Detection, genetic characterization and reassortment analysis of Tribeč virus
Katarína Loziaková Peňazziová, Petra Schusterová, Soňa Pivka, Zuzana Pačanská, Tomáš Szemes, Boris Klempa, Tomáš Csank

TL;DR
Researchers identified and genetically analyzed Tribeč virus in ticks from Slovakia, revealing its genetic diversity and potential for reassortment.
Contribution
A newly developed RT-PCR assay enabled detection of Tribeč virus in ticks, and whole-genome sequencing revealed genetic variability and reassortment.
Findings
Tribeč virus was detected in Ixodes ricinus ticks collected from a goat using a new RT-PCR assay.
Whole-genome sequencing showed high sequence identity with other TRBV and Muko virus strains, but segment 4 suggests reassortment.
Phylogenetic analysis revealed distinct clades for TRBV, with segment 10 showing the highest variability.
Abstract
This study reports the detection and genetic characterization of Tribeč virus (TRBV), a tick-borne orbivirus of zoonotic relevance. The virus was identified in Ixodes ricinus ticks collected from a goat using a newly developed RT-PCR assay. Goats grazing in the study area showed seroconversion, with low titre neutralization antibody response. The new isolate, designated 16.C/2016/Dubrava/SVK, was successfully cultured in Vero E6 cells and detected by immunofluorescence. Replication efficiency varied across rodent, human, and bovine cell lines. Whole-genome sequencing revealed that most genome segments shared high sequence identity with other TRBV strains and with Muko virus (MUKV) strains from Japan. In contrast, phylogenetic analysis based on the VP4 outer capsid protein indicated that the TRBV clade is more closely related to Kemerovo viruses from Russia. Segment 10 showed the highest…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
