Isolation and identification of Pseudoxanthomonas winnipegensis from blood culture by MALDI-MS
Satomi Takei, Tatsuya Nagasawa, Yuji Sekiguchi, Junya Fujimura, Kanae Teramoto, Mitsuru Wakita, Teruo Kirikae, Yuki Uehara, Tatsuya Tada, Yoko Tabe

TL;DR
This paper describes the first isolation of Pseudoxanthomonas winnipegensis from human blood in Japan and shows how MALDI-MS can help identify these rare bacteria quickly.
Contribution
The study introduces MALDI-MS marker peaks for rapid identification of Pseudoxanthomonas species in clinical settings.
Findings
P. winnipegensis was first isolated from a human blood sample in Japan in 2022.
MALDI-MS using ribosomal proteins L29, L33, and CspA can reliably identify Pseudoxanthomonas species.
Routine microbiological tests struggle to accurately identify Pseudoxanthomonas species.
Abstract
Pseudoxanthomonas is a genus primarily isolated from environmental samples and causes opportunistic infections. In this study, we conducted a detailed investigation of P. winnipegensis, which was isolated for the first time from a blood sample in Japan in 2022, and evaluated five Pseudoxanthomonas species, including P. kaohsiungensis, P. japonensis, P. mexicana, P. spadix and P. winnipegensis, that cause human infections. However, it is difficult to identify accurately by routine microbiological testing. Our analysis revealed that matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) classification is feasible when using the mass peaks corresponding to ribosomal proteins L29 and L33, as well as the cold-shock protein CspA, as marker peaks. These findings indicate the potential of MALDI-MS for the rapid and reliable detection of Pseudoxanthomonas species in routine…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
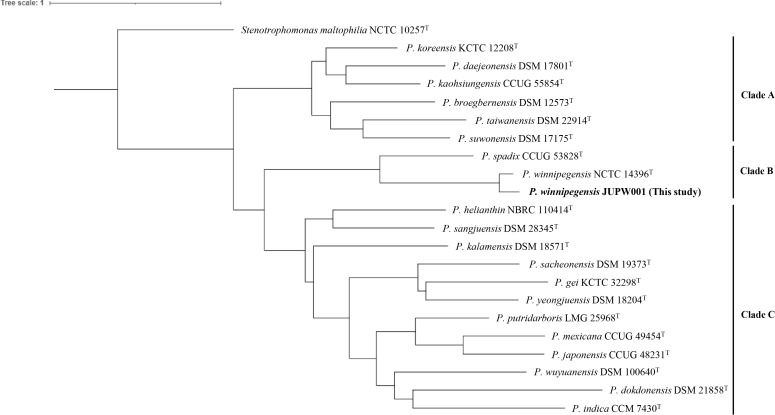 Figure 1
Figure 1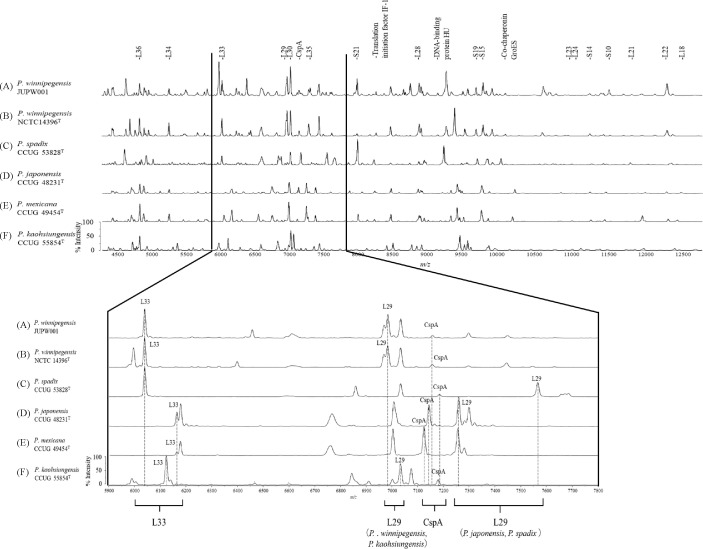 Figure 2
Figure 2 Figure 3
Figure 3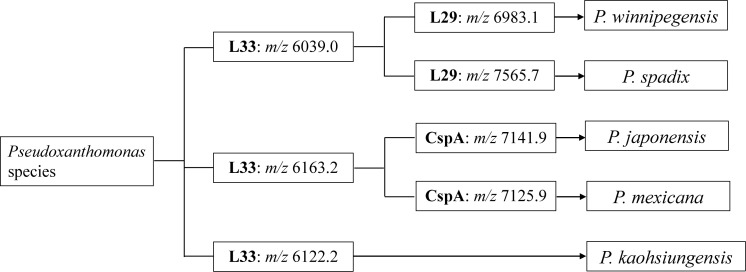 Figure 4
Figure 4| Strains | MicrobialTrack | MALDI biotyper | ||
|---|---|---|---|---|
| Closest species (Reliability) | Annotated ribosomal proteins | Annotated proteins except for ribosomal proteins | Closest species (score values) | |
| 24 | 11 | |||
| 21 | 17 | |||
| 19 | 8 | |||
| 24 | 17 | |||
| 23 | 19 | |||
| 28 | 19 | |||
| Biomarker protein | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Calculated masses ( | Average | SE | Peak numbers (n=5) | Calculated masses ( | Average | SE | Peak numbers (n=5) | Calculated masses ( | Average | SE | Peak numbers (n=5) | Calculated masses ( | Average | SE | Peak numbers (n=5) | Calculated masses ( | Average | SE | Peak numbers (n=5) | Calculated masses ( | Average | SE | Peak numbers (n=5) | |
| L36 | 4842.9 | 4842.9 | 0 | 5 | 4842.9 | 4842.7 | 0.16 | 5 | 4842.9 | 4842.9 | 0 | 5 | 4842.9 | 4842.9 | 0 | 5 | 4842.9 | 4842.9 | 0 | 5 | 4842.9 | 4842.9 | 0 | 5 |
| L34 | 5271.2 | 5270.4 | 0.23 | 5 | 5271.2 | 5271.4 | 0.09 | 5 | 5271.2 | 5270.1 | 0.48 | 5 | 5267.2 | 5267.4 | 0.05 | 5 | 5267.2 | 5267.3 | 0.03 | 5 | 5324.3 | 5324.6 | 0.11 | 5 |
| L33 | 6039.0 | 6039.9 | 0.31 | 5 | 6039.0 | 6040.6 | 0.16 | 5 | 6039.0 | 6039.0 | 0.10 | 5 | 6163.2 | 6163.5 | 0.06 | 5 | 6163.2 | 6163.2 | 0.06 | 5 | 6122.2 | 6123.6 | 0.09 | 5 |
| L32 | 6968.8 | NA | NA | 0 | 6968.8 | NA | NA | 0 | 6857.6 | 6859.2 | 0.35 | 5 | 7014.8 | NA | NA | 0 | 7002.8 | 7004.2 | 0.06 | 5 | 7000.8 | 7001.5 | 0.15 | 5 |
| L29 | 6983.1 | 6980.0 | 0.48 | 5 | 6983.1 | 6983.6 | 0.16 | 5 | 7565.7 | 7565.3 | 0.07 | 5 | 7257.5 | 7257.9 | 0.09 | 5 | 7296.6 | NA | NA | 0 | 7032.1 | 7032.5 | 0.15 | 5 |
| L30 | 7033.2 | 7033.6 | 0.21 | 5 | 7033.2 | 7033.3 | 0.18 | 5 | 7033.2 | 7032.8 | 0.11 | 5 | 7005.2 | 7005.9 | 0.14 | 5 | 7005.2 | NA | NA | 0 | 7074.2 | 7074.7 | 0.18 | 5 |
| CspA | 7155.9 | 7159.0 | 0.10 | 5 | 7155.9 | 7157.9 | 0.54 | 5 | 7183.9 | 7185.1 | 0.12 | 5 | 7141.9 | 7142.2 | 0.12 | 5 | 7125.9 | 7126.1 | 0.07 | 5 | 7178.9 | 7179.8 | 0.27 | 5 |
| L35 | 7296.6 | 7296.3 | 0.12 | 5 | 7296.6 | 7297.5 | 0.24 | 5 | 7296.6 | 7296.3 | 0.14 | 5 | 7296.6 | 7297.6 | 0.07 | 5 | 7282.6 | 7282.2 | 0.05 | 5 | 7368.7 | 7369.5 | 0.17 | 5 |
| Translation initiation factor IF-1 | 8273.6 | 8273.8 | 0.19 | 5 | 8273.6 | 8273.2 | 0.16 | 5 | 8245.6 | 8244.5 | 0.14 | 5 | 8245.6 | 8245.6 | 0.14 | 5 | 8245.6 | 8244.7 | 0.12 | 5 | 8285.6 | 8285.6 | 0.06 | 5 |
| S21 | 7997.8 | 7996.2 | 0.20 | 5 | 7997.8 | 7997.8 | 0.12 | 5 | 8483.8 | 8484.2 | 0.25 | 5 | 8483.8 | 8484.1 | 0.13 | 5 | 8483.8 | 8483.1 | 0.08 | 5 | 8423.3 | 8422.9 | 0.05 | 5 |
| L28 | 8897.2 | 8897.0 | 0.24 | 5 | 8897.2 | 8897.2 | 0.00 | 5 | 8897.2 | 8897.2 | 0.00 | 5 | 8878.2 | 8878.2 | 0.00 | 5 | 8895.3 | 8894.9 | 0.38 | 5 | 8785.0 | 8785.0 | 0.00 | 5 |
| L27 | 8898.1 | NA | NA | 0 | 8898.1 | NA | NA | 0 | 8997.3 | NA | NA | 0 | 8937.3 | 8938.0 | 0.27 | 5 | 8967.3 | 8967.1 | 0.08 | 5 | 8858.1 | 8858.0 | 0.07 | 5 |
| DNA-binding protein HU | 9289.6 | 9292.7 | 0.33 | 5 | 9289.6 | 9289.8 | 0.05 | 5 | 9259.5 | 9260.0 | 0.15 | 5 | 9444.9 | 9445.7 | 0.17 | 5 | 9444.9 | 9444.1 | 0.06 | 5 | 9484.0 | 9484.6 | 0.09 | 5 |
| S19 | 9823.5 | 9826.4 | 0.24 | 5 | 9823.5 | 9824.0 | 0.07 | 5 | 9895.6 | 9895.2 | 0.24 | 5 | 9795.5 | 9797.6 | 0.17 | 5 | 9795.5 | 9796.3 | 0.14 | 5 | 9595.3 | 9596.7 | 0.02 | 5 |
| S15 | 9954.5 | 9955.6 | 0.24 | 5 | 9954.5 | 9954.0 | 0.13 | 5 | 9879.3 | NA | NA | 0 | 9813.2 | NA | NA | 0 | 9811.2 | NA | NA | 0 | 9902.3 | 9903.0 | 0.04 | 5 |
| Co-chaperonin GroES | 10146.7 | 10148.8 | 0.54 | 5 | 10146.7 | 10146.5 | 0.05 | 5 | 10085.7 | 10086.8 | 0.19 | 5 | 10276.8 | 10278.4 | 0.16 | 5 | 10247.8 | 10247.8 | 0.19 | 5 | 10204.7 | 10205.7 | 0.18 | 5 |
| L23 | 11014.5 | 11016.4 | 0.17 | 5 | 11014.5 | 11012.2 | 0.54 | 4 | 11002.4 | 11002.9 | NA | 1 | 11111.7 | NA | NA | 0 | 11111.7 | 11113.0 | 0.42 | 5 | 10879.4 | 10879.0 | 0.40 | 2 |
| L24 | 11059.6 | 11059.8 | 0.30 | 5 | 11059.6 | NA | NA | 0 | 11113.7 | 11114.1 | 0.28 | 5 | 11073.6 | NA | NA | 0 | 11115.7 | NA | NA | 0 | 11113.7 | 11113.9 | 0.20 | 5 |
| S14 | 11372.2 | 11373.1 | 0.35 | 5 | 11372.2 | 11370.8 | 0.24 | 5 | 11400.2 | 11400.4 | 0.44 | 5 | 11368.2 | 11370.1 | 0.32 | 5 | 11386.2 | 11385.1 | 0.42 | 5 | 11235.0 | 11235.6 | 0.20 | 5 |
| S10 | 11572.3 | 11572.3 | 0.29 | 5 | 11572.3 | 11570.6 | 0.18 | 4 | 11572.3 | 11572.1 | 0.43 | 5 | 11600.4 | 11603.4 | 0.40 | 5 | 11584.4 | 11585.6 | 0.35 | 5 | 11512.3 | 11512.7 | 0.22 | 5 |
| L21 | 11860.7 | 11862.9 | 0.36 | 5 | 11860.7 | 11860.2 | 0.28 | 5 | 11870.7 | 11872.0 | NA | 1 | 10574.4 | NA | NA | 0 | 11475.3 | 11475.8 | 0.72 | 5 | 11314.1 | 11315.1 | 0.23 | 5 |
| L22 | 12317.4 | 12318.4 | 0.36 | 5 | 12317.4 | 12316.2 | 0.19 | 5 | 12224.3 | 12224.3 | NA | 1 | 12172.4 | 12171.1 | 0.60 | 2 | 12172.5 | NA | NA | 0 | 12018.2 | 12019.1 | 0.31 | 5 |
| L18 | 12572.4 | 12574.8 | 0.46 | 5 | 12572.4 | 12572.1 | 0.25 | 5 | 12641.5 | 12641.6 | 1.48 | 3 | 12687.5 | 12690.0 | 0.53 | 5 | 12641.5 | 12637.8 | 0.54 | 5 | 12596.5 | 12596.5 | 0.39 | 5 |
- —Japan Society for the Promotion of Science10.13039/501100001691
- —Kurozumi Medical Foundation10.13039/501100008660
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Identification and Susceptibility Testing · Insects and Parasite Interactions · Aquaculture disease management and microbiota
Introduction
1
Members of the genus Pseudoxanthomonas were, first described in 2000 by Finkmann et al. and are known to be Gram-negative, aerobic, rod-shaped, motile and oxidase-positive bacteria (Finkmann et al., 2000). To date, the genus Pseudoxanthomonas comprises 20 validly published species, which have been isolated from a variety of environmental sources, including soil and water samples1.
To our knowledge, only four reports have described Pseudoxanthomonas species isolated from human clinical samples. P. kaohsiungensis was isolated from a blood culture in Taiwan in 2003 (Kuo and Lee, 2018), P. japonensis from blood culture in Sweden in 19872, P. mexicana from a urine sample in Germany in 2002 (Thierry et al., 2004), and P. winnipegensis from respiratory sample in Canada in 2013 (Bernard et al., 2020).
In clinical laboratories, Pseudoxanthomonas species are identified using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS), but some species cannot be correctly identified because they are not registered in the database (Wang et al., 2014; Bernard et al., 2020). Although 16S rRNA genes and whole-genomes sequencing can identify Pseudoxanthomonas species correctly, they are difficult to use routinely due to its high cost and time-consuming procedures (Cutiño-Jiménez et al., 2020). The susceptibility patterns of Pseudoxanthomonas species are unclear, therefore it is important to accumulate surveillance data from clinical laboratories (Thierry et al., 2004; Bernard et al., 2020).
GPMsDB-dbtk is a new software for genomically predicting the theoretical protein mass database for mass spectrometry and MicrobialTrack is a software for identifying bacterial strains by MALDI-MS data (Sekiguchi et al., 2023; Nakagawa et al., 2025). The predicted proteins were experimentally validated to be the correspond proteins (Sun et al., 2006; Teramoto et al., 2019). In this study, we analyzed species identification using MicrobialTrack and evaluated three biomarker peaks, including L29, L33 and CspA, annotated by GPMsDB-dbtk to distinguish five Pseudoxthanthomonas species.
In this study, we first isolated P. winnipegensis in blood cultures that caused bacteremia. We attempted to identify five Pseudoxanthomonas species, including P. kaohsiungensis, P. japonensis, P. mexicana, P. spadix and P. winnipegensis that cause infections against humans by MALDI-MS peaks.
Materials and methods
2
Bacterial strains
2.1
P. winnipegensis strain JUPW001 was isolated from a blood sample of a 17-year-old woman patient at Juntendo University Hospital, Tokyo, Japan in September 2022. The patient was admitted for suspected central line-associated bloodstream infection due to recurrent fever and elevated C-reactive protein (CRP). Treatment with piperacillin/tazobactam (4.5 g/day) was started and her symptoms improved 2 days after onset. Two sets of blood cultures taken at the time of fever were positive after 36 hours. Three days after treatment, blood cultures were negative. The samples of the positive blood culture were stained and were cultured on AccuRate™ separated Sheep Blood Agar/Chocolate Agar EXII (Shimadzu Diagnostics Co., Kyoto, Japan) and BTB lactose agar (Eiken Chemical Co. Ltd., Tokyo, Japan) at 35°C. The type strain of P. winnipegensis NCTC 14396^T^ was obtained from National Collection of Type Cultures (Salisbury, UK). Type strains of P. kaohsiungensis CCUG 55854^T^, P. mexicana CCUG 49454^T^, P. japonensis CCUG 48231^T^, and P. spadix CCUG 53828^T^ were obtained from the Culture Collection, University of Göteborg (CCUG), Sweden. All isolates were cultured aerobically at 35°C on 5% sheep blood agar plates (Becton, Dickinson Diagnostic Systems, MD, USA) or Luria-Bertani broth under aerobic conditions at 35 °C.
Whole-genome sequencing
2.2
The genomic DNA of the clinical isolate was extracted using DNeasy blood and tissue kits (Qiagen, Tokyo, Japan) and Genomic-tips 20/G (Qiagen). For short-read sequencing, DNA library was prepared using Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA, USA). The genomes were sequenced on the Illumina MiniSeq platform (300 cycles). The raw reads were trimmed and assembled using CLC Genomic Workbench version 10.0.1 (CLC bio, Aarhus, Denmark). For long-read sequencing, DNA library was prepared using Native Barcoding Kit 24 V14 SQK-NBD 114.24 (Oxford Nanopore Technologies, Oxford, UK). Sequencing was performed on R10.4.1 flowcell using MinION Mk1B (Oxford Nanopore Technologies). MinKNOW (version 24.02.8) and Guppy (version 7.3.11) (Oxford Nanopore Technologies) were used for base calling and adapter trimming of raw data. The sequences determined by MiniSeq and MinION were assembled using Unicycler v0.5.0 (Wick et al., 2017). The genome relatedness of the relevant strains was estimated using an average nucleotide identity (ANI) calculator (Yoon et al., 2017) and a Type (Strain) Genome Sever3. ANI values were calculated using reference genomes from P. winnipegensis (NCTC 14396^T^; genome accession number GCF_004283755). Virulence factors were detected by the virulence factor database (VFDB4) and MacSyfinder v2.0 (Abby et al., 2014) for secretion systems (SS).
Phylogenetic analysis
2.3
Genome completeness and contamination were assessed using CheckM2 v1.0.1 with lineage_wf and default settings (Parks et al., 2015). A phylogenetic tree was constructed using the kSNP4 software based on pangenome SNPs5 (Gardner et al., 2015), and visualized using iTol ver.66. The strains of P. broegbernensis (DSM 12573^T^; NZ_JACHGU010000001), P. daejeonensis (DSM 17801^T^; NZ_PDWN01000010), P. dokdonensis (DSM 21858^T^; NZ_LDJL01000001), P. gei (KCTC 32298^T^; NZ_QOVG01000010), P. helianthin (NBRC 110414^T^; NZ_JAGKTC010000001), P. indica (CCM 7430^T^; NZ_BMCL01000001), P. japonensis (CCUG 48231^T^; NZ_PDWW01000010), P. kalamensis (DSM 18571^T^; NZ_PDWQ01000001), P. kaohsiungensis (CCUG 55854^T^; NZ_PDWO01000010), P. koreensis (KCTC 12208^T^; NZ_PDWM01000010), P. mexicana (CCUG 49454^T^; NZ_PDWV01000100), P. putridarboris (LMG 25968^T^; NZ_JBBWWT010000001), P. sacheonensis (DSM 19373^T^; NZ_PDWS01000010), P. sangjuensis (DSM 28345^T^; NZ_PDWR01000010), P. spadix (CCUG 53828^T^; NZ_RDQN01000001), P. suwonensis (DSM 17175^T^; NZ_PDWP01000010), P. taiwanensis (DSM 22914^T^; NZ_PDWK01000100), P. winnipegensis (NCTC 14396^T^; NZ_SHMH01000001), P. wuyuanensis (DSM 100640^T^; NZ_PDWU01000010), P. yeongjuensis (DSM 18204^T^; NZ_PDWT01000010), Stenotrophomonas maltophilia (NCTC 10257^T^; GCF_900186865.1) were used as reference strains.
Calculation of the theoretical mass of Pseudoxanthomonas winnipegensis for MALDI-MS proteotyping
2.4
Theoretical masses of proteins encoded in the genomes of Pseudoxanthomonas were calculated for the following genomes as part of the development of a genomically predicted protein mass database toolkit (GPMsDB-tk): P. japonensis (CCUG 48231^T^; NZ_PDWW01000010), P. kaohsiungensis (CCUG 55854^T^; NZ_PDWO01000010), P. mexicana (CCUG 49454^T^; NZ_PDWV01000100), P. spadix (CCUG 53828^T^; NZ_RDQN01000001), and P. winnipegensis (NCTC 14396^T^; NZ_SHMH01000001) (Sekiguchi et al., 2023). The genome sequences were obtained from the NCBI database7. The prediction of genes from the genomes obtained in this study was performed using GPMsDB-dbtk v1.0.18 (Sekiguchi et al., 2023).
Bacterial sample preparation for MALDI-MS
2.5
Alpha-cyano-4-hydroxycinnamic acid (CHCA) was used as a matrix. To prepare this matrix solution, 10 mg of 4-CHCA was dissolved in 1 mL of solvent consisting of 1% (v/v) trifluoroacetic acid, 35% (v/v) ethanol, 15% (v/v) acetonitrile, and milliQ water. A full loop of bacterial cells was dispersed in 200 μL of distilled water in a microtube and mixed with 800 μL of ethanol. The suspensions were briefly vortexed and centrifuged at 15, 000 g for 2 min. The pellets were then dried for 5 min. The pellets were suspended in 50 μL of 70% formic acid, vortexed, suspended in 50 μL of acetonitrile, and centrifuged at 15, 000 g for 2 min. Supernatants were analyzed by MALDI-MS according to the manufacturer’s instruction.
MALDI-MS measurement
2.6
MALDI-MS measurements were performed in positive linear mode using MALDI-8020 RUO (Shimadzu Corporation, Kyoto, Japan) and Microflex LT/SH (Bruker Daltonics, Germany) equipped with a 200 Hz Nd: YAG laser (355 nm) and 60 Hz nitrogen laser (337 nm), respectively. Before sample analysis, the MALDI-MS instrument was mass-calibrated externally using six peaks with m/z 4365.4, 5381.4, 6411.6, 7274.0, 8369.8, and 10300.1 from Escherichia coli DH5α. More than five individual mass spectra were acquired for each bacterial extract in the range of m/z 2, 000-20, 000. The assignment of the peak was performed using eMSTAT Solution™ software (Shimadzu Corporation). Species identifications were performed by the MicrobialTrack software v1.1.0 (Shimadzu Corporation) (Sekiguchi et al., 2023; Nakagawa et al., 2025) and MBT Compass 4.1 with Microflex LT/SH (Bruker Daltonics).
Cluster analysis for Pseudoxanthomonas type strains using L29, L33 and CspA
2.7
For biomarker validation, the theoretical mass of L29, L33 and CspA was calculated for 15 Pseudoxanthomonas type strains: P. broegbernensis (DSM 12573^T^; NZ_JACHGU010000001), P. daejeonensis (DSM 17801^T^; NZ_PDWN01000010), P. dokdonensis (DSM 21858^T^; NZ_LDJL01000001), P. gei (KCTC 32298^T^; NZ_QOVG01000010), P. helianthin (NBRC 110414^T^; NZ_JAGKTC010000001), P. indica (CCM 7430^T^; NZ_BMCL01000001), P. kalamensis (DSM 18571^T^; NZ_PDWQ01000001), P. koreensis (KCTC 12208^T^; NZ_PDWM01000010), P. putridarboris (LMG 25968^T^; NZ_JBBWWT010000001), P. sacheonensis (DSM 19373^T^; NZ_PDWS01000010), P. sangjuensis (DSM 28345^T^; NZ_PDWR01000010), P. suwonensis (DSM 17175^T^; NZ_PDWP01000010), P. taiwanensis (DSM 22914^T^; NZ_PDWK01000100), P. wuyuanensis (DSM 100640^T^; NZ_PDWU01000010), and P. yeongjuensis (DSM 18204^T^; NZ_PDWT01000010). A phylogenetic tree was constructed using SRplot with the unweighted pair group method with arithmetic mean (UPGMA) (Tang et al., 2023).
Accession numbers
2.8
The whole-genome sequence of P. winnipegensis JUPW001 obtained in this study has been deposited in GenBank under the accession number AP0447399.
Results
3
Identification of P. winnipegensis JUPW001
3.1
Samples of the positive blood culture were stained as Gram-negative rod. After 24 h of incubation, a large number of oxidase-positive and yellow-colored colonies were observed on the agars. The isolate was identified as “Pseudoxanthomonas species” using MALDI-MS with MBT Compass 4.1 by Microflex LT/SH (Bruker Daltonics).
The ANI value using whole-genome sequence of the isolate was 97.4% identical to the sequence of P. winnipegensis (NCTC 14396^T^; NZ_SHMH01000001). Therefore, the isolate was confirmed to be P. winnipegensis and designated as P. winnipegensis JUPW001.
P. winnipegensis JUPW001 had flagellum, type 1 secretion system (T1SS), T2SS, T4SS, T5SS and type IVa pilus (T4aP). In particular, T4SS included VirB2 to VirB6 and VirB8 to VirB11.
Phylogenic analysis
3.2
As shown in Figure 1, the phylogeny was separated by the genus level between Pseudoxanthomonas and Stenotrophomonas. The phylogenetic tree revealed three clades: A, B, and C. P. winnipegensis JUPW001 belonged to clade B and the species was close to P. spadix (Figure 1). In clade A, P. kaohsiungensis (GenBank accession no. NZ_PDWO01000010) was isolated from humans (Kuo and Lee, 2018). Other previously reported clinical isolates, P. japonensis (accession no. NZ_PDWW01000010) and P. mexicana (accession no. NZ_PDWV01000100), belonged to clade C (Thierry et al., 2004). The other species were mainly isolated from soil or water environments.
Phylogenetic tree of one clinical strain and 20 type strains of Pseudoxanthomonas species. Phylogenetic trees were constructed using kSNP4 software based on pangenome SNPs (footnote 5) (Gardner et al., 2015) and visualized using iTol ver.6 (footnote 6).
MALDI-MS analysis of Pseudoxanthomonas species
3.3
The bacterial identification for Pseudoxanthomas species was performed using MicrobialTrack, which is a software for peak matching by predicted mass values using the annotated protein sequences. The MicrobialTrack analysis revealed that P. winnipegensis JUPW001, P. winnipegensis NCTC 14396^T^, P. spadix CCUG 53828^T^, P. japonensis CCUG 48231^T^, P. mexicana CCUG 49454^T^, and P. kaohsiungensis CCUG 55854^T^ were correctly identified with high reliability (Table 1). In this analysis, the number of ribosomal proteins peaks in each strain were from 19 to 28 and annotated proteins except for ribosomal proteins were from 8 to 19. In contrast, Microflex LT/SH analysis using the Biotyper database correctly identified P. kaohsiungensis, P. mexicana, and P. spadix at the species level, whereas P. japonensis was misidentified and P. winnipegensis was classified only at the genus level (Table 1).
Biomarker proteins to distinguish Pseudoxanthomonas species
3.4
The candidate mass peaks of MALDI-MS analysis for the five type strains, including P. kaohsiungensis, P. japonensis, P. mexicana, P. spadix and P. winnipegensis, are shown in Figure 2. In the MALDI-MS profiles, 21 major mass peaks were commonly detected and successfully annotated with predicted protein names in P. winnipegensis JUPW001 and NCTC 14396^T^ (Table 2). Of these 21 annotated peaks, 17 were predicted as ribosomal subunit proteins, and the remaining four were co-chaperonin GroES, cold shock-like protein (CspA), DNA-binding protein HU and translation initiation factor IF-1. As shown in Table 2, compared to the theoretical mass peaks of P. winnipegensis NCTC 14396^T^, most of the peaks were different from the theoretical mass peaks of P. spadix CCUG 53828^T^, P. japonensis CCUG 48231^T^, P. mexicana CCUG 49454^T^, and P. kaohsiungensis CCUG 55854^T^. To distinguish five Pseudoxanthomonas at the species level using MALDI-MS, the combination of appropriate peaks of ribosomal L29, L33 and CspA were selected as biomarkers.
Representative mass spectra of Pseudoxanthomonas species, including P. winnipegensis JUPW001 (A), P. winnipegensis NCTC 14396T(B), P. spadix CCUG 53828T(C), P. japonensis CCUG 48231T(D), P. mexicana CCUG 49454T(E), and P. kaohsiungensis CCUG 55854T(F). Upper figure indicates mass spectra from m/z 4, 500 to 12, 500. The annotated peaks indicate the assigned peaks based on the calculated masses within the tolerance at 500 ppm. Lower figure indicates amplifications of the m/z 5, 900 to 7, 800 section of mass spectra and variations of m/z values for peaks ribosomal L29, L33 and CspA.
As shown in Figure 2 and Table 2, the corresponding theoretical peaks of ribosomal protein L29 were m/z 6983.1 for P. winnipegensis NCTC 14396^T^; m/z 7565.7 for P. spadix CCUG 48231^T^; m/z 7257.5 for P. japonensis CCUG 48231^T^ and m/z 7032.1 for P. kaohsiungensis CCUG 55854^T^ (Figure 2; Table 2). The peak at m/z 7296.6 for L29 of P. mexicana CCUG 49454^T^ were not detected, although the theoretical peaks were calculated by GPMsDB-dbtk. The corresponding theoretical peaks of the ribosomal protein L33 were m/z 6039.0 for P. winnipegensis NCTC 14396^T^ and P. spadix CCUG 48231^T^; m/z 6163.2 for P. japonensis CCUG 48231^T^ and P. mexicana CCUG 49454^T^; m/z 6122.2 for P. kaohsiungensis CCUG 55854^T^ (Figure 2; Table 2). The corresponding theoretical peaks of CspA were m/z 7155.9 for P. winnipegensis NCTC 14396^T^; m/z 7183.9 for P. spadix CCUG 48231^T^; m/z 7141.9 for P. japonensis CCUG 48231^T^; m/z 7125.9 for P. mexicana CCUG 49454^T^ and m/z 7178.9 for P. kaohsiungensis CCUG 55854^T^ (Figure 2; Table 2).
Compared to the amino acid sequence of L29 in P. winnipegensis, there were 6 amino acid substitutions in P. spadix, 8 in P. japonensis, 8 in P. mexicana and 11 in P. kaohsiungensis (Figure 3A). Compared to the amino acid sequence of L33 in P. winnipegensis, there were 5 substitutions in P. japonensis, 5 in P. mexicana, and 6 in P. kaohsiungensis (Figure 3B). Compared to the amino acid sequence of CspA of P. winnipegensis, there were 1 substitution in P. spadix, 4 in P. japonensis, 3 in P. mexicana, and 10 in P. kaohsiungensis (Figure 3C).
Alignment of amino acid sequences of the ribosomal protein L29 (A), L33 (B) and CspA (C) of Pseudoxanthomonas species. Amino acid substitutions are shaded in gray.
MALDI-MS proteotyping was able to distinguish P. winnipegensis, P. spadix, P. japonensis, P. mexicana and P. kaohsiungensis using the three biomarkers, including L29, L33 and CspA.
Cluster analysis for 20 Pseudoxanthomonas type strains
3.5
Cluster analysis using L29, L33 and CspA revealed that 20 Pseudoxanthomonas type strains were distinguished by the three theoretical mass peaks (Supplementary Table S1 and Supplementary Figure S1). The close cluster such as P. japonensis, P. putridarboris and P. wuyuanensis were theoretically separated by L29 or CspA, whereas P. sacheonensis and P. yeongjuensis were theoretically separated by CspA (Supplementary Figure S1).
Discussion
4
Pseudoxanthomonas is a relatively recently characterized genus and reports of human infection remain rare. Accurate and rapid diagnosis of Pseudoxanthomonas infections in immunocompromised patients is critical, because it is known that the species have several virulence factors, including T1SS, T2SS, T3SS, T4SS and T4aP. Especially, T4SS plays crucial roles in pathogens in the delivery of effector proteins (Green and Mecsas, 2016; Costa et al., 2024). The T4SS signature protein, VirB4, in Xanthomonadaceae is known to kill other bacterial cells and have advantage to competitive growth in mixed bacterial communities (Souza et al., 2015). VirB4 is also known the only ubiquitous protein with recognizable homologs in all known T4SS (Guglielmini et al., 2014). The T4SS effector protein, VirB10, in S. maltophilia is known to reduce apoptotic activity and promote its growth (Nas et al., 2019). P. winnipegensis JUPW001 harbored both of VirB4 and VirB10 protein.
The biomarker peaks of L29, L33 and CspA are useful for distinguishing Pseudoxanthomonas at the species level. Our study suggests that at least five Pseudoxanthomonas species can be separated using MALDI-MS, even if MicrobialTrack is not installed. Figure 4 shows the workflow for the rapid identification of Pseudoxanthomonas species by MALDI-MS, describing L33 for three groups: P. winnipegensis/P. spadix, P. japonensis/P. mexicana and P. kaohsiungensis, L29 for P. winnipegensis and P. spadix, and CspA for P. japonensis and P. mexicana. Analysis using the theoretical masses of the other 15 Pseudoxanthomonas species revealed that it is possible to classify 20 Pseudoxanthomonas species, including P. kaohsiungensis, P. japonensis, P. mexicana, P. spadix and P. winnipegensis, using the three biomarker peaks (Supplementary Table S1 and Supplementary Figure S1).
Workflow for the identification of phylotypes of five Pseudoxanthomonas species by MALDI-MS proteotyping.
The identification of MALDI-MS proteotyping using the MicrobialTrack will be useful for species lacking mass spectral reference libraries such as Pseudoxanthomonas species. Previous study on the P. winnipegensis identification using MALDI-MS have shown that the misidentification as P. spadix with low scores ranging from 1.22 to 1.38 in Biotyper analysis (Bernard et al., 2020). Most of Pseudoxanthomonas species can identify at the genus-level, and only three species, including P. indica, P. mexicana and P. spadix, can identify at the species-level using MALDI reference libraries of MBT Compass 4.1 installed in Biotyper (Table 1). It is possible to identify species-level of Pseudoxanthomonas using MicrobialTrack if the exact genome sequences and the enough quality of the measured MALDI spectra are obtained from tested bacteria.
This study has a few limitations: first, only one clinical strain was analyzed obtained in this study, and five of 20 species were tested using MALDI-MS. More data on clinical isolates of Pseudoxanthomonas species should be collected and the prospective validations are important.
In conclusion, this study reports the first case of human infection caused by P. winnipegensis and highlights the potential utility of MALDI-MS proteotyping for rapid and accurate species-level identification of Pseudoxanthomonas clinical isolates.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abby S. S. Néron B. Ménager H. Touchon M. Rocha E. P. (2014). Mac Sy Finder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. Plo S One 9, e 110726. doi: 10.1371/journal.pone.0110726, PMID: 25330359 PMC 4201578 · doi ↗ · pubmed ↗
- 2Bernard K. A. Vachon A. Pacheco A. L. Burdz T. Wiebe D. Beniac D. R. . (2020). Pseudoxanthomonas winnipegensis sp. nov., derived from human clinical materials and recovered from cystic fibrosis and other patient types in Canada, and emendation of Pseudoxanthomonas spadix Young et al., 2007. Int. J. Syst. Evol. Microbiol. 70, 6313–6322. doi: 10.1099/ijsem.0.004533, PMID: 33118921 · doi ↗ · pubmed ↗
- 3Costa T. R. D. Patkowski J. B. MacéK. Christie P. J. Waksman G. (2024). Structural and functional diversity of type IV secretion systems. Nat. Rev. Microbiol. 22, 170–185. doi: 10.1038/s 41579-023-00974-3, PMID: 37814112 PMC 11290344 · doi ↗ · pubmed ↗
- 4Cutiño-Jiménez A. M. Menck C. F. M. Cambas Y. T. Díaz-Pérez J. C. (2020). Protein signatures to identify the different genera within the Xanthomonadaceae family. Braz. J. Microbiol. 51, 1515–1526. doi: 10.1007/s 42770-020-00304-2, PMID: 32488841 PMC 7688752 · doi ↗ · pubmed ↗
- 5Finkmann W. Altendorf K. Stackebrandt E. Lipski A. (2000). Characterization of N 2O-producing Xanthomonas-like isolates from biofilters as Stenotrophomonas nitritireducens sp. nov., Luteimonas mephitis gen. nov., sp. nov. and Pseudoxanthomonas broegbernensis gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 50 Pt 1, 273–282. doi: 10.1099/00207713-50-1-273, PMID: 10826814 · doi ↗ · pubmed ↗
- 6Gardner S. N. Slezak T. Hall B. G. (2015). k SNP 3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv 271, PMID: 25913206 · doi ↗ · pubmed ↗
- 7Green E. R. Mecsas J. (2016). Bacterial secretion systems: an overview. Microbiol. Spectr. 4. doi: 10.1128/microbiolspec.VMBF-0012-2015, PMID: 26999395 PMC 4804464 · doi ↗ · pubmed ↗
- 8Guglielmini J. Néron B. Abby S. S. Garcillán-Barcia M. P. de la Cruz F. Rocha E. P. (2014). Key components of the eight classes of type IV secretion systems involved in bacterial conjugation or protein secretion. Nucleic Acids Res. 42, 5715–5727. doi: 10.1093/nar/gku 194, PMID: 24623814 PMC 4027160 · doi ↗ · pubmed ↗