5.4 Hz, 2H), 7.36–7.12 (m, 4H), 3.67 (s, 2H), 2.98 (dd, J = 22.2, 10.8 Hz, 1H), 2.02–1.89 (m, J = 6.2 Hz, 1H), 1.54–1.33 (m, 2H), 0.86 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 168.2, 167.9 (d, J = 3.7 Hz), 139.7, 139.2, 137.1, 131.3, 130.7, 130.4, 129.7, 129.3, 128.8, 114.3, 114.0, 110.2, 46.0 (d, J = 127.4 Hz), 42.0, 35.8, 26.5 (d, J = 14.5 Hz), 23.2, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.97. HRMS (ESI^–^) calculated for C_20_H_22_Cl_2_N_2_O_5_P^–^ [M-H]^−^ 471.0649, found 471.0649.
[1-({4-[2-(3,4-Dichlorophenyl)acetamido]-3-fluorophenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (157)
Compound 157 was synthesized over two steps according to the general procedure C (Step 2), using 154 (200 mg, 0.36 mmol) and bromotrimethylsilane (480 μL, 3.65 mmol) in DCM (6 mL). The TMS ester was cleaved using MeOH (6 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (89.5 mg, 50%). ^1^H NMR (500 MHz, DMSO) δ 10.17 (s, 1H), 9.92 (s, 1H), 7.73–7.64 (m, 2H), 7.63–7.55 (m, 2H), 7.32 (dd, J = 8.3, 1.9 Hz, 1H), 7.19 (dd, J = 8.8, 1.5 Hz, 1H), 3.72 (s, 2H), 2.95 (ddd, J = 22.3, 11.2, 2.3 Hz, 1H), 1.95 (ddd, J = 15.7, 9.9, 3.7 Hz, 1H), 1.52–1.35 (m, 2H), 0.85 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 168.6, 168.2 (d, J = 4.9 Hz), 153.7 (d, J = 243.0 Hz), 137.1, 137.0, 131.4, 130.8, 130.5, 129.8, 129.3, 124.8, 120.6 (d, J = 12.4 Hz), 114.5, 106.2 (d, J = 24.8 Hz), 46.2 (d, J = 126.6 Hz), 41.2, 35.7, 26.5 (d, J = 14.7 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.49. ^19^F NMR (470 MHz, DMSO) δ – 22.79. HRMS (ESI^–^) calculated for C_20_H_21_Cl_2_FN_2_O_5_P^–^ [M-H]^−^ 489.0555, found 489.0554.
[1-({4-[2-(3,4-Dichlorophenyl)acetamido]-3-(trifluoromethyl)phenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (158)
Compound 158 was synthesized over two steps according to the general procedure C (Step 2), using 155 (129 mg, 0.22 mmol) and bromotrimethylsilane (285 μL, 2.20 mmol) in DCM (4 mL). The TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (61 mg, 51%). ^1^H NMR (500 MHz, DMSO) δ 10.33 (s, 1H), 9.77 (s, 1H), 8.14 (d, J = 1.7 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.64–7.55 (m, 2H), 7.40–7.25 (m, 2H), 3.69 (s, 2H), 3.01–2.91 (m, 1H), 2.04–1.91 (m, 1H), 1.53–1.38 (m, 2H), 0.86 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 169.4, 168.5 (d, J = 4.4 Hz), 138.0, 137.0, 131.2, 131.2, 130.8, 130.4, 129.7, 129.6, 129.3, 125.3 (q, J = 29.0 Hz), 123.4 (q, J = 273.4 Hz), 122.8, 116.1 (q, J = 5.0 Hz), 46.3 (d, J = 125.9 Hz), 41.0, 35.6, 26.5 (d, J = 14.5 Hz), 23.2, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.22. ^19^F NMR (470 MHz, DMSO) δ −59.73. HRMS (ESI^–^) calculated for C_21_H_21_Cl_2_F_3_N_2_O_5_P^–^ [M-H]^−^ 539.0523, found 539.0512.
{1-[(4-{[(3,4-Dichlorophenyl)methyl]sulfonamido}phenyl)amino]-4-methyl-1-oxopentan-2-yl}phosphonic
Acid (159)
Compound 159 was synthesized according to the general procedure C (Step 2), using 156 (60 mg, 0.11 mmol) and bromotrimethylsilane (70 μL, 0.53 mmol) in DCM (2 mL). The TMS ester was cleaved using MeOH (2 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (18.5 mg, 34%). ^1^H NMR (500 MHz, DMSO) δ 9.96 (s, 1H), 9.72 (s, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.58 (d, J = 8.7 Hz, 2H), 7.50 (d, J = 1.9 Hz, 1H), 7.24 (dd, J = 8.3, 2.0 Hz, 1H), 7.12 (d, J = 8.8 Hz, 2H), 4.48 (s, 2H), 2.96 (dd, J = 22.1, 11.2 Hz, 1H), 1.96 (s, 1H), 1.44 (dd, J = 26.4, 14.6 Hz, 2H), 0.87 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7, 135.9, 132.8, 132.8, 131.3, 131.1, 130.9, 130.9, 130.5, 120.4, 119.8, 55.2, 46.0 (d, J = 132.8 Hz), 35.8, 26.5 (d, J = 14.3 Hz), 23.3, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.93. HRMS (ESI^–^) calculated for C_19_H_22_Cl_2_N_2_O_6_PS^–^ [M-H]^−^ 507.0319, found 507.0320.