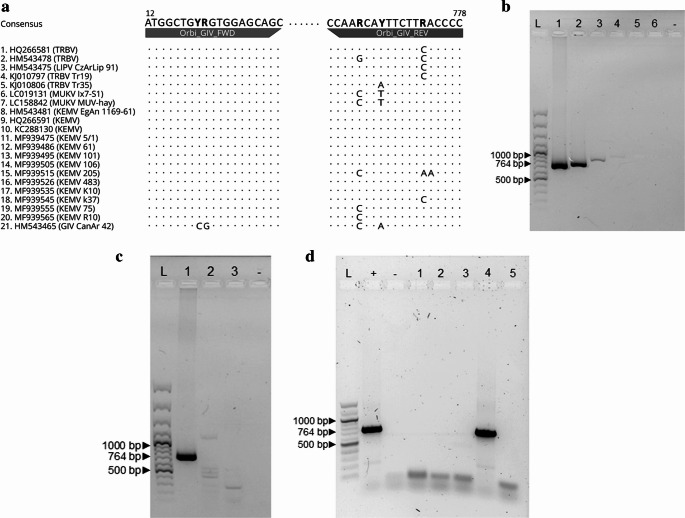 Figure 1
Figure 1 Figure 1
Figure 1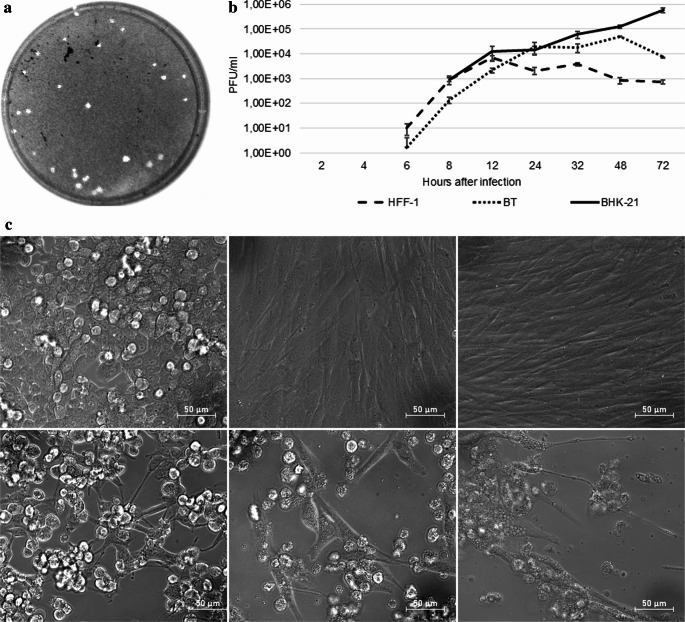 Figure 2
Figure 2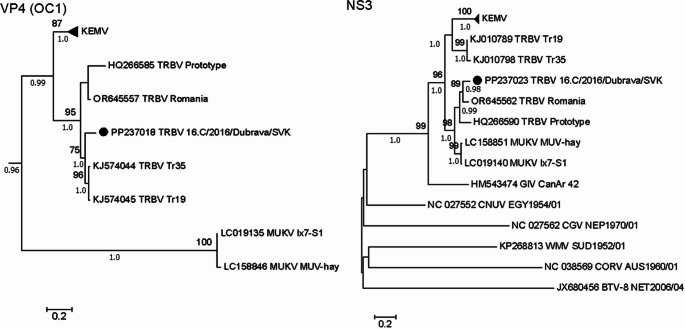 Figure 3
Figure 3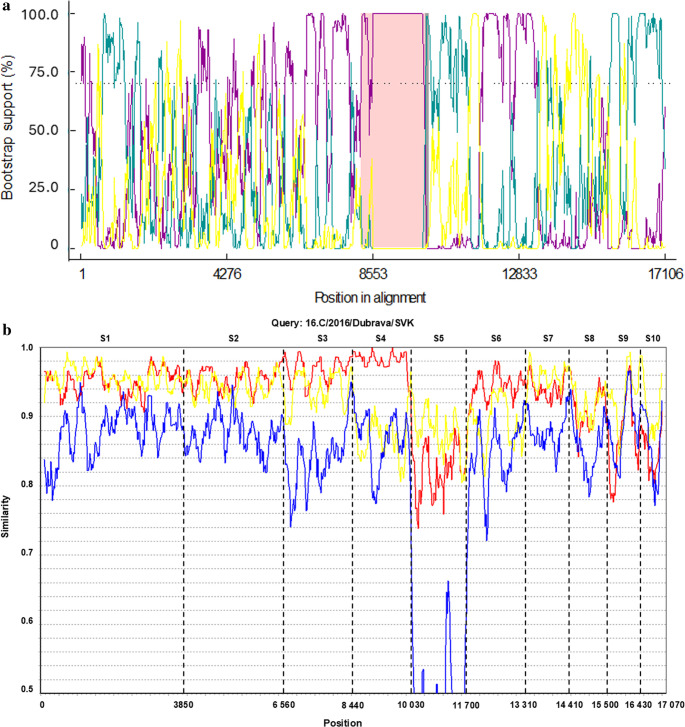 Figure 4
Figure 4- —University of Veterinary Medicine and Pharmacy
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVector-Borne Animal Diseases · Viral Infections and Vectors · Vector-borne infectious diseases
Introduction
Orbiviruses (genus Orbivirus, family Sedoreoviridae) are a diverse group of double-stranded RNA (dsRNA) viruses with segmented genomes. According to the 2022 virus taxonomy release, the genus Orbivirus comprises twenty-two recognized species. Among these, the tick-borne species include Orbivirus chenudaense (formerly Chenuda virus), Orbivirus chobarense (Chobar Gorge virus), Orbivirus wadmedaniense (Wad Medani virus), Orbivirus saintcroixense (St. Croix River virus), and Orbivirus magninsulae (Great Island virus; GIV) [1].
GIV comprises at least 36 serotypes, of which Tribeč virus (TRBV), Lipovník virus (LIPV), and Kemerovo virus (KEMV) have reported zoonotic potential [2–5]. These viruses are maintained in nature through biological transmission between ticks and reservoir hosts, primarily rodents. Under natural conditions, their main vectors are Ixodes ricinus [6–8], Ixodes persulcatus and Ixodes pavlovskyi [2, 9–12]. However, virus detection has also been reported in Haemaphysalis punctata [13]. Additionally, under laboratory conditions, Dermacentor marginatus has demonstrated the ability to transmit the virus [14].
