The Catalytic Asymmetric Mukaiyama–Michael Reaction of Silyl Ketene Acetals with Cyclic Enones: Short Routes to Jasmonates
Ruigang Xu, Hui Zhou, Han Yong Bae, Vijay N. Wakchaure, Lorenzo Baldinelli, Isaac F. Leach, Giovanni Bistoni, Philip Kraft, Benjamin List

TL;DR
A new catalyst enables efficient and selective synthesis of jasmonates, important compounds in plant signaling and fragrance chemistry.
Contribution
A silylium imidodiphosphorimidate catalyst enables high-yield, enantioselective Mukaiyama–Michael reactions for streamlined jasmonate synthesis.
Findings
The reaction achieves up to 98% yield and >99:1 enantiomeric ratio with 0.05 mol% catalyst loading.
The method allows scalable synthesis of jasmonates and related compounds with high enantioselectivity.
Computational studies explain the origin of the observed enantioselectivity.
Abstract
A silylium imidodiphosphorimidate (IDPi) Lewis acid catalyst enables a broadly applicable organocatalytic asymmetric Mukaiyama–Michael addition of moderately electrophilic cycloenones with enol silanes, affording 1,4-adducts in up to 98% yield and >99:1 e.r. At 0.05 mol % catalyst loading, the reaction scales to 167 g of product with 96% catalyst recovery. The method accommodates a wide range of enones and silylated nucleophiles, allowing streamlined access to key jasmonates and related valuable targets. Computational studies elucidated the origin of the enantioselectivity in this reaction. This platform thus provides a versatile entry to structurally complex chiral scaffolds with direct relevance to plant signaling, fragrance chemistry, aromatherapy, and pharmaceutical sciences.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
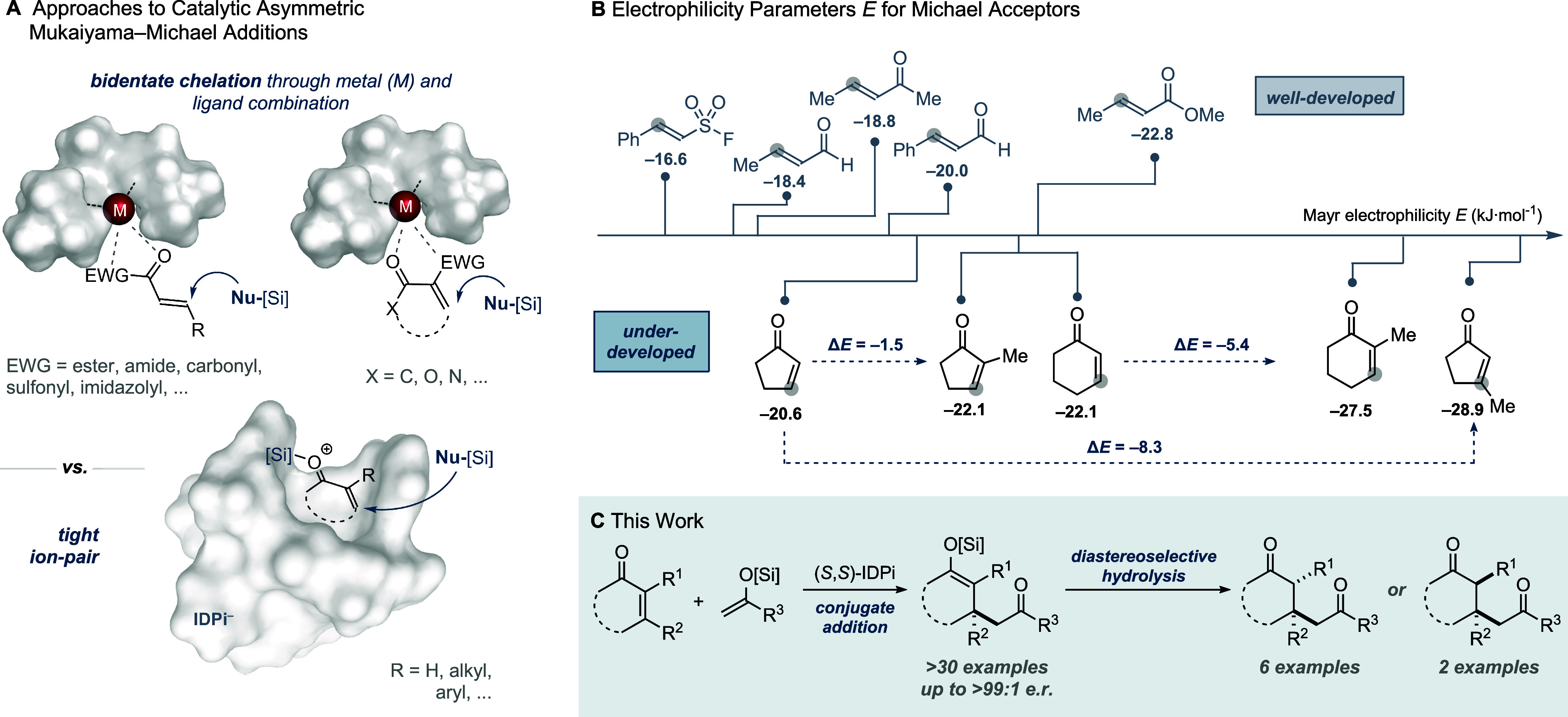 Figure 1
Figure 1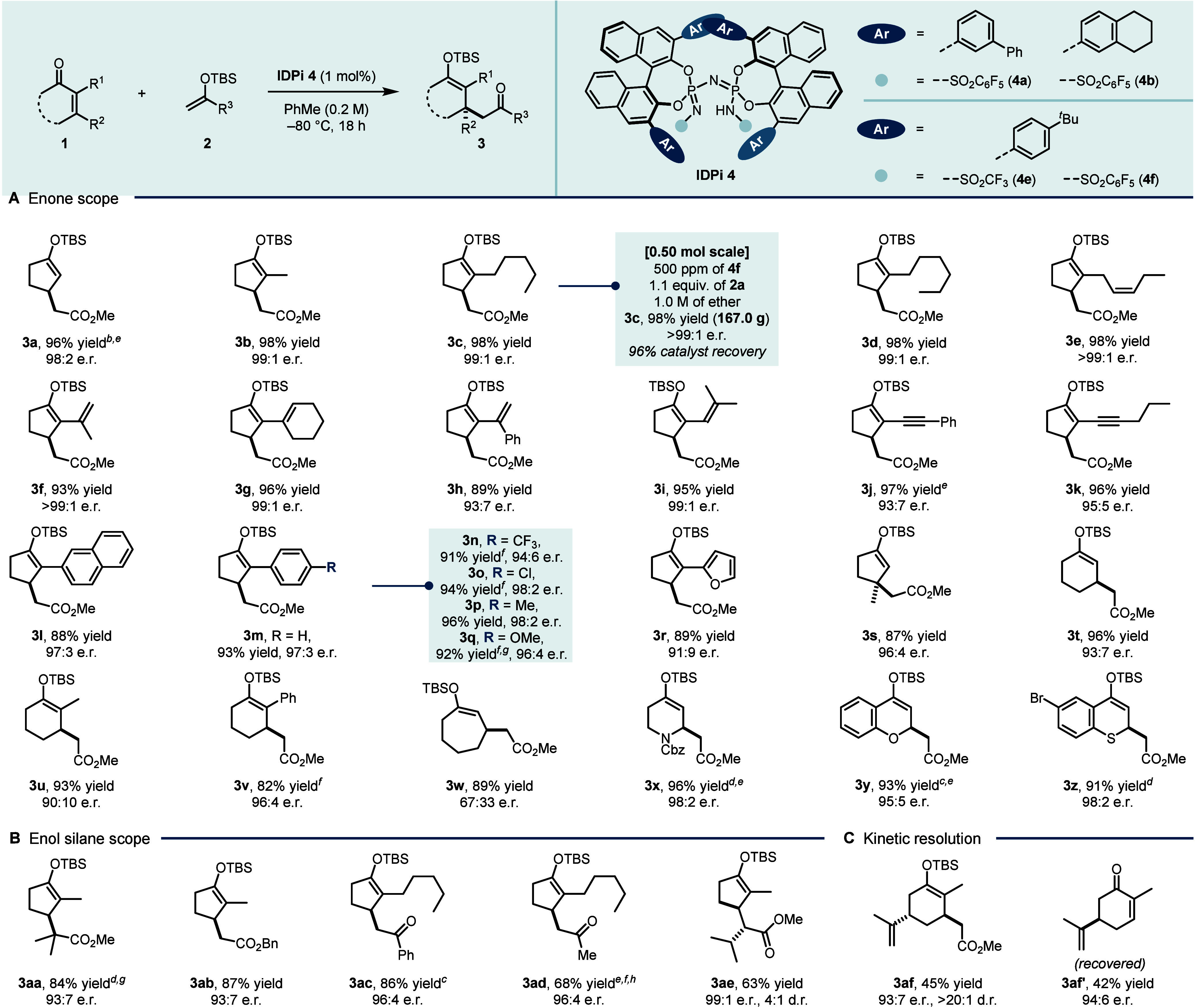 Figure 2
Figure 2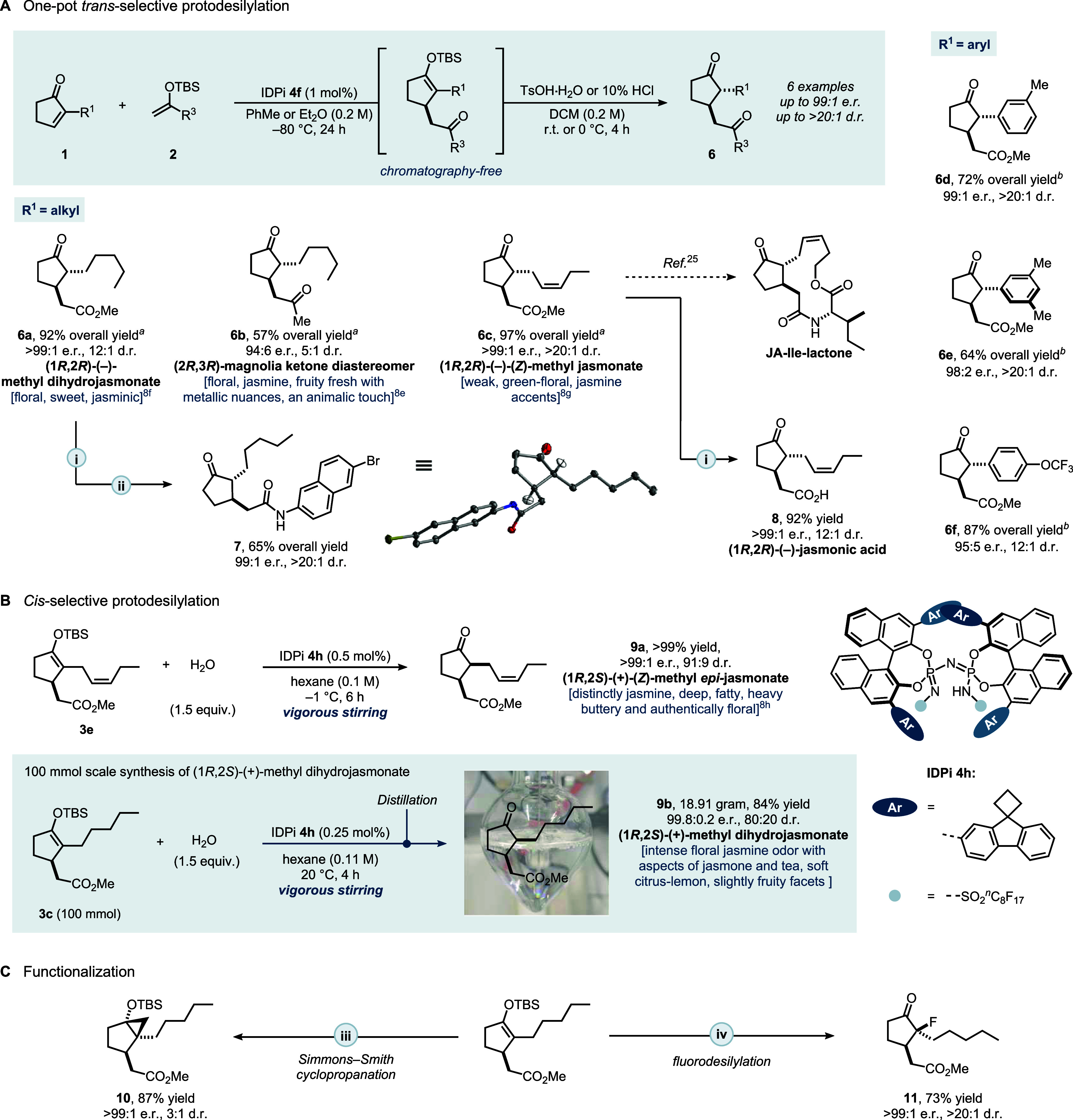 Figure 3
Figure 3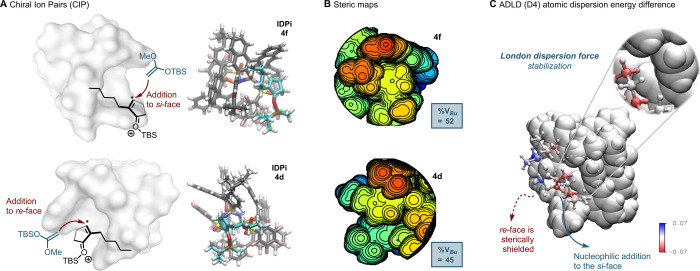 Figure 4
Figure 4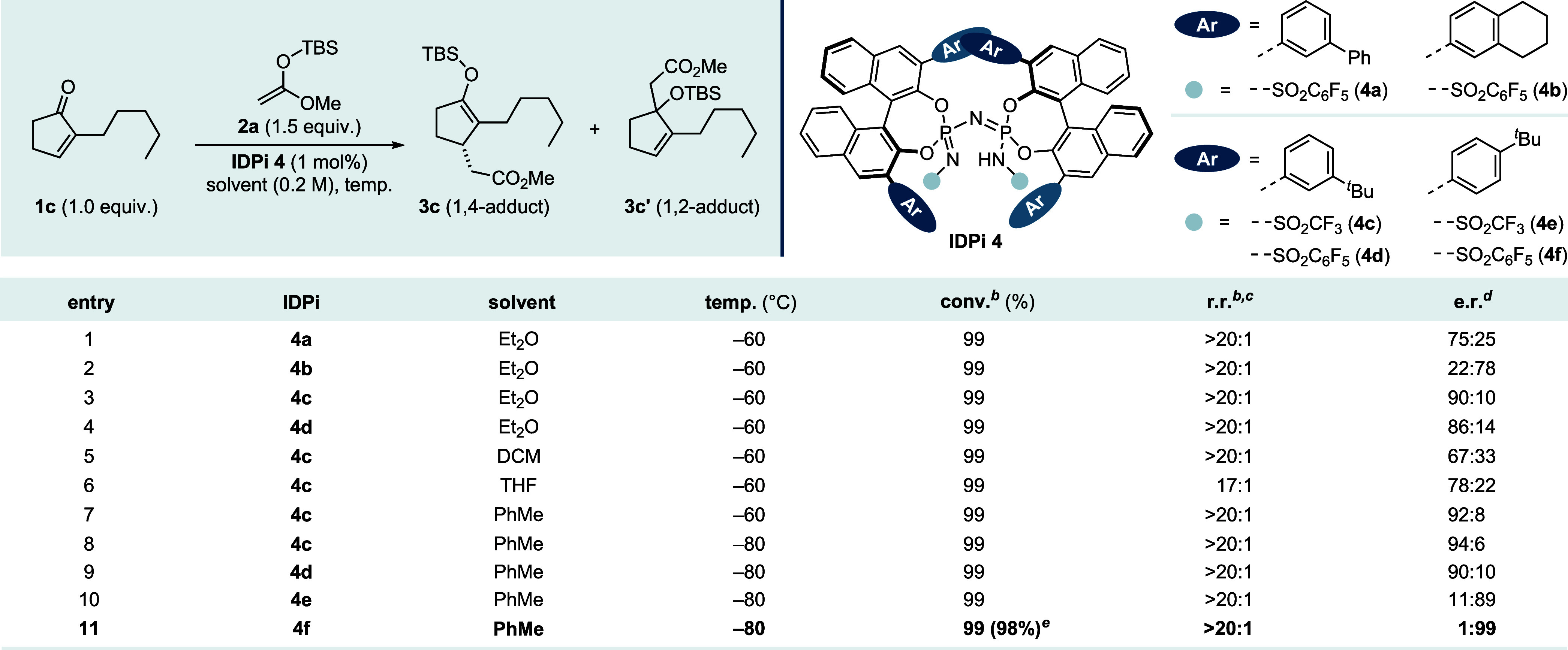 Figure 5