{1-[(4-Benzoylphenyl)amino]-4-methyl-1-oxopentan-2-yl}phosphonic
Acid (189)
Compound 189 was synthesized over two steps according to the general procedure C using 180 (195 mg, 0.52 mmol) and triethyl phosphite (2 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 7/3 to 3/7) to give diethyl phosphonate as transparent oil (132 mg, 59%). The product obtained was then treated with bromotrimethylsilane (275 μL, 2.08 mmol) in DCM (6 mL). TMS ester was cleaved using MeOH (6 mL). Solvents were concentrated in vacuo and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (90 mg, 80%; 47% over two steps). ^1^H NMR (500 MHz, DMSO) δ 10.37 (s, 1H), 7.81–7.77 (m, 2H), 7.75–7.69 (m, 4H), 7.66 (ddd, J = 8.7, 2.5, 1.2 Hz, 1H), 7.55 (dd, J = 10.6, 4.6 Hz, 2H), 3.06 (ddd, J = 22.5, 11.2, 2.5 Hz, 1H), 2.03–1.93 (m, 1H), 1.55–1.39 (m, 2H), 0.87 (d, J = 5.8 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 194.6, 168.7 (d, J = 4.9 Hz), 143.6, 137.7, 132.3, 131.2, 129.4, 128.5, 118.3, 46.3 (d, J = 126.0 Hz), 35.7, 26.6 (d, J = 14.4 Hz), 23.2, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.32. HRMS (ESI^–^) calculated for C_19_H_21_NO_5_P^–^ [M-H]^−^ 374.1163, found 374.1163.
{1-[(4-Benzylphenyl)amino]-4-methyl-1-oxopentan-2-yl}phosphonic
Acid (190)
Compound 190 was synthesized over two steps according to the general procedure C using 181 (158 mg, 0.44 mmol) and triethyl phosphite (1 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 7/3 to 3/7) to give diethyl phosphonate as transparent oil (80 mg, 44%). The product obtained was then treated with bromotrimethylsilane (125 μL, 0.95 mmol) in DCM (4 mL). TMS ester was cleaved using MeOH (4 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (35.8 mg, 52%; 23% over two steps). ^1^H NMR (500 MHz, DMSO) δ 9.86 (s, 1H), 7.50 (d, J = 8.4 Hz, 2H), 7.29–7.24 (m, 2H), 7.21–7.11 (m, 5H), 3.87 (s, 2H), 2.95 (dd, J = 21.6, 10.5 Hz, 1H), 2.00–1.89 (m, 1H), 1.52–1.34 (m, 2H), 0.85 (d, J = 6.1 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.7, 141.6, 137.5, 135.8, 128.9, 128.6, 128.4, 125.9, 119.2, 46.0 (d, J = 127.0 Hz), 40.5, 35.8, 26.5 (d, J = 14.5 Hz), 23.2, 21.4. ^31^P NMR (202 MHz, DMSO) δ 20.03. HRMS (ESI^–^) calculated for C_19_H_23_NO_4_P^–^ [M-H]^−^ 360.1370, found 360.1375.
[1-({4-[(3,4-Dichlorobenzyl)thio]phenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (191)
Compound 191 was synthesized over two steps according to the general procedure C using 182 (142 mg, 0.31 mmol) and triethyl phosphite (1.5 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 8/2 to 3/7) to give diethyl phosphonate as transparent oil (84 mg, 53%). The product obtained was then treated with bromotrimethylsilane (106 μL, 0.80 mmol) in DCM (3 mL). The TMS ester was cleaved using MeOH (3 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (65 mg, 87%; 46% over two steps). ^1^H NMR (500 MHz, DMSO) δ 9.99 (s, 1H), 7.53 (dd, J = 9.3, 7.5 Hz, 4H), 7.32–7.17 (m, 3H), 4.15 (s, 2H), 2.95 (ddd, J = 22.4, 11.3, 2.6 Hz, 1H), 2.01–1.88 (m, 1H), 1.52–1.35 (m, 2H), 0.85 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.9 (d, J = 4.6 Hz), 139.6, 138.5, 130.9, 130.8, 130.7, 130.4, 129.4, 129.1, 127.7, 119.5, 46.1 (d, J = 126.3 Hz), 36.6, 35.7, 26.4 (d, J = 14.7 Hz), 23.2, 21.4. ^31^P NMR (202 MHz, DMSO) δ 19.68. HRMS (ESI^–^) calculated for C_19_H_21_Cl_2_NO_4_PS^–^ [M-H]^−^ 460.0311, found 460.0317.