Rodents are considered the primary reservoirs of tick-borne orbiviruses; however, serological studies indicate a broader host range, including birds [15, 16], hares [17], goats [6], and both domestic and wild ruminants [18, 19]. Although animal diseases caused by the GIV serogroup have not been reported under natural conditions, experimental studies indicate that these viruses can induce neuropathology in susceptible animal models. In rhesus macaques, intracerebral inoculation with TRBV or KEMV was associated with inflammatory changes in the brainstem and meninges, as well as virus detection in cerebrospinal fluid [20, 21]. In suckling mice, intracerebral inoculation with TRBV resulted in encephalitis-like clinical signs by day 4 post-infection, whereas subcutaneous inoculation caused inapparent infection without clinical signs [22]. In interferon-α/β receptor-deficient (IFNAR^−/−^) mice, low-titre (≤ 10² plaque forming units; PFU) KEMV infection caused no clinical signs. In contrast, higher titre (10³–10⁴ PFU) infection was associated with viraemia and clinical manifestations [23]. In humans, KEMV, LIPV, and TRBV infections are most often asymptomatic; however, previous reports have linked these viruses to cases of neurological disorders of unknown aetiology [4, 5].
The genome of tick-borne orbiviruses consists of ten linear dsRNA segments arranged in order of decreasing molecular weight. It encodes seven structural proteins (VP1, VP2, VP3, VP4, VP5, VP6/VP6a, VP7) and five non-structural proteins (NS1, NS2, NS3/NS3a, NS4, NS5). Most genome segments encode a single protein through one open reading frame (ORF) [1, 24–26]. The exceptions are segments 9 and 10, which encode NS4 and NS5 proteins via secondary small ORFs [26, 27].
The orbivirus core particle is enclosed within a double-shelled icosahedral capsid. The inner shell comprises a sub-core and core-surface layers formed by VP2(T2) and VP7(T13) proteins, respectively. The outer shell is formed by VP4(OC1) and VP5(OC2) proteins, which mediate virion attachment and penetration into host cells. Serogroups are defined based on similarities in VP2(T2) and VP7(T13), whereas serotype classification primarily relies on VP4(OC1) and VP5(OC2). These outer capsid proteins are under immune selection pressure in the host system. Within the core lumen, the genomic dsRNA segments are closely associated with multiple transcription complexes composed of three structural proteins—VP1 (RNA-dependent RNA polymerase; RdRp), VP3 (capping enzyme; Cap), and VP6 (helicase; Hel). These complexes are anchored to the inner surface of the sub-core [1, 28].
Reassortment is an evolutionary mechanism in segmented RNA viruses, where viruses of the same species simultaneously infect a host cell [29]. During the co-infection, their genome segments can be mixed and packed into newly assembled progeny virions. This process results in genetically distinct strains derived from the parental viruses. Novel genome segment combinations may lead to new phenotypes and significantly impact immune evasion, host or vector range, transmissibility, and virulence or pathogenicity [30–33]. While reassortment has been extensively studied in some segmented RNA viruses, such as influenza viruses and rotaviruses, it remains poorly characterized in tick-borne orbiviruses. To date, evidence is largely limited to laboratory observations [34, 35].
Given the limited knowledge, our study aimed the detection and isolation of TRBV from ticks collected from grazing goats. The new isolate was genetically characterized and analysed for evidence of reassortment. Its replication dynamics were evaluated in rodent, bovine, and human cell lines. In addition, serological investigation was performed in natural host.
Materials and methods
Animals and blood collection
In April 2016, a small-scale study was conducted to investigate the occurrence of tick-borne orbiviruses. Viruses were screened in ticks collected from clinically healthy goats that grazed annually on pasture from late March to mid-October. Goats were selected because domestic ruminants can serve as sentinel animals for exposure to tick-borne viruses in endemic areas [6]. The animals were housed on a small backyard farm in Dúbrava locality (Prešov county).
At the time of blood sampling in 2016, 11 animals were available: five adult goats and six kids. However, later during the year, four kids were removed from the herd while one buck was imported, resulting in eight animals available for sampling in 2017. In 2018, one additional goat was imported, and nine animals were sampled. The number, age, and sex of the goats in each year are summarized in Table 1.Table 1. Screening of goat serum samples for TRBV neutralizing antibodiesAnimalSexAge in the year of first blood collection2016201720181F4 y-1:201:102M3 m-1:10-3M3 m-NANA4F3 y-1:101:105M3 m---6F3 y-1:20-7M3 m-NANA8F1 y--1:109F1 y-1:201:1010F3 m-NANA11F3 m-NANA12M3 yNA-1:1013F2 yNANA-F female, M male, y year, m months, - – negative, NA not available
Blood was collected annually in April from the jugular vein (vena jugularis) by the local veterinarian. After clotting, samples were transported to the laboratory. Serum was separated by centrifugation at 1500 relative centrifugal force (RCF) at 4 °C and stored at −20 °C for later use in the neutralization test.
