Development of Prostate-Specific Lysosome-Targeting Degraders
Deqin Cai, Xuankun Chen, Yaxian Zhou, Malick Bio Idrissou, Reinier Hernandez, Weiping Tang

TL;DR
This paper introduces a new method to selectively degrade proteins in prostate cancer cells using a prostate-specific receptor and lysosome-targeting degraders.
Contribution
The novel contribution is the development of prostate-specific lysosome-targeting degraders (PTACs) that can degrade extracellular and membrane proteins in prostate cancer cells.
Findings
PTACs can selectively and potently degrade extracellular and membrane proteins via the lysosome pathway in prostate cancer cells.
Ctx-L3 and Atz-L5 achieved DC50 values of 4.3 pM for EGFR and 2 pM for PD-L1 in LNCaP cells.
PTACs can degrade PD-L1 using both antibody- and small-molecule-based formats, showing platform versatility.
Abstract
Targeted protein degradation (TPD) technologies have emerged as transformative therapeutic modality for treating cancers and other diseases. While significant progress has been achieved in intracellular protein degradation, degradation of membrane proteins and extracellular targets remains in an early stage. In this study, we developed a prostate-specific lysosome-targeting degradation strategy using a prostate-specific membrane antigen (PSMA) as a lysosome-targeting receptor (LTR). We demonstrated that both extracellular and membrane proteins can be selectively degraded in prostate cancer cells via the lysosome pathway. These PSMA TArgeting Chimeras (PTACs) were shown to facilitate lysosomal degradation in a selective, potent, rapid, and sustained manner. Notably, Ctx-L3 and Atz-L5 exhibited exceptional degradation potencies in LNCaP cells, with DC50 values of 4.3 pM for EGFR and 2 pM…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1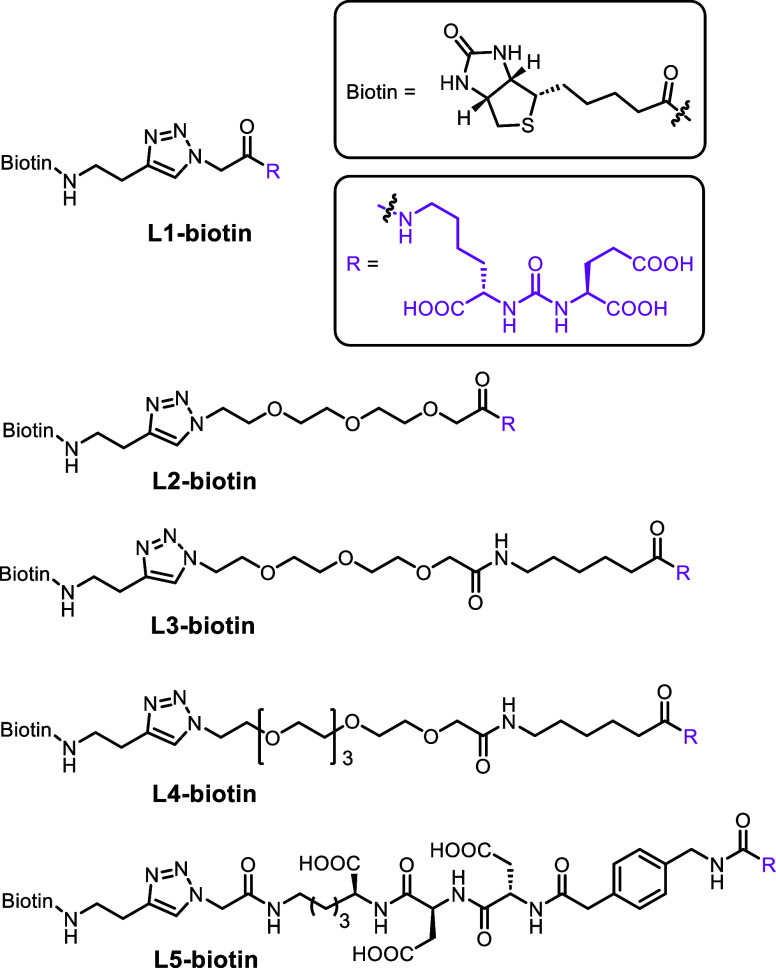 2
2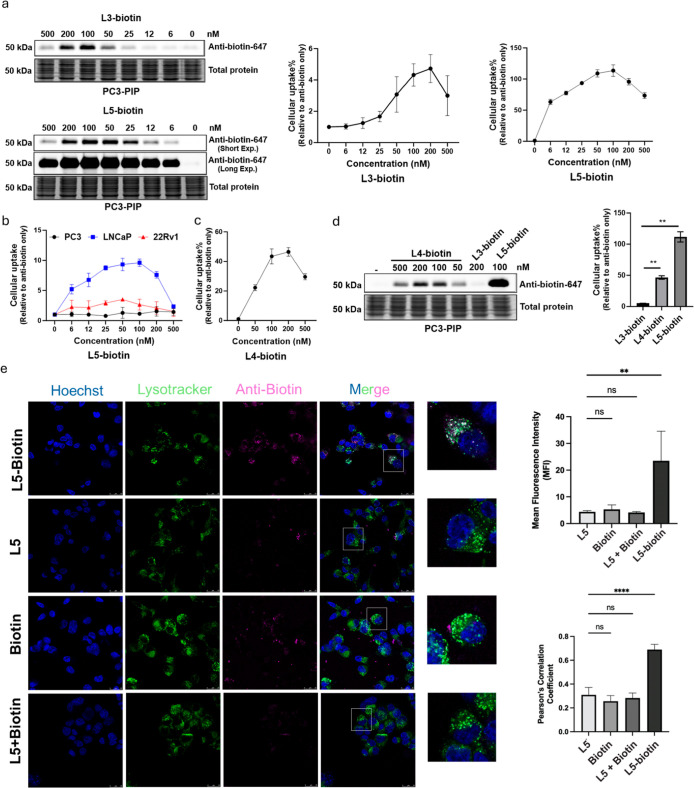 3
3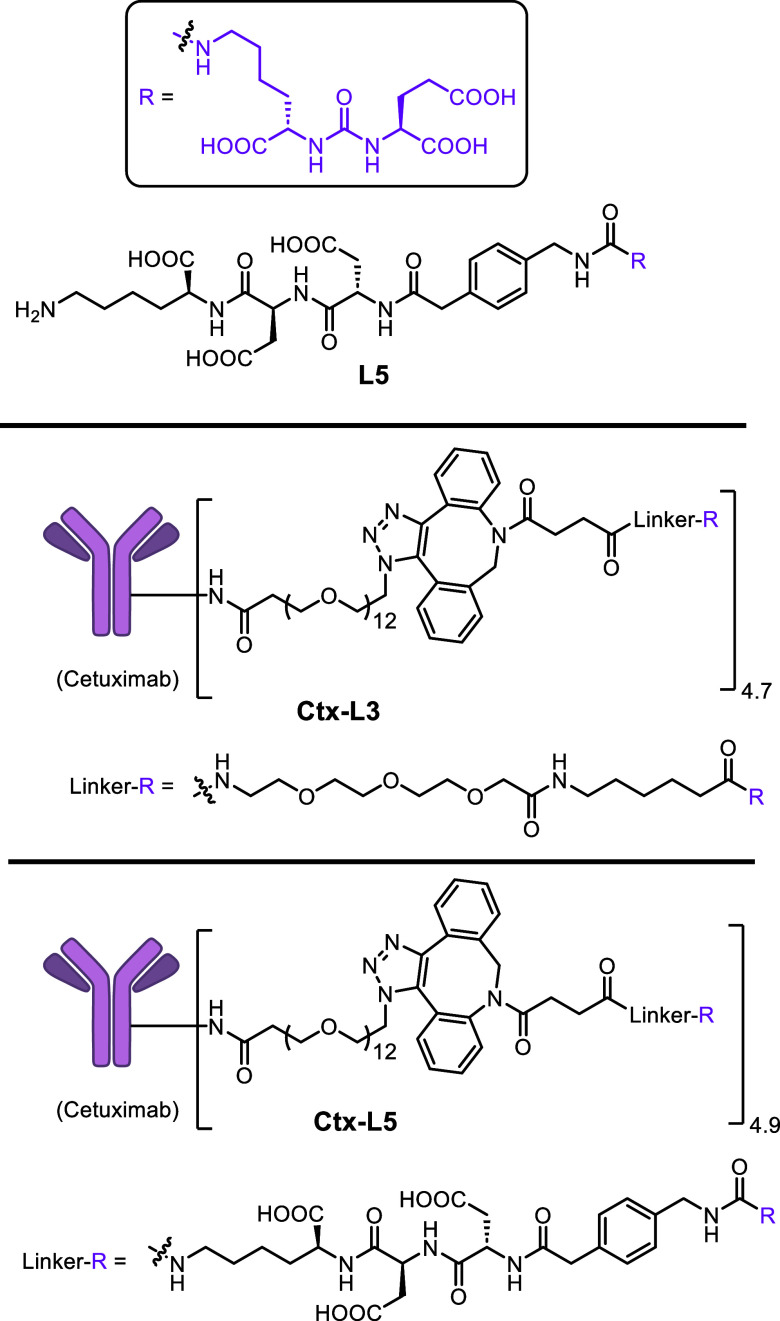 4
4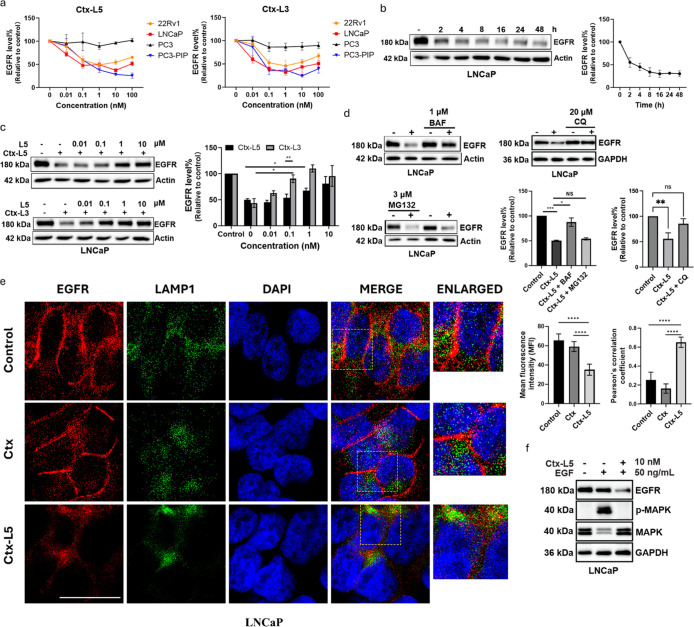 5
5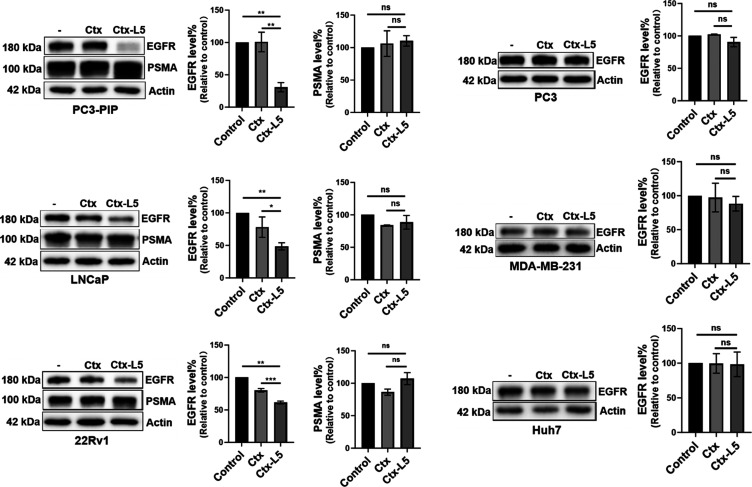 6
6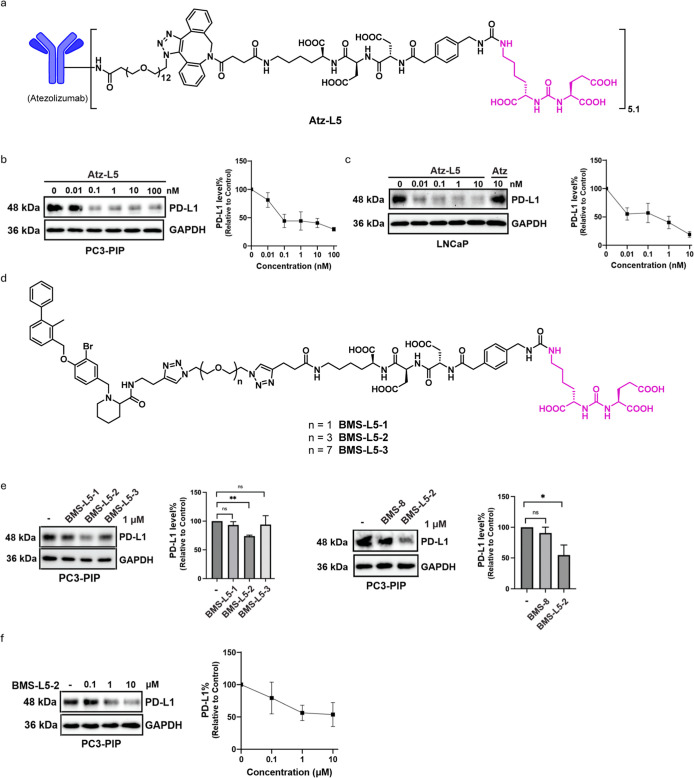 7
7- —National Cancer Institute10.13039/100000054
- —National Institute of General Medical Sciences10.13039/100000057
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtein Degradation and Inhibitors · Click Chemistry and Applications · Histone Deacetylase Inhibitors Research
Introduction
Protein degradation is essential for maintaining cellular homeostasis, primarily mediated by the ubiquitin-proteasome system (UPS) and the lysosomal degradation pathway. ?−? ? Proteolysis targeting chimeras (PROTACs) and monovalent molecular glue degraders leverage the UPS by inducing the proximity between protein of interest (POI) and E3 ligase in cytoplasm, leading to POI ubiquitination and subsequent proteasomal degradation.? PROTAC ARV-471 has advanced to a phase III clinical trials for the treatment of advanced breast cancer. ?,? Unlike classical occupation-driven enzyme inhibition or ligand blocking, TPD can eliminate the entire protein including both the enzymatic and nonenzymatic functions. ?,? This novel mechanism of action holds the potential to overcome resistance to conventional treatments and provides a promising avenue for discovering drugs targeting challenging or undruggable proteins.? However, the proteasomal pathway is limited to degrading POIs with cytosolic domains.
To extend the targeted protein degradation paradigm to extracellular proteins, lysosome-targeting chimeras (LYTACs) have been developed. ?−? ? ? LYTACs are designed to bind extracellular or membrane proteins and direct them to lysosomes for degradation via lysosomal targeting receptor (LTR)-mediated endocytosis, offering a complementary approach to proteasomal degradation. Several LTRs have been identified,? including cation-independent mannose-6-phosphate receptor (CI-M6PR), ?,?−? ? ? ? ? asialoglycoprotein receptor (ASGPR), ?−? ? macrophage galactose-type lectin 1 (MGL1),? integrin, ?−? ? scavenger receptors (SRs),? cytokine receptors,? mannose receptor (CD206),? glucagon-like peptide-1 receptor (GLP-1R),? low-density lipoprotein receptor-related protein (LRP-1),? glucose transporter 1 (GLUT1),? transferrin receptor,? and folate receptor.? Other innovative strategies ?,? have also been developed for extracellular or transmembrane protein degradation, such as lysosome-based antibody-based PROTAC (AbTAC),? proteasome and lysosome-based proteolysis-targeting antibodies (PROTABs),? covalent nanobody-based PROTAC strategy (GlueTAC),? and multivalent nanobody-targeting chimeras (mNbTACs).?
Despite these significant advancements, most LTRs identified thus far are expressed across multiple tissues, with ASGPR being the notable exception, as reported by us and others. ?−? ? This lack of specificity poses challenges in minimizing on-target and off-tissue side effects and achieving precise therapeutic outcomes. Developing degraders with high tissue specificity is crucial to maximizing therapeutic efficacy while minimizing unintended side effects. This is particularly important in targeting diseases where localized protein degradation is essential, such as tissue-specific cancers.
Prostate-specific membrane antigen (PSMA) is overexpressed in most of prostate cancer cells, correlating with disease progression and poor prognosis, while its expression in normal prostate tissue is minimal, often undetectable.? PSMA is a transmembrane glycoprotein, consisting of a 24-amino acid transmembrane region, a 19-amino acid N-terminal cytoplasmic sequence, and a large extracellular domain comprising 707 amino acids. ?,? PSMA facilitates the internalization of targeting molecules and their payloads, making it as an ideal molecular target for diagnostic imaging and targeted therapies. ?,?−? ? ? Notably, PSMA has already been clinically validated through the FDA approval of [^68^Ga]Ga-PSMA-11 as a PET imaging agent and [^177^Lu]Lu-PSMA-617 (Pluvicto) as a targeted radioligand therapy for advanced prostate cancer.
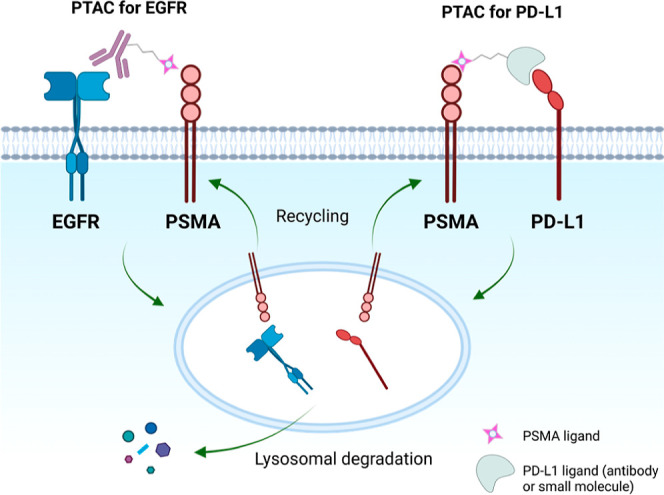
In this study, we report the development of PSMA TArgeting Chimeras (PTACs) as a versatile platform for selective degradation of extracellular and membrane proteins in PSMA-positive prostate cancer cells (Figure). The work started with the uptake of soluble antibiotin mediated by biotinylated PSMA ligands. The utility of the platform was subsequently demonstrated through the selective degradation of the membrane protein epidermal growth factor receptor (EGFR). The PTAC approach was further extended to the degradation of PD-L1 in both antibody- and small molecule-based formats. Antibody-based PTACs were prepared by conjugating the PSMA ligands Glu-urea-Lys (EuK) ?,? to antibody targeting the POI, while small-molecule-based PTACs were synthesized by linking the PD-L1 ligand BMS-8 with the EuK ligand. Chimeras incorporating PSMA ligands with varying affinities were evaluated to assess their impact on the POI degradation efficiency. Our results suggest that PTACs hold significant promise as a technology for both research purposes and therapeutic applications in the study and treatment of prostate-related diseases.
Schematic illustration of PSMA TArgeting Chimeras (PTACs) used in the current study for TPD of membrane proteins EGFR and PD-L1 via lysosomal degradation pathways.
Results and Discussion
Uptake of Fluorescent Antibiotin-647 by a Biotinylated PSMA
Ligand with Various Linkers in Different Prostate Cancer Cell Lines
We began with investigating the uptake of antibiotin-647 using biotinylated PSMA ligands. Five distinct biotinylated PSMA ligands were designed and synthesized, as shown in Figure. The synthetic route and procedure are provided in the Supporting Information. Initially, biotinylated PSMA ligands with linkers of three different lengths were synthesized: L1-biotin, L2-biotin, and L3-biotin. No uptake was observed with L1-biotin and L2-biotin, while uptake by L3-biotin bearing a longer linker was observed only in PC3-PIP cells, a PSMA-overexpressing variant of the PC3 cell line, in a dose-dependent manner (Figuresa and S1).
Biotinylated PSMA ligands with various linkers.
*Uptake of fluorescent antibiotin-647 in prostate cancer cells treated with L3-biotin, L4-biotin, or L5-biotin. (a) Dose–response of antibiotin-647 uptake after 24 h of treatment with L3-biotin or L5-biotin in PC3-PIP cells (n = 3). (b) Dose–response of antibiotin-647 uptake after 24 h of treatment with L5-biotin in LNCaP, 22Rv1, and PC3 cells (n = 3). (c) Dose–response of antibiotin-647 uptake after 24 h of treatment with L4-biotin in PC3-PIP cells (n = 3). (d) The maximum uptake of antibiotin-647 (24 h) treated with L3-biotin (200 nM), L4-biotin (200 nM), and L5-biotin (100 nM) in PC3-PIP cells (n = 3). (e) Cellular uptake and lysosome colocalization of antibiotin-647 (100 nM) in the presence of L5-biotin (100 nM), free L5 (100 nM), biotin (100 nM), and biotin (100 nM) + free L5 (100 nM) in LNCaP cells for 4 h by immunofluorescent staining. Scale bar: 25 μm. The intracellular fluorescence intensity is presented as mean fluorescence intensity (MFI) (n = 9). The colocalization was analyzed by Pearson’s correlation coefficients (n = 9). Data are presented as mean ± SD. The statistical significance was assessed using an unpaired two-tailed t-test, **P <0.01, ***P <0.0001, NS: not significant.
These observations are not surprising, since it has been shown that the linker attached to the PSMA ligand could significantly affect its binding affinity to PSMA. ?−? ? Notably, a ∼20 Å deep funnel exists between the ligand binding site and the protein surface.? In addition to linker length, other parameters can also significantly enhance the binding affinity. For example, a PSMA ligand that contains a short peptide Asp-Asp-Cys and an aromatic motif exhibits a 30-fold higher binding affinity to PMPA,? a known PSMA ligand with a K _ i _ of 0.3 nM.? In contrast, the PSMA ligand bearing a linker of a linear aliphatic motif has a K _ i _ value of only 8.84 nM.? Based on these, we synthesized biotinylated PSMA ligand L5-biotin, with a short peptide Asp-Asp-Lys and an aromatic motif. The uptake of antibiotin-647 by L5-biotin was clearly observed in all PSMA positive cell lines: PC3-PIP, LNCaP, and 22Rv1 (Figurea,b). The uptake efficiency is dose-dependent and corelated to the PSMA expression level (LNCaP > 22Rv1 > PC3, Figuresb, S2, and S3).
To further investigate the effect of linker structure on uptake efficiency, we synthesized L4-biotin, which has the same linker length as L5-biotin. L4-biotin exhibited significantly better uptake efficiency than the previous linear structure, L3-biotin, in a dose-dependent manner (Figurec). The improvement is likely due to the longer linker present in L4-biotin. However, at a concentration of 200 nM, L4-biotin achieved only ∼40% of the uptake of L5-biotin (Figurea,d), which was further confirmed by co-localization data (Figuree). Remarkably, uptake induced by L5-biotin could be observed as low as at 6 nM. Additionally, a hook effect? was observed for all three compounds: L3-biotin, L4-biotin, and L5-biotin. Notably, L5-biotin, with its higher binding affinity, exhibits a broader effective concentration range.
Degradation of EGFR Mediated by PTACs
Given the differential PSMA expression across prostate cancer cell lines, it is important to note that PC3-PIP cells (engineered to overexpress PSMA) exhibit the highest PSMA levels, followed by LNCaP (endogenous expression) and 22Rv1 (low expression), while PC3 cells are essentially PSMA-negative. In this study, we initially employed PC3-PIP cells to establish mechanistic insights and subsequently focused on LNCaP cells to ensure greater clinical relevance. After demonstrating the uptake efficiency of a model target protein mediated by PTACs bearing PSMA ligands with varying binding affinities, we initiated our investigation into the development of degraders targeting therapeutically relevant proteins, such as EGFR. EGFR is an oncogenic driver overexpressed in many cancers, including prostate cancer. Commercially available reagent N_3_–PEG_12_-C_3_–OSu was used to conjugate with cetuximab (Ctx), an FDA-approved EGFR blocking antibody, via reaction with lysine residues on the antibody to produce azide-modified Ctx. L3-DBCO and L5-DBCO were then reacted with azide-modified Ctx to generate Ctx-L3 and Ctx-L5, respectively, through the copper-free Click reaction (Figures and S4–S5). Interestingly, Ctx-L3 and Ctx-L5 showed similar degradation efficiencies for EGFR across all three PSMA positive cell lines. Minimal degradation of EGFR was observed in PSMA-negative PC3 cells (Figuresa and S6). The D max (maximum degradation) is positively corelated with PSMA expression levels (PC3-PIP > LNCaP > 22Rv1), reaching approximately 75% in PC3-PIP cells. The degradation of EGFR induced by Ctx-L3 and Ctx-L5 was dose-dependent.
Structure of PSMA ligand L5 and cetuximab-PSMA ligand conjugates with two different linkers. L5 was used in the competitive study of EGFR degradation induced by PTACs.
*PTACs mediate the lysosomal degradation of EGFR by recruiting PSMA. (a) Dose–response of EGFR degradation after 24 h of treatment with Ctx-L5 and Ctx-L3 in four prostate cancer cell lines: PC3-PIP, LNCaP, 22Rv1, and PC3 (n = 3). (b) Time course of EGFR degradation induced by Ctx-L5 (10 nM) in LNCaP cells (n = 3). (c) Inhibition of EGFR degradation induced by Ctx-L3 (10 nM) and Ctx-L5 (10 nM) in the presence of free ligand L5 (0.01–10 μM) in LNCaP cells for 7 h (n = 3). (d) Inhibition of EGFR degradation induced by Ctx-L5 (10 nM for BAF and MG132, or 1 nM for CQ) in the presence of Bafilomycin A1 (BAF, 1 μM), MG132 (3 μM), and CQ (20 μM) in LNCaP cells for 6 h (n = 3). (e) Immunofluorescent staining of EGFR degradation and lysosome colocalization after treatment of Ctx (10 nM) and Ctx-L5 (10 nM) for 24 h in LNCaP cells. Scale bar: 25 μm. The intracellular fluorescence intensity is presented as mean fluorescence intensity (MFI) (n = 9). The colocalization was analyzed by Pearson’s correlation coefficients (n = 9). (f) Downregulation of EGFR, MAPK phosphorylation, and MAPK in LNCaP cells (n = 3). Data are presented as mean ± SD. The statistical significance was assessed using an unpaired two-tailed t-test, *P <0.05, **P <0.01, ***P <0.001, ***P <0.0001, NS: not significant.
Notably, the DC_50_ (concentration required for 50% degradation) for both Ctx-L3 and Ctx-L5 was below 100 pM in all three PSMA-positive cell lines, with the DC_50_ of Ctx-L3 in LNCaP as low as 4.3 pM, which is one of the most potent DC_50_ values for LYTACs and related degraders reported to date. The DC_50_ and D max values of Ctx-L3 and Ctx-L5 across PC3-PIP, LNCaP, and 22Rv1 cells are summarized in Table S1.
Despite the noticeable difference in uptake efficiency between L3-biotin and L5-biotin, Ctx-L3 and Ctx-L5 demonstrate a similar effective concentration range for degradation, with Ctx-L3 even showing potency slightly better than that of Ctx-L5. These results indicated the degradation efficiency is not solely dependent on the binding affinity of the LTR ligand. This phenomenon may be attributed to the formation of relatively more unproductive binary complexes (degrader-PSMA) with a high-affinity PSMA ligand, compared to the productive ternary complexes required for effective degradation in these two scenarios.?
Our time course study indicated that Ctx-L5 exerts a rapid and sustained effect on EGFR degradation in LNCaP cells at 10 nM. Approximately 50% of EGFR degradation was observed 2 h post-treatment and the maximal degradation persisted for at least 48 h (Figureb).
To further investigate the role of PSMA in degrader-induced EGFR degradation, Ctx-L3 and Ctx-L5 were cotreated with an increasing amount of free PSMA ligand L5 in LNCaP cells. We observed that the degradation of EGFR induced by PTACs was abolished by L5 in a dose-dependent manner. These results suggest that PSMA is involved in mediating EGFR degradation. Interestingly, degrader Ctx-L5, with a higher-affinity ligand, is much less sensitive to competition from L5 (Figurec). The binding affinity of the PTACs was supported by heterologous competition binding assays using [^177^Lu]Lu-PSMA-617 as the tracer ligand (Figure S7). The results suggested that PTACs with higher binding affinities have an enhanced ability to resist competition from endogenous ligands.
To investigate the mechanistic pathways of EGFR degradation, cells were treated with the lysosomal inhibitor Bafilomycin A1 (BAF), Chloroquine (CQ), and proteasome inhibitor MG132. Treatment with BAF or CQ significantly reduced EGFR degradation in the presence of Ctx-L5, while MG132 had no effect (Figured). These results confirm that Ctx-L5-induced EGFR degradation occurs via the lysosomal pathway rather than the proteasomal pathway. In addition, pretreatment with known endocytosis inhibitors, including chlorpromazine, methyl-β-cyclodextrin, or the chaperone-mediated autophagy (CMA) regulator VER-155008, applied at concentrations as reported,? did not appreciably affect Ctx-L5-induced EGFR degradation (Figure S8). While these data support PSMA-dependent internalization and subsequent lysosomal degradation, the precise endocytic pathways involved warrant future investigation.
Confocal imaging further validated the degradation effect of EGFR induced by Ctx-L5 in the LNCaP cells. Degrader-treated cells showed a marked reduction of EGFR compared to control and Ctx-treated groups. Additionally, a distinct translocation of EGFR from the cell surface into the cytoplasm was observed after 24 h of Ctx-L5 treatment. To track EGFR postinternalization, costaining of EGFR and lysosomal marker LAMP1 revealed that EGFR predominantly colocalized with lysosomes following degrader incubation (Figuree). These findings demonstrate that PTACs facilitate the delivery of membrane proteins to lysosomes for degradation.
To assess the downstream functional consequences, we pretreated LNCaP cells with Ctx-L5 (10 nM, 6 h) followed by EGF stimulation (50 ng/mL, 30 min, Figuref). Compared with the control, EGF treatment alone robustly increased the p-MAPK level and concomitantly decreased total MAPK, while EGFR levels remained largely unchanged. In contrast, Ctx-L5 treatment resulted in pronounced EGFR reduction and abolished the EGF-induced p-MAPK response, with total MAPK unchanged. These findings suggest that EGFR degradation triggered by Ctx-L5 may attenuate EGF-induced signaling, consistent with an on-target PTAC mechanism.
Prostate Cancer Selectivity of PTACs Targeting EGFR
To investigate the selectivity of Ctx-L5 for EGFR degradation in PSMA-positive cancer cells, the potency of the degrader was compared across prostate cancer cells PC3-PIP, LNCaP, 22Rv1, and PC3, as well as two nonprostate cancer cell lines, MDA-MB-231 and Huh7. As expected, degradation was observed in the following PSMA-positive cancer cell lines: PC3-PIP, LNCaP, and 22Rv1. The total PSMA level was not affected by the degrader (Figure S9). In contrast, no degradation occurred in PSMA-negative cancer cell lines: PC3, MDA-MB-231, and Huh7 (Figure). These results indicate that PTACs can selectively degrade EGFR in PSMA-positive prostate cancer cell lines.
*Selectivity of EGFR degradation (24 h) induced by Ctx (10 nM) and Ctx-L5 (10 nM) in PSMA-positive and -negative cell lines, and PSMA levels following EGFR degradation in PSMA-positive cancer cell lines. PSMA-positive cancer cell lines: PC3-PIP, LNCaP, 22Rv1; PSMA-negative cancer cell lines: PC3, MDA-MB-231, and Huh7. Data are presented as mean ± SD. The statistical significance was assessed using an unpaired two-tailed t-test, *P <0.01, NS: not significant.
Degradation of PD-L1 Mediated by PTACs
To demonstrate the versatility of the PTAC platform, we extended its application to PD-L1, a membrane protein critical for tumor immune evasion. Atezolizumab (Atz), an FDA-approved PD-L1 antibody, was conjugated with L5 via copper-free click chemistry to generate Atz-L5 (Figurea). Atz-L5 induced potent PD-L1 degradation in PC3-PIP cells, with a DC_50_ of 18 pM and D max of 70% at 10 nM for 24 h (Figureb). Notably, Atz-L5 exhibited even more efficient PD-L1 degradation in LNCaP cells, with a DC_50_ of 2 pM and a D max of 81% for 24 h (Figurec).
*Degradation of PD-L1 mediate by both antibody- and small molecule-based PTACs. (a) Structure of the atezolizumab-L5 conjugate (Atz-L5). (b) Dose–response of PD-L1 degradation induced by Atz-L5 with indicated concentration in PC3-PIP cells for 24 h, DC50 = 18 pM, D max = 70% (n = 3). (c) Dose–response of PD-L1 degradation induced by Atz-L5 with indicated concentration in LNCaP cells for 24 h, DC50 = 2 pM, D max = 81% (n = 3). (d) The structures of BMS-L5–1, BMS-L5–2, and BMS-L5–3. (e) PD-L1 degradation induced by BMS-L5–1, BMS-L5–2, BMS-L5–3, and BMS-8 at 1 μM in PC3-PIP cells for 24 h (n = 3). (f) Dose–response of PD-L1 degradation after 24 h of treatment with BMS-L5–2 with indicated concentration in PC3-PIP cells for 24 h (n = 3). Data are presented as mean ± SD. The statistical significance was assessed using an unpaired two-tailed t-test, *P <0.05, *P <0.01, NS: not significant.
We further applied the PTAC strategy to small-molecule degraders by linking PD-L1 binder BMS-8 with PSMA ligand L5 through PEG linkers of different lengths, generating BMS-L5–1, BMS-L5–2, and BMS-L5–3 (Figured). Among them, BMS-L5–2 showed the highest activity, achieving 47% degradation at 10 μM (Figuree and f).
These results confirm the versatility of the PTAC platform across both antibody conjugates and small-molecule ligands. Notably, while both antibody- and small-molecule-based PTACs enabled PD-L1 degradation, the small-molecule conjugates required substantially higher concentrations to achieve comparable effects, indicating that the intrinsic properties and compatibility of the POI ligand can markedly influence degradation efficiency. Given the role of PD-L1 in immune evasion, these findings suggest that PTAC-mediated PD-L1 degradation could potentially enhance immune recognition of prostate cancer cells, providing a therapeutic rationale for further exploration.
Conclusions
In summary, this work successfully demonstrated that PSMA can function as an LTR, enabling applications in tissue-specific TPD. PTACs were shown to facilitate rapid and selective lysosomal degradation of both extracellular antibiotin model protein target and therapeutically relevant membrane proteins, such as EGFR and PD-L1, in PSMA-positive prostate cancer cells. Two PTACs for EGFR degradation, Ctx-L3 and Ctx-L5, bearing ligands with different PSMA binding affinities, exhibited comparable degradation efficiencies despite their differing affinities. However, PTACs with higher binding affinities demonstrate superior resistance to competition from LTR ligands, indicating that they may be less susceptible to interference from endogenous PSMA substrates. This characteristic enhances their potential for therapeutic applications, where competition with naturally occurring PSMA substrates could otherwise diminish efficacy. Notably, the DC_50_ values for both PTACs are all below 100 pM, with one of them as low as 4.3 pM, among the lowest reported for LYTACs and related degraders to date. PTACs for PD-L1 degradation were demonstrated in both antibody- and small-molecule-based formats, enriching this versatile platform to an additional target and degrader modality. By utilizing PSMA as an LTR, this study extends the strategy of degradation of extracellular and membrane proteins and demonstrates the potential of PTACs as a tool for prostate cancer-selective degraders. These findings broaden the therapeutic landscape for TPD, paving the way for the development of innovative treatments for PSMA-positive cancers and other diseases.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bard J. A. M.Goodall E. A.Greene E. R.Jonsson E.Dong K. C.Martin A.Structure and Function of the 26S Proteasome Annu. Rev. Biochem.20188769772410.1146/annurev-biochem-062917-01193129652515 PMC 6422034 · doi ↗ · pubmed ↗
- 2Luzio J. P.Pryor P. R.Bright N. A.Lysosomes: fusion and function Nat. Rev. Mol. Cell Biol.20078862263210.1038/nrm 221717637737 · doi ↗ · pubmed ↗
- 3Ciechanover A.Proteolysis: from the lysosome to ubiquitin and the proteasome Nat. Rev. Mol. Cell Biol.200561798710.1038/nrm 155215688069 · doi ↗ · pubmed ↗
- 4Bekes M.Langley D. R.Crews C. M.PROTAC targeted protein degraders: the past is prologue Nat. Rev. Drug Discovery 202221318120010.1038/s 41573-021-00371-635042991 PMC 8765495 · doi ↗ · pubmed ↗
- 5Hamilton E.Ma C. X.De Laurentiis M.Iwata H.Hurvitz S. A.Wander S. A.Danso M. A.Lu D. R.Perkins J.Liu Y.257Ti P VERITAC-2: A global, randomized phase III study of ARV-471, a Proteolysis T Argeting Chimera (PROTAC) estrogen receptor (ER) degrader, vs fulvestrant in ER+/human epidermal growth factor receptor 2 (HER 2)- advanced breast cancer ESMO Open 20238110144510.1016/j.esmoop.2023.101445 · doi ↗
- 6Hamilton E. P.Ma C.Laurentiis M.Iwata H.Hurvitz S. A.Wander S. A.Danso M.Lu D. R.Smith J. P.Liu Y.VERITAC-2: a Phase III study of vepdegestrant, a PROTAC ER degrader, versus fulvestrant in ER+/HER 2- advanced breast cancer Future Oncol.2024202447245510.1080/14796694.2024.237753039072356 PMC 11524203 · doi ↗ · pubmed ↗
- 7Sun D.Zhang J.Dong G.He S.Sheng C.Blocking Non-enzymatic Functions by PROTAC-Mediated Targeted Protein Degradation J. Med. Chem.20226521142761428810.1021/acs.jmedchem.2c 0115936306471 · doi ↗ · pubmed ↗
- 8Zhao L.Zhao J.Zhong K.Tong A.Jia D.Targeted protein degradation: mechanisms, strategies and application Signal Transduct. Targeted Ther.20227111310.1038/s 41392-022-00966-4PMC 897743535379777 · doi ↗ · pubmed ↗