[1-({4-[(3,4-Dichlorobenzyl)oxy]phenyl}amino)-4-methyl-1-oxopentan-2-yl]phosphonic
Acid (192)
Compound 192 was synthesized over two steps according to the general procedure C using 183 (102 mg, 0.23 mmol) and triethyl phosphite (1 mL). The resultant oil was purified by column chromatography (Hex/EtOAc = 8/2 to 2/8) to give diethyl phosphonate as transparent oil (47 mg, 41%). The product obtained was then treated with bromotrimethylsilane (60 μL, 0.44 mmol) in DCM (2 mL). The TMS ester was cleaved using MeOH (2 mL). Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (20.5 mg, 53%; 22% over two steps). ^1^H NMR (500 MHz, DMSO) δ 9.80 (s, 1H), 7.70 (d, J = 1.9 Hz, 1H), 7.65 (d, J = 8.3 Hz, 1H), 7.51 (d, J = 9.0 Hz, 2H), 7.42 (dd, J = 8.3, 1.9 Hz, 1H), 6.94 (d, J = 9.0 Hz, 2H), 5.08 (s, 2H), 2.92 (dd, J = 21.5, 10.5 Hz, 1H), 2.00–1.88 (m, J = 10.9, 4.5 Hz, 1H), 1.53–1.34 (m, 2H), 0.86 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ 167.4 (d, J = 1.4 Hz), 153.6, 138.6, 133.2, 131.1, 130.7, 130.3, 129.4, 127.8, 120.5, 114.8, 67.8, 45.9 (d, J = 127.3 Hz), 35.8, 26.4 (d, J = 14.9 Hz), 23.2, 21.4. ^31^P NMR (202 MHz, DMSO) δ 20.11. HRMS (ESI^–^) calculated for C_19_H_21_Cl_2_NO_5_P^–^ [M-H]^−^ 444.0540, found 444.0545.
1-[4-(3,4-Dichlorophenylthio)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (90)
Compound 90 was synthesized according to general procedure C (step 2), using 76 (96 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (35 mg, 41%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.22 (s, 1H), 7.71 (d, J = 8.7 Hz, 2H), 7.53 (d, J = 8.5 Hz, 1H), 7.45 (d, J = 8.7 Hz, 2H), 7.32 (d, J = 2.2 Hz, 1H), 7.03 (dd, J = 8.5, 2.2 Hz, 1H), 3.00 (ddd, J = 22.5, 11.3, 2.6 Hz, 1H), 2.05–1.82 (m, 1H), 1.58–1.22 (m, 2H), 0.85 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.7 (d, J = 4.9 Hz), 141.0, 139.8, 135.4, 132.3, 131.6, 128.8, 128.8, 127.8, 124.3, 120.7, 46.6 (d, J = 126.3 Hz), 36.1 (d, J = 4.0 Hz), 26.9 (d, J = 14.7 Hz), 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 19.52. HRMS (ESI^–^) calculated for C_18_H_19_Cl_2_NO_4_PS [M-H]^−^ 446.0155, found 446.0157.
1-[4-(3,4-Dichlorophenoxy)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (91)
Compound 91 was synthesized according to general procedure C (step 2), using 77 (93 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (25 mg, 30%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.05 (s, 1H), 7.67 (d, J = 8.9 Hz, 2H), 7.59 (d, J = 8.9 Hz, 1H), 7.22 (d, J = 2.8 Hz, 1H), 7.06 (d, J = 8.9 Hz, 2H), 6.95 (dd, J = 8.9, 2.8 Hz, 1H), 2.98 (ddd, J = 22.4, 11.3, 2.6 Hz, 1H), 1.98 (tdd, J = 11.8, 7.3, 4.2 Hz, 1H), 1.55–1.37 (m, 2H), 0.88 (d, J = 6.3 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.2 (d, J = 4.7 Hz), 157.8, 150.7, 136.8, 132.4, 132.0, 125.1, 121.2, 120.6, 119.7, 118.2, 46.5 (d, J = 126.9 Hz), 36.2 (d, J = 4.0 Hz), 26.9 (d, J = 14.8 Hz), 23.7, 21.8.