Tick collection and pooling
The animals were examined for tick infestation only during the first blood collection. The ticks were not pooled according to the animals from which they were collected. Ticks were stored at −80 °C until sorting. Overall, 102 Ixodes ricinus ticks were divided based on sex, development stage and engorgement [non-engorged female (n = 42); engorged female (n = 15); males (n = 15); non-engorged nymphs (n = 10) and engorged nymphs (n = 20)] into 33 pools. The pools contained up to 10 unfed or 5 engorged nymphs, up to 5 unfed adults and the engorged females were processed individually. Pool sizes were standardized to limit cytotoxicity of tick homogenates and to reduce the amount of host blood in engorged ticks, which may otherwise compromise downstream examinations.
Processing of tick homogenates
Ticks were washed in 70% ethanol for 10 min, then twice rinsed in nuclease free water. Ticks were homogenized in tubes with three 2.8 mm stainless steel beads (Bertin Technologies, France) and 700 µl of Eagle´s Minimal Essential Medium (BIOWEST, France – EMEM) supplemented with 1% foetal bovine serum (Biochrom AG, Germany – FBS), 1% of penicillin (6 g/l) and streptomycin (10 g/l) mixture (BIOWEST – P/S), 0.1% of gentamicin sulphate (50 mg/ml, BIOWEST – GNM), 1% of amphotericin B (250 µg/ml, BIOWEST – AMF) and 1% non-essential amino acids (PAN BIOTECH – NEAA) by Precellys 24 Dual (Bertin Technologies) at 6000 RPM with 3 × 10 s rounds and 15 s pause. Homogenates were clarified by centrifugation (4000 RCF for 10 min at 4 °C), aliquoted and stored at −80 °C until RNA extraction and virus isolation.
Cell lines
Baby hamster kidney (BHK-21 C-13, ATCC CCL-10), human foreskin fibroblast (HFF-1, ATCC SCRC-1041) and bovine turbinate (BT, ATCC CRL-1390) cells were cultivated in Dulbecco´s minimal essential medium (BIOWEST, France – DMEM) and African green monkey kidney epithelial cells (Vero E6, ATCC CRL-1586) in EMEM. The media were supplemented by 10% FBS, 1% of 200 mM stable glutamine (200 mM, PAN BIOTECH, Germany), 1% of P/S, 0.1% of GNM, 1% of AMF and 1% NEAA. All cell cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO₂.
Virus isolation
One-day-old Vero E6 monolayer, cultivated in a T25 flask (Greiner Bio One, Germany), was inoculated with 200 µl of the tick suspension that tested positive by reverse transcription polymerase chain reaction (RT-PCR). Viruses were allowed to adsorb for 1 h at 37 °C under 5% CO₂ atmosphere. The monolayer was washed once with EMEM and fresh 1% EMEM with antibiotics was added. Cells were maintained at 37 °C with 5% CO₂ and monitored daily for cytopathic effect (CPE). The monolayer exhibiting CPE was freeze/thawed once to release cell-associated virus. Cultures were then centrifuged at 4000 RCF for 10 min at 4 °C. The supernatants were aliquoted and stored at −80 °C until further analysis.