Figure 5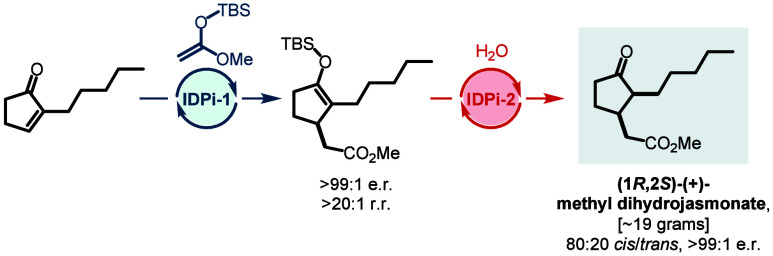 Figure 6
Figure 6- —Werner Siemens-Stiftung10.13039/100016964
- —European Union?s Horizon 2020 research and innovation programNA
- —Deutsche Forschungs-gemeinschaft (DFG, German Research Foundation)NA
- —Deutsche Forschungs-gemeinschaft (DFG, German Research Foundation)NA
- —Max Planck SocietyNA
- —Symrise AG, HolzmindenNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAsymmetric Synthesis and Catalysis · Cyclopropane Reaction Mechanisms · N-Heterocyclic Carbenes in Organic and Inorganic Chemistry
The Mukaiyama–Michael addition of enol silanes to α,β-unsaturated carbonyl compounds constitutes a fundamental and widely employed method for C–C bond formation. Since Mukaiyama’s seminal enantioselective addition of thioester-derived enol silanes to enones using a chiral titanium catalyst,? numerous catalytic asymmetric variants have been developed, particularly via iminium-ion and Lewis acid activation. ?−? ? ? In metal-catalyzed systems, high enantioselectivity is typically achieved when the acceptor can chelate bidentately to the metal center.? This chelation preferentially aligns the substrate to expose one reactive enantioface within the chiral environment, while significantly lowering its LUMO energy, thereby facilitating selective nucleophilic attack (FigureA). In contrast, nonchelating, weakly coordinating α,β-unsaturated acceptors, particularly cyclic enones and α-alkyl-substituted cycloenones,? typically display moderate to poor reactivity and limited stereocontrol under metal-based catalytic conditions. Moreover, substitution at the α- or β-position markedly decreases the electrophilicity, as indicated by the Mayr electrophilicity parameter E (FigureB). The retarding effect of α- and β-substituted cycloenones can be rationalized by the combined effects of steric constraints around the Michael acceptor and the electron inductive effect of the substituent. ?,? Consequently, a general enantioselective Mukaiyama–Michael addition to α-substituted cyclic enones remains elusive.
In 2018 we disclosed a silylium imidodiphosphorimidate (IDPi) Lewis acid catalyzed Mukaiyama–Michael addition of silyl ketene acetals to α,β-unsaturated esters.? In this system, a silylium species is paired with a chiral counteranion to form a tight, well-organized ion pair that engages the acceptor, obviates the need for chelation, and enables precise enantiocontrol within a confined chiral pocket.? Given that a general, catalytic asymmetric Mukaiyama–Michael addition to cyclic enones would represent a powerful tool in natural product synthesis, we initiated a research program toward this goal.? Here we report the fruition of these investigations with a silylium IDPi-catalyzed and broadly applicable and scalable Mukaiyama–Michael reaction followed by a diastereoselective protodesilylation sequence (FigureC). Notably, the enol silane intermediate enables stereodivergent hydrolysis, providing access to either diastereomer under kinetic or thermodynamic control.
We began our investigation using 2-pentylcyclopentenone 1c and commercially available silyl ketene acetal (SKA) 2a under cryogenic conditions. Moderately acidic chiral Brønsted acids, including CPA, ?,? IDP,? iIDP ?,? families, failed to promote productive conjugate addition, whereas a strongly acidic catalyst (DSI)? favored silicon–hydrogen exchange rather than C–C bond formation (see Supporting Information (SI)). IDPi 4a, characterized by its high acidity and an exceptionally confined chiral microenvironment ?−? ? ? ? ? ? enabled quantitative formation of 1,4-adduct 3c with excellent regioselectivity (1,4/1,2 >20:1) and a moderate enantiomeric ratio of 75:25 (Table, entry 1). Among the IDPi variants evaluated (entries 2–8), catalyst 4c (Ar = 3-^ t ^Bu-C_6_H_4_) provided superior enantiocontrol, affording the product with up to 94:6 e.r. (entry 8). Ultimately, IDPi 4f bearing a p-^ t ^Bu phenyl substituent at the 3,3′-positions and a pentafluorophenylsulfonyl core, was identified as optimal. It not only induced a reversal of enantiofacial selectivity but also delivered the 1,4-adduct with excellent regio- and enantioselectivity (1:99 e.r.) in nearly quantitative yield (entry 11). On this basis, we investigated how the steric properties of different silyl groups in the SKA affect the reactivity, stereo- and chemoselectivity (see SI).
With the optimal reaction conditions in hand, we explored the generality of the conjugate addition (FigureA). Cycloenones bearing alkyl, alkenyl, alkynyl, and aryl substituents reacted smoothly to provide the corresponding 1,4-adducts in high yields and typically around 95:5 e.r. (3a–q). A heteroaryl example was also tolerated, furnishing product 3r with good enantioselectivity. Notably, 3-methyl cyclopentenone also proved to be a competent substrate, affording enol silane 3s, featuring a quaternary stereocenter, in 87% yield with high regio- and enantioselectivity. However, substituting the methyl group with an n-butyl moiety resulted in a preference for the corresponding 1,2-addition (1,2/1,4 = 62:38; see SI). Furthermore, extension to six- and seven-membered cycloenones afforded products 3t–w in moderate yields with diminished regio- and enantioselectivity. Remarkably, heterocyclic enones containing nitrogen, oxygen or sulfur were well tolerated, delivering products 3x–z in good yields and high enantioselectivities (95:5 to 98:2 e.r.). These results demonstrate the broad scope and versatility of the IDPi-catalyzed asymmetric conjugate addition for constructing structurally and stereochemically complex products, and further underscore the potential of silylium IDPi Lewis acid catalysis as a powerful platform for enantioselective synthesis. We next evaluated both silyl ketene acetals and enol silanes (FigureB). Under the standard conditions, sterically hindered SKA 2b, bearing two methyl substituents, showed poor conversion. Increasing the reaction temperature to −50 °C effectively addressed this limitation, affording the desired 1,4-adduct 3aa in 84% yield with excellent enantioselectivity (93:7 e.r.).
Despite this success, certain combinations of enone electrophiles and silylated nucleophiles proved challenging, showing diminished reactivity or leading to competing side processes such as silicon–hydrogen exchange or undesired 1,2-addition (see SI). Notably, rac-carvone underwent a kinetic resolution with catalyst 4f to furnish the corresponding 1,4-adduct 3af with high diastereo- and enantioselectivity. The unreacted enantiomer, (R)-carvone 3af′, was recovered in 42% yield with 94:6 e.r., highlighting the remarkable potential of the methodology also for kinetic resolution.
To demonstrate the synthetic practicality and scalability of our approach, the conjugate addition was performed on a hectogram scale. Under slightly modified conditions, product 3c was isolated by distillation in high yield (167.0 g, 98% yield) and excellent enantiopurity, with the catalyst recovered in 96% yield from the organic phase after chromatographic purification and acidification, further highlighting the process’s potential industrial relevance.
We subsequently applied our newly established methodology to the trans-selective protodesilylation of Michael adducts, enabling streamlined access to bioactive pharmaceuticals and perfumery ingredients? without the need for intermediate chromatographic purification. By carefully tuning the acidity and steric profile of the proton source to each silylated adduct, we successfully developed a thermodynamically controlled trans-selective protonation pathway. Thus, treatment of silyl ether 3c with 10 vol% aqueous HCl in THF at 0 °C selectively furnished trans-jasmonates, (1R,2R)-(−)-methyl dihydrojasmonate (6a) with a floral, sweet jasminic odor in agreement to ref.,? (2R,3R)-configured Magnolia Ketone (6b) with a fruity-fresh floral nuances jasmine odor and in contrast to ref.? additional metallic and an animalic touch, and finally (1R,2R)-(−)-(Z)-methyl jasmonate (6c) with only a weak green-floral odor and very faint jasmine accentsthe latter in contrast to the literature,? in which it was described very weak to odorless with a 240 ppb threshold. These products were obtained in good to high yields with moderate to excellent diastereo- and enantioselectivities (FigureA). Subsequent LiOH-mediated hydrolysis in THF/H_2_O smoothly transformed methyl ester 6c to the corresponding acid 8 while retaining its enantiomeric purity. Amidation of methyl dihydrojasmonate 6a provided derivative 7, whose absolute configuration was confirmed by single-crystal X-ray diffraction. Furthermore, JA-Ile-lactone, a biologically active regulator of plant growth, can be accessed from 6c in a few steps.? For substrates bearing electron-rich aryl substituents, the milder HCl protocol proved insufficient to achieve complete hydrolysis. Instead, treatment with TsOH·H_2_O enabled clean protodesilylation, exclusively affording trans-products 6d–f with diastereomeric ratios ranging from 12:1 to >20:1.
In parallel, we investigated the kinetically controlled cis- selective protodesilylation. Two commercially valuable perfumery ingredients, (1R,2S)-(+)-methyl dihydrojasmonate (9b) and (1R,2S)-(+)-(Z)-methyl epi-jasmonate (9a), the olfactory properties of which critically depend on the absolute and relative stereochemistry,? were targeted by protonation of enol silanes 3c and 3e, respectively (FigureB). An extensive catalyst screening identified IDPi 4h as optimal (see SI for details), with water serving as an effective proton source. Under these conditions, enol silane 3e cleanly converted to cis-product 9a in excellent yield with high diastereomeric ratio. Control experiments further indicated that the stereochemical configuration of 4h is the primary determinant of the diastereoselectivity (see SI). Notably, scale-up to a decagram synthesis of (1R,2S)-(+)-methyl dihydrojasmonate (9b) was achieved in a mechanical agitation perfumery reactor (2000 rpm stirring) without any loss in selectivity. Following careful workup and fractional distillation, the desired product (9b) was isolated in high yield with an 80:20 d.r. and >99:1 e.r. Interestingly, the undersired 20% of the trans-configured diastereomer (1R,2S)-(−)-9b did not deteriorate the olfactory properties of the mixture which was found to possess an intense floral jasmine odor with aspects of jasmone and tea, and soft citrus-lemon as well as slightly fruity facets. This particular mixture was even found superior to the pure (1R,2R)-(+)-enantiomer. Overall, these results demonstrate that, by judicious choice of catalyst and proton source, our methodology can be switched between thermodynamic and kinetic protonation pathways, enabling stereodivergent access to both perfumery ingredients and bioactive compounds under operationally simple and scalable conditions.
To further diversify the enantioenriched enol silanes, downstream transformations were demonstrated (FigureC). Simmons–Smith cyclopropanation of 3c furnished bicyclo[3.1.0]hexane 10 (retaining e.r.), and fluorodesilylation with Selectfluor delivered α-fluorocyclopentanone 11 in good yield with high stereoselectivity.
We next turned our attention to the origin of the enantioselectivity. For the IDPi catalysts, formation of the chiral ion-pair intermediate (CIP) is endothermic, whereas the ensuing C–C bond forming step proceeds with virtually no barrier (see SI for details). We therefore optimized and analyzed the CIPs of 4d and 4f with the 2-pentylcyclopentenone–TBS cation to rationalize enantiodiscrimination and the meta/para tert-butyl effect (FigureA). Steric maps generated using Cavallo’s web tool? reveal that, in each CIP, attack on the less hindered face selectively furnishes the major enantiomer (FigureB), in full agreement with the experimental outcomes. Electrostatic analyses further indicate substantial catalyst–substrate interactions within the ion pair (Figure S11). In addition, utilizing our atomic decomposition of London dispersion energy method, ADLD (D4), ?,? we uncovered that the pentyl chain plays a larger stabilizing role in the formation of the CIP in 4f as compared to 4d (FigureC). The role of the alkyl chain can be quantified by taking the difference between its atomic London dispersion contributions in catalyst 4f vs. 4d (−2.8 kcal·mol^–1^). Red atoms indicate where 4f provides stronger dispersion stabilization, and the blue where it is weaker. This enhanced dispersion stabilization is consistent with the higher enantioselectivity observed for 4f (>99:1 e.r.) relative to 4d (10:90 e.r.).
We have developed a scalable catalytic platform for the asymmetric Mukaiyama–Michael reaction of moderately electrophilic cycloenones. The reaction is enabled by chiral silylium ion-based Lewis acids, affording 1,4-adducts with high diastereo- and enantioselectivity across a broad range of substrates and enol silanes. Careful olfactory analysis of the jasmonate products allowed to confirm and complement the literature data on this commercially important class of odorants, and computational studies highlight the pivotal influence of the chiral ion pair on enantioinduction. This methodology offers a versatile route to structurally complex chiral scaffolds, with direct applicability to Haute Parfumerie and bioactive pharmaceuticals, even potential application in aromatherapy.?
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Narasaka K.Soai K.Aikawa Y.Mukaiyama T.The Michael Reaction of Silyl Enol Ethers with α,β-Unsaturated Ketones and Acetals in the Presence of Titanium Tetraalkoxide and Titanium Tetrachloride Bull. Chem. Soc. Jpn.19764977978310.1246/bcsj.49.779 · doi ↗
- 2a Brown S. P.Goodwin N. C.Mac Millan D. W. C.The First Enantioselective Organocatalytic Mukaiyama-Michael Reaction: A Direct Method for the Synthesis of Enantioenriched γ-Butenolide Architecture J. Am. Chem. Soc.20031251192119410.1021/ja 029095 q 12553821 · doi ↗ · pubmed ↗
- 3a Frias M.Mas-BallestéR.Arias S.Alvarado C.Alemán J.Asymmetric Synthesis of Rauhut-Currier Type Products by A Regioselective Mukaiyama Reaction under Bifunctional Catalysis J. Am. Chem. Soc.201713967267910.1021/jacs.6b 0785128004935 · doi ↗ · pubmed ↗
- 4Gatzenmeier T.Kaib P. S. J.Lingnau J. B.Goddard R.List B.The Catalytic Asymmetric Mukaiyama-Michael Reaction of Silyl Ketene Acetals with α,β-Unsaturated Methyl Esters Angew. Chem., Int. Ed.2018572464246810.1002/anie.20171208829232022 · doi ↗ · pubmed ↗
- 5a Evans D. A.Rovis T.Kozlowski M. C.Downey C. W.Tedrow J. S.Enantioselective Lewis Acid Catalyzed Michael Reactions of Alkylidene Malonates. Catalysis by C 2-Symmetric Bis(oxazoline) Copper(II) Complexes in the Synthesis of Chiral, Differentiated Glutarate Esters J. Am. Chem. Soc.20001229134914210.1021/ja 002246+ · doi ↗
- 6Waulters-Kline Q.Gangadurai C.Thorat S. S.Renner A. C.Sibi M. P.Mukaiyama-Michael Reaction: Enantioselective Strategies and Applications in Total Synthesis Org. Biomol. Chem.2025238609864010.1039/D 5OB 01134 E 40926694 · doi ↗ · pubmed ↗
- 7a Mayer R. J.Allihn P. W. A.Hampel N.Mayer P.Sieber S. A.Ofial A. R.Electrophilic Reactivities of Cyclic Enones and α,β-Unsaturated Lactones Chem. Sci.2021124850486510.1039/D 0SC 06628 A 34163736 PMC 8179571 · doi ↗ · pubmed ↗
- 8a Kraft P.Bajgrowicz J. A.Denis C.Fráter G.Odds and Trends: Recent Developments in the Chemistry of Odorants Angew. Chem., Int. Ed.2000392980301010.1002/1521-3773(20000901)39:17<2980::AID-ANIE 2980>3.0.CO;2-#11028024 · doi ↗ · pubmed ↗