^31^P NMR (202 MHz, DMSO) δ ppm: 19.95. HRMS (ESI^–^) calculated for C_18_H_19_Cl_2_NO_5_P [M-H]^−^ 430.0383, found 430.0382.
1-[4-(3,4-Dichlorophenylcarbamoyl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (93)
Compound 93 was synthesized according to general procedure C (step 2), using 79 (98 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (22 mg, 25%). ^1^H NMR (500 MHz, DMSO) δ ppm: 10.34 (s, 2H), 8.13 (d, J = 2.4 Hz, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.81–7.64 (m, 3H), 7.58 (d, J = 8.8 Hz, 1H), 3.02 (dd, J = 22.2, 10.6 Hz, 1H), 1.95 (ddd, J = 15.4, 10.0, 3.4 Hz, 1H), 1.52–1.34 (m, 2H), 0.85 (s, 3H), 0.83 (s, 3H). ^13^C NMR (126 MHz, DMSO) δ ppm: 169.0 (d, J = 6.0 Hz), 165.6, 143.3, 139.9, 131.3, 131.0, 129.2, 128.5, 125.3, 121.8, 120.6, 118.6, 46.7 (d, J = 127.3 Hz), 36.2 (d, J = 4.1 Hz), 27.0 (d, J = 14.3 Hz), 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 19.39. HRMS (ESI^–^) calculated for C_19_H_20_Cl_2_N_2_O_5_P [M-H]^−^ 457.0492, found 457.0493.
1-[4-(3,4-Dichlorophenylsulfamoyl)phenylcarbamoyl]-3-methylbutylphosphonic
Acid (92)
Compound 92 was synthesized according to general procedure C (step 2), using 78 (105 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as a white solid (42 mg, 45%). ^1^H NMR (500 MHz, Acetone) δ ppm: 9.88 (s, 1H), 9.33 (s, 1H), 7.85 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 8.7 Hz, 2H), 7.45 (t, J = 6.0 Hz, 2H), 7.25–7.19 (m, 1H), 3.25–3.08 (m, 1H), 2.22–2.10 (m, 1H), 1.65–1.54 (m, 2H), 0.87 (d, J = 14.3 Hz, 6H). ^13^C NMR (126 MHz, Acetone) δ ppm: 168.0 (d, J = 5.0 Hz), 143.4, 138.2, 133.6, 132.2, 131.1, 128.3, 127.0, 121.6, 120.0, 119.2, 46.4 (d, J = 128.8 Hz), 35.5 (d, J = 5.0 Hz), 26.7 (d, J = 14.5 Hz), 22.5, 20.8. ^31^P NMR (202 MHz, Acetone) δ ppm: 23.91. HRMS (ESI^–^) calculated for C_18_H_20_Cl_2_N_2_O_6_PS [M-H]^−^ 493.0162, found 493.0161.
1-{4-[(3,4-Dichlorophenylthio)methyl]phenylcarbamoyl}-3-methylbutylphosphonic
Acid (164)
Compound 164 was synthesized according to general procedure C (step 2), using 163 (98 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (16 mg, 18%). ^1^H NMR (500 MHz, DMSO) δ ppm: 9.98 (s, 1H), 7.60 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 8.5, 4.6 Hz, 3H), 7.32–7.24 (m, 3H), 4.27 (s, 2H), 2.97 (ddd, J = 22.4, 11.3, 2.7 Hz, 1H), 1.96 (tdd, J = 11.6, 7.2, 4.1 Hz, 1H), 1.51–1.32 (m, 2H), 0.86 (d, J = 6.4 Hz, 6H). ^13^C NMR (126 MHz, DMSO) δ ppm: 168.3 (d, J = 5.6 Hz), 139.1, 138.2, 132.0, 131.4, 131.2, 129.6, 129.5, 128.5, 128.4, 119.4, 46.5 (d, J = 126.7 Hz), 40.5, 36.2 (d, J = 16.6 Hz), 26.9 (d, J = 15.0 Hz), 23.7, 21.8. ^31^P NMR (202 MHz, DMSO) δ ppm: 19.89. HRMS (ESI^–^) calculated for C_19_H_21_Cl_2_NO_4_PS [M-H]^−^ 460.0311, found 460.0311.
1-{4-[(3,4-Dichlorophenylamino)methyl]phenylcarbamoyl}-3-methylbutylphosphonic
Acid (169)
Compound 169 was synthesized according to general procedure C (step 2), using 168 (115 mg, 0.19 mmol), bromotrimethylsilane (250 μL, 1.9 mmol) and DCM (15 mL). The reaction was stirred at r.t. overnight. The crude product was purified using preparative HPLC (CH_3_CN (HCOOH 0.05%)/H_2_O (HCOOH 0.05%) = 1/9 to 10/0). The product was obtained as white solid (31 mg, 37%). ^1^H NMR (500 MHz, Acetone) δ ppm: 9.56 (s, 1H), 7.50 (dd, J = 8.4, 1.6 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 2.7 Hz, 1H), 6.49 (dd, J = 8.8, 2.7 Hz, 1H), 4.18 (s, 2H), 3.16–3.00 (m, 1H), 2.08–1.94 (m, 1H), 1.46 (dd, J = 11.9, 5.9 Hz, 2H), 0.74 (d, J = 6.1 Hz, 3H), 0.73 (d, J = 6.1 Hz, 3H). ^13^C NMR (126 MHz, Acetone) δ ppm: 167.8 (d, J = 4.7 Hz), 148.9–147.7 (m), 138.9–136.7 (m), 135.1–133.3 (m), 131.9, 130.5, 127.7, 119.7, 118.0, 113.6, 113.0, 46.6, 46.2 (d, J = 128.8 Hz), 35.8 (d, J = 3.9 Hz), 26.8 (d, J = 14.7 Hz), 22.6, 21.0. ^31^P NMR (202 MHz, Acetone) δ ppm: 24.39. HRMS (ESI^–^) calculated for C_19_H_22_Cl_2_N_2_O_4_P [M-H]^−^ 443.0670, found 443.0697.
In Vitro Inhibition Assays (LasB, MMPs, TACE,
anad COX-1)
All in vitro inhibition assays were performed as described previously.? The TACE inhibitor screening kit were purchased from Sigma-Aldrich (Saint Louis, MO), the COX-1 kit from Abcam (Cambridge, UK). MMPs along with the SensoLyte 520 Generic MMP Activity Kit Fluorimetric were purchased from AnaSpec (Fremont, CA, USA), and the fluorometric cyclooxygenase 1 (COX1) inhibitor assay kit was purchased from Abcam (Cambridge, UK). The assays were performed according to the guidelines of the respective manufacturer. Fluorescence signals were measured using a CLARIOstar plate reader (BMG Labtech, Ortenberg, Germany). Pulmonary surfactant (poractant alfa), which is an extract of natural porcine lung surfactant, was purchased from Creative BioMart (Shirley, NY, USA). DPPC was purchased from Lipoid (Ludgwigshafen, Germany). LasB was expressed as described previously. ?,?
Murine Survival Model with Instilled LasB
Procedures involving mice were approved by the local ethical committee (Paris-Nord/No 121) and by the French Ministry of Education and Research (agreement number 28050–2020102015484235). Eight-week-old male C57BL/6 mice were purchased from Janvier (Le Genest-Saint-Isle, France. Mice were anaesthetised using intramuscular injection of ketamine 500 and xylazine 2% in 0.9% NaCl (20/10/70). 40 μg of purified LasB were instilled intranasally in a final volume of 40 μL. In groups with LasB inhibitor treatment, LasB was preincubated with inhibitors at a 1/10 molar ratio at 37 °C for 1 h before intranasal instillation (300 μM of compound vs 30 μM of enzyme). Finally, the survival of the mice was monitored over 7 d.
Lipophilicity Determination (logD7.4)
LogD_7.4_ was analyzed using an HPLC-based method. The UV retention time of reference compounds with known logD_7.4_ was determined and plotted toward their logD_7.4_. Linear regression was used to determine LogD_7.4_ of unknown compounds. Analysis was performed using a Vanquish Flex HPLC system with variable wavelength detector (Thermo Fisher, Dreieich, Germany) with the following conditions: EC150/2 NUCLEODUR C18 Pyramid column, 5 μM (Macherey Nagel, Düren, Germany); eluent A: 50 mM NH_4_OAc pH 7.4, eluent B: acetonitrile, and flow: 0.6 mL/min. The gradient was set to 0–100% B from 0 to 2.5 min, 100% B from 2.5 to 3.0 min, 100–0% B from 3.0 to 3.2 min, and 0% B from 3.2–5.0.
X-ray Crystallography
For LasB cocrystallization trials, the protein was purified from culture supernatant of P. aeruginosa PA14 (for 35 and 141), or recombinantly expressed in Eschierichia coli (for 21), as described previously. ?,? The amino acid sequence of the recombinantly expressed LasB differs from PA14 in four positions and corresponds to the LasB sequence of P. aeruginosa PAO1. There is no difference between both protein variants in terms of protein activity and susceptibility to inhibitors, and the four mutations are not located in the vicinity of the LasB active (compound binding) site.? The Met128Val variant of LasB was constructed via Gibson assembly using the primers listed in Table S7, and the protein was recombinantly expressed in E. coli and purified under the same reported conditions as the wild type. LasB was concentrated to ∼ 5 mg/mL after size-exclusion chromatography using a HiLoad Superdex S200 16/600 column and incubated with a final concentration of 1 mM (600 μM for 35) compound in 10 mM Tris pH 8.0 and 2 mM CaCl_2_ for 1 h on ice prior to crystallization. Co-crystallization trials for all compounds were carried out using commercially available screens (NeXtal) and prepared in sitting-drop SwissSCI plates by a Gryphon crystallization robot. Crystals were observed in a variety of conditions after approximately one moth incubation at 293 K. Crystals of wild-type LasB in complex with 21 appeared in a well solution of 0.1 M sodium citrate pH 5.5 and 35% (v/v) 2-ethoxyethanol and cocrystals of the LasB Met128Val variant were obtained in the condition 0.1 M sodium chloride, 0.1 M HEPES pH 7.5, 1.6 M ammonium sulfate. Diffraction quality cocrystals for 141 were observed in the condition 0.01 M trisodium citrate and 33% (w/v) PEG 6000 and crystals for 35 in complex with LasB appeared in a well solution of 0.1 M HEPES pH 6.5 and 2.4 M ammonium sulfate. Protein crystals were cryo-protected with 32% glycerol and flash-frozen in liquid nitrogen. Diffraction data were collected at 100 K using ESRF beamline ID23–1 for LasB in complex with 35 and at beamline P11 at Petra III (DESY) for the other tree compounds.? Data were processed using Aimless and Pointless, both implemented in CCP4 and the structures were determined by molecular replacement using PHASER. ?−? ? Structures 8CR3, 1EZM and 6FZX were used as search models. ?,?,? Further processing of the data was performed using AutoBuild, the structure was manually rebuilt in COOT and the refinement was carried out using Phenix.refine. ?,? The refined structures of LasB in complex with 21, 141 and 35, as well as the LasB Met128Val structure in complex with 21 were deposited in the Protein Data Bank (PDB) as entries 9FRY, 9GMV, 9FRZ and 9FS0, respectively. Figures were generated in PyMOL by Schrödinger (version 3.1.6.1).
Culturing of HepG2, A549, and HEK293 Cells
The human hepatocellular carcinoma cell line HepG2, the lung adenocarcinoma cell line A549 and Human Embryonic Kidney (HEK) 293 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM), containing 10% Fetal Bovine Serum (FBS) and 1% penicillin-streptomycin mixture. Cells were maintained according to standard cell-culture procedures.
Cytotoxicity Assay
An MTT-based assay was employed to evaluate the viability of HepG2, HEK293 and A549 cells after challenge with selected LasB inhibitors and performed as described previously.?
Metabolic and Plasma Stability
Murine liver S9 and plasma stability as well as profiling of stability in mouse, rat and minipig liver microsomes and plasma was performed as described previously.?
Calu-3 Permeability
Compound permeability was assessed in vitro with Calu-3 HTB-55 cell line (ATCC) as described previously.?
Protein Binding Assays
PPB was determined using the Rapid Equilibrium Dialysis (RED) system (Thermo Fisher Scientific, Waltham MA, USA). Compounds were diluted to 10 μM in murine (CD-1) plasma (Neo Biotech, Nanterre, France) and added to the respective chamber according to the manufacturer’s protocol, followed by addition of PBS pH 7.4 to the opposite chamber. Samples were taken immediately after addition to the plate as well as after 2, 4, and 5 h by mixing 10 μL with 80 μL ice-cold acetonitrile containing 12.5 nM diphenhydramine as internal standard, followed by addition of 10 μL plasma to samples taken from PBS and vice versa. Samples were stored on ice until the end of the incubation, and precipitated protein was removed by centrifugation (15 min, 4 °C, 4,000 g, 2 centrifugation steps). Concentration of the remaining test compound at the different time points was analyzed by HPLC-MS/MS (TSQ Quantum Access MAX, Thermo Fisher, Dreieich, Germany). The amount of compound bound to protein was calculated using the equation PPB [%] = 100–100(amount in buffer chamber/amount in plasma chamber).
Surfactant protein binding was determined similarly using 1% porcine lung surfactant in FRET assay buffer instead of plasma. The mixture was incubated for 8 h.
Bacterial Growth Inhibition Assay
Assays regarding the determination of the MIC were performed as described recently for P. aeruginosa PA14.?
Production of Culture Supernatants
The culture supernatants of wild type PAO1 and PAO1 ΔlasB as well as the strains used for the screening across isolates were produced as previously reported.
Biological Evaluation of 35 via an A549 Cell-Based Assay
The A549 human lung adenocarcinoma cell line was cultivated and maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal bovine serum (FBS) and 1% penicillin-streptomycin mixture, following standard cell-culture protocols.
2.5 × 10^3^ cells per well were seeded into a 96-well flat-bottom plate (Corning Costar) and incubated at 37 °C with 5% CO_2_ for 24 h. The next day, various concentrations of the compound 35 were tested against 10% (v/v) P. aeruginosa PAO1 (DSM 22644, ATCC 15692) culture supernatant, which was diluted in DMEM, starting from 5 μM. Initially, the compound was dissolved in 99.9% dimethyl sulfoxide (DMSO), and a final assay concentration of 0.5% (v/v) DMSO was applied. Furthermore, 10% (v/v) ΔlasB PAO1 was introduced to the cells to verify the toxic effect attributed to LasB. A control group was included, consisting of cells treated solely with DMEM, without any additional agents. Subsequently, the plates were incubated at 37 °C with 5% CO_2_ for 24 h before conducting the MTT assay.
MTT Assay
To assess cell viability, the content of the wells was aspirated, followed by a single wash with 100 μL of phosphate buffered saline (PBS) at pH 7.4. A solution of 5 mg of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) per milliliter of PBS was prepared and then diluted to a 10% (v/v) concentration in DMEM. Subsequently, 100 μL of the MTT solution was added to each well, and the plates were incubated at 37 °C for 2 h with 5% CO_2_. Following the incubation period, the MTT solution was carefully aspirated, and the formed crystals were dissolved by the addition of 150 μL per well of MTT lysis buffer (composed of 250 mL DMSO, 25 g SDS, and 1.25 mL acetic acid). The plates were once again incubated for 30 min at 37 °C with 5% CO_2_ before the measurement was performed using a PHERAstar microplate reader. The optical density was measured at 550 nm for the test samples and at 620 nm for the blank. Finally, the obtained data were statistically analyzed and graphically presented using GraphPad Prism 9.
Assessment of 35 Inhibitory Effects Against Clinical Isolates
of P. aeruginosa Using a FRET-Based Inhibition Assay
Bacterial culture supernatants were prepared from eight clinical isolates of P. aeruginosa following overnight growth in lysogeny broth medium (LB). After approximately 18 h of growth, the cultures were centrifuged for 10 min at 5,000 rpm and filtered through 0.2 μm nonpyrogenic sterile filters. The fluorogenic substrate 2-Aminobenzoyl-Ala-Gly-Leu-Ala-4-nitrobenzylamide, sourced from Peptides International (Louisville, KY, USA), was employed in this study.
Fluorescence intensity measurements were conducted at 37 °C for a duration of 60 min using a CLARIOstar microplate reader (BMG Labtech, Ortenberg, Germany). The excitation and emission wavelengths utilized were 340 ± 15 nm and 415 ± 20 nm, respectively. The measurements were performed in black 384-well microtiter plates from Greiner Bio-One (Kremsmünster, Austria). The experimental setup involved a final volume of 50 μL, which included varying concentrations of 35 dissolved in DMSO and mixed with assay buffer (comprising 50 mM Tris, pH 7.2, 2.5 mM CaCl_2_, 0.075% Pluronic F-127, and 5% DMSO), along with bacterial culture supernatant at a concentration where linear LasB activity was observed, and the substrate at a final concentration of 150 μM.
Each sample was duplicated in the multiwell plate, and controls without supernatant/compound were included to facilitate blank correction. After subtracting the blank values, the FRET signal of the samples was plotted at various time points, with measurements taken every 2 min, using GraphPad Prism 9.
Zebrafish Embryo Toxicity
The experiment was performed according to a procedure described in the literature? with minor modifications using zebrafish embryos of the AB wild-type line at 2 days post fertilization (dpf). A detailed protocol has been given in our recent publication.?
In Vivo Pharmacokinetic Studies
For pharmacokinetic experiments, outbred male CD-1 mice (Charles River, Netherlands), 4 weeks old, were used. The animal studies were conducted in accordance with the recommendations of the European Community (Directive 2010/63/EU, first January 2013). All animal procedures were performed in strict accordance with the German regulations of the Society for Laboratory Animal Science (GV- SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). Animals were excluded from further analysis if sacrifice was necessary according to the human end points established by the ethical board. All experiments were approved by the ethical board of the Niedersächsisches Landesamt für Verbraucherschutz and Lebensmittelsicherheit, Oldenburg, Germany.
Compounds 23, 141, 21, 130, 81, 82 and 195 were dissolved in 2% DMSO, 1% Tyloxapol and 97% PBS. The compounds were administered intratracheally (IT) at 0.25 mg/kg per compound as cassette. Up to 5 compounds were dosed per cassette. Before administration, mice were anesthetized using ketamine 100 mg/kg and xylazine 10 mg/kg intraperitoneally. Mice (n = 3 per time point) were euthanatized at t= 0.5, 2, and 5 h post administration. Blood was collected from the heart. 207, 21, 141, 81, 82, 130, 209, 212, 211, 206, 35 and 138 were administered at 2 mg/kg intravenously per compound as cassette. Up to 5 compounds were dosed per cassette intravenously, n = 2 mice were used per cassette. Blood was collected from the lateral tail vein at time points t= 0.25, 0.5, 1, and 3 h post administration. At 5 h post administration, mice were euthanatized, and blood was collected from the heart. 35 was subjected to a focused PK study and was administered 30 mg/kg SC. At time points t= 0.25, 0.5, 1, 2, 4, 8, and 24 h post administration, mice (t= 3 mice per time point) were euthanatized and blood was collected from the heart. For all PK studies, whole blood was collected into Eppendorf tubes coated with 0.5 M EDTA and immediately spun down at 13,000 rpm at 4 °C for 10 min. The plasma was transferred into a new Eppendorf tube and then stored at −80 °C until analysis. Then, a broncheoalveolar lavage was conducted using isotonic sodium chloride solution for all PK studies. For all PK studies, lung, kidney and liver were aseptically removed and homogenized using a Polytron (Kinematica) in isotonic sodium chloride solution. Organ samples were aliquoted into Eppendorf tubes and stored at −80 °C until analysis. Moreover, spontaneous urine was also collected.
All PK plasma samples were analyzed via HPLC-MS/MS using an Agilent 1290 Infinity II HPLC system and coupled to an AB Sciex QTrap6500+ mass spectrometer as described previously.? Mass spectrometric conditions can be found in Table S8. Urea was used to enable calculation of epithelial lining fluid (ELF) concentrations. Peak areas of each sample and of the corresponding internal standard were analyzed using MultiQuant 3.0 software (AB Sciex). Peak areas of the respective sample were normalized to the internal standard peak area. Peaks of PK samples were quantified using the calibration curve. The accuracy of the calibration curve was determined using QCs independently prepared on different days. PK parameters were determined using a noncompartmental analysis with PKSolver.? ELF concentrations were calculated using the following formula: ?,?
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Boucher H. W.Talbot G. H.Bradley J. S.Edwards J. E.Gilbert D.Rice L. B.Scheld M.Spellberg B.Bartlett J.Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America.Clin. Infect. Dis.20094811210.1086/59501119035777 · doi ↗ · pubmed ↗
- 2World Health Organization . WHO Bacterial Priority Pathogens List. Bacterial pathogens of public health importance to guide research, development and strategies to prevent and control antimicrobial resistance; World Health Organization: Geneva 2024.
- 3Pang Z.Raudonis R.Glick B. R.Lin T.-J.Cheng Z.Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies Biotechnol. Adv.20193717719210.1016/j.biotechadv.2018.11.01330500353 · doi ↗ · pubmed ↗
- 4Ventola C. L.The antibiotic resistance crisis: part 1: causes and threats.Pharm. Ther.201540277283 PMC 437852125859123 · pubmed ↗
- 5Miethke M.Pieroni M.Weber T.Brönstrup M.Hammann P.Halby L.Arimondo P. B.Glaser P.Aigle B.Bode H. B.Moreira R.Li Y.Luzhetskyy A.Medema M. H.Pernodet J.-L.Stadler M.Tormo J. R.Genilloud O.Truman A. W.Weissman K. J.Takano E.Sabatini S.Stegmann E.Brötz-Oesterhelt H.Wohlleben W.Seemann M.Empting M.Hirsch A. K. H.Loretz B.Lehr C.-M.Titz A.Herrmann J.Jaeger T.Alt S.Hesterkamp T.Winterhalter M.Schiefer A.Pfarr K.Hoerauf A.Graz H.Graz M.Lindvall M.Ramurthy S.Karlén A.van Dongen M.Petkovic H.Keller A.Peyrane F.Donadio S.Fraisse L.Piddock L. J. V.Gilbert I. · doi ↗ · pubmed ↗
- 6Rasko D. A.Sperandio V.Anti-virulence strategies to combat bacteria-mediated disease.Nat. Rev. Drug Discovery 2010911712810.1038/nrd 301320081869 · doi ↗ · pubmed ↗
- 7Clatworthy A. E.Pierson E.Hung D. T.Targeting virulence: a new paradigm for antimicrobial therapy Nat. Chem. Biol.2007354154810.1038/nchembio.2007.2417710100 · doi ↗ · pubmed ↗
- 8Dickey S. W.Cheung G. Y. C.Otto M.Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance Nat. Rev. Drug Discovery 20171645747110.1038/nrd.2017.2328337021 PMC 11849574 · doi ↗ · pubmed ↗