Primer design
Twenty-one RNA sequences of segment 1 from TRBV, LIPV, MUKV, KEMV, and GIV were downloaded from the National Center for Biotechnology Information database (Fig. 1a). Complete coding sequences were aligned using Clustal Omega 1.2.2 [36] within the Geneious 9.1.8 software (Biomatters, New Zealand). Based on the prototype TRBV strain segment 1 (HQ266581), the primers were designed to flank a 767-base pair (bp) amplicon. Owing to variability in the binding sites, degenerate bases were included. The primers were named and sequenced as follows: Orbi_GIV_FWD 5´-ATGGCTGYRGTGGAGCAGC-3´ and Orbi_GIV_REV 5´-CCCCARTTCTTYACRAACC-3´. The primers were designed using Primer 3 v2.3.4 [37–39] within Geneious 9.1.8 software.Fig. 1. Detection of TRBV 16.C/2016/Dubrava/SVK by RT-PCR, immunofluorescence and isolation. (a) Scheme of the annealing site of primers designed for detection of TRBV, LIPV, MUKV, KEMV and GIV. The accession numbers and strain designation of GIV group orbiviruses used for primer design are depicted. Bold letters in the primer sequences are degenerated nucleotides. (b) Sensitivity of the RT-PCR tested with the prototype TRBV strain (HQ266581): L – GeneRuler 100 bp Plus DNA Ladder (Thermo Scientific, Lithuania); 1–1.5 × 10^5^ PFU/ml; 2–1.5 × 10^4^ PFU/ml; 3–1.5 × 10^3^ PFU/ml; 4–1.5 × 10^2^ PFU/ml; 5–1.5 × 10^1^ PFU/ml; 6–1.5 × 10^0^ PFU/ml; - – negative control (water). (c) Specificity of the RT-PCR: L − 100 bp Plus DNA Ladder; 1 – prototype TRBV strain; 2 – Uukuniemi virus strain 37.C/2020/Dubrava/SVK (PX853966); 3 – TBEV strain Hypr (Ref-SKU: 008 N-EVA442); - – negative control (water). (d) DNA band of expected size in agarose gel after RT-PCR examination of ticks for orbiviruses: L – 100 bp Plus DNA Ladder; + – positive control (prototype TRBV strain); - – negative control (water); lines 1, 2, 3 and 5 – negative tick samples from this study; 4 – positive sample 16.C/2016/Dubrava/SVK. (e) The RT-PCR positive sample caused CPE in Vero E6 cells 3 days after infection (EC Plan-Neoflural 10×/0.3 Ph 1, Zeiss, Germany). Left – negative control (1% EMEM), right – 16.C/2016/Dubrava/SVK sample. (f) TRBV antigen was detected 3 days post infection by immunofluorescence in Vero E6 cells. The virus antigen was localized in the cytoplasm and perinuclearly (LD Plan-Neoflural 40×/0.6 Korr Ph 2 M27, Zeiss)
RT-PCR sensitivity and specificity
Sensitivity of the RT-PCR reaction was measured using the prototype TRBV propagated in Vero E6 cells. One hundred microlitres of virus culture containing 1.5 × 10^5^ PFU/ml was used for RNA extraction with addition of 1 µg of RNA carrier following the manufacturer instructions (INDICAL bioscience, Germany). The RNA was eluted in 50 µl of DEPC (Sigma Aldrich, United States) treated deionized water. Ten microlitres of RNA was used for complementary DNA (cDNA) synthesis using the LunaScript RT SuperMix (New England Biolabs, United States) according to the manufacturer’s protocol. The synthesized cDNA from the virus stock represented dilution 10^0^, which was 10-fold diluted in DEPC treated deionized water down to 10^− 5^, which represented 10^0^ PFU/ml. Orbivirus RNA was detected by conventional PCR using the DreamTaq™ Green PCR Master Mix (Thermo Fisher Scientific, Lithuania). The PCR mix contained 1 µl from each dilution of cDNA and 200 nM final concentration of each primer. The thermal profile was the following: 95 °C/60 seconds; 40 cycles of 95 °C/45 seconds, 55 °C/30 seconds, 72 °C/45 seconds and a final elongation at 72 °C/10 minutes. The specificity of the RT-PCR was tested with cDNA of other tick-borne viruses, namely Uukuniemi virus strain 37.C/2020/Dubrava/SVK (PX853966) and Hypr strain of tick-borne encephalitis virus [deposited in the European Virus Archive (EVAg), Ref-SKU: 008 N-EVA442). The results of the PCRs were visualized by electrophoresis in 1% agarose and gel stained with intercalating dye (GelRed™, Biotium, United States) using GEL LOGIC 100 (Eastman Kodak Company, United States) imaging system.
Virus RNA detection in tick homogenates
RNA was extracted using the IndiSpin Pathogen Kit (INDICAL bioscience, Germany) without RNA carrier from 100 µl of tick homogenates. The cDNA synthesis, the PCR and electrophoresis was detected as described above.
Immunofluorescence
Viral antigen from the newly isolated TRBV was detected in Vero E6 cells cultivated on coverslips and infected using the method described earlier. Cells were fixed with ice cold methanol at −20 °C for 10 min at the onset of CPE. Nonspecific binding sites were blocked with 20% normal mouse serum in 0.2% Triton X-100 in phosphate-buffered saline (Tx-PBS) for 60 min at room temperature.
The blocking solution was replaced with mouse TRBV hyperimmune serum [40], diluted 1:50 in 0.5% solution of bovine serum albumin in PBS. Cover slips were incubated overnight at 4 °C on a plate rocker. Cells were washed thrice with Tx-PBS, followed by incubation with the Alexa Fluor^®^ 488 conjugated secondary antibody (goat anti-mouse IgG H&L, ab150113, Abcam) for 30 min at 37 °C. After additional three washes, the cell nuclei were stained with Hoechst 33,342 (Cell Signaling Technologies, United States).
Neutralization test
In total, 28 goat serum samples were tested for the presence of neutralization antibodies against the prototype TRBV. Accordingly, 50 µl of heat-inactivated (56 °C for 30 min) sera were serially diluted two-fold in 1% EMEM on flat-bottom microtitration plates (Greiner Bio One, Germany), with the dilutions ranging from 1:5 to 1:320. TRBV hyperimmune serum [40] and normal goat serum (BIOWEST, France) were used as the positive and negative controls, respectively. Next, 50 µl of virus suspension containing 100 tissue culture infectious dose 50% (TCID_50_) of the prototype TRBV (kindly provided by Professor Gerhard Döbler, Institut für Mikrobiologie der Bundeswehr, München, Germany) was added to each serum dilution. This resulted in a final dilution ranging from 1:10 to 1:640.
Each sample included a cytotoxicity control, where 50 µl of 1% EMEM was added instead of the virus. After 1 h of incubation at 37 °C, 1 × 10^4^ Vero E6 cells in 10% EMEM were added to each well. The plates were incubated for 3 days at 37 °C and 5% CO_2_. Neutralization was assessed by inverted light microscopy. Only samples without cytotoxicity were evaluated. The highest dilution showing complete neutralization was recorded as the neutralization titre. The infectious dose was controlled by back-titration of the virus inoculum. Each sample was tested in duplicates.
Replication of TRBV in human, bovine, and rodent cells
Baby hamster kidney (BHK-21), human foreskin fibroblast (HFF-1), and bovine turbinate (BT) cells were seeded in six-well plates at a density of 6 × 10^4^ cell/cm^2^. The replication was analysed at 2, 4, 6, 8, 12, 24, 32, 48, and 72 h post-infection. For each time point, one plate containing three tests and three negative control wells was used.
After overnight cultivation, the medium was removed and cells were infected with 250 µl of the new isolate at a multiplicity of infection of 1. The control wells received an equal volume of cultivation medium. After 1 h of adsorption, the inoculum was discarded and each well was washed with excess ice-cold DMEM. Next, 3 ml of cultivation medium was added into each well. At each time point, the medium from each test and control wells was collected, centrifuged at 4000 RCF for 10 min at 4 °C, aliquoted (1 ml), and stored at −80 °C until plaque formation test.
Plaque formation test
Overnight cultivated Vero E6 cells seeded on 6 well plates at density of 4 × 10^4^ cells/cm^2^ were infected with 250 µl of supernatants from certain time points. After one hour adsorption the inoculum was discarded and replaced with 3 ml of 2% carboxymethylcellulose (Sigma-Aldrich, United States) in 1% EMEM. Cells were fixed with 10% formalin after three days and stained with 0.1% crystal violet. The amount of infectious virus progeny at each time point was expressed as the mean PFU/ml of a triplicate.
Genetic and bioinformatic analysis
Whole-genome sequencing of the TRBV isolate propagated in Vero E6 cells was performed using next-generation sequencing [28]. Libraries were prepared with the Nextera Library Prep Kit (Illumina, United States) according to the manufacturer’s protocol and sequenced on an Illumina Miseq platform using the Miseq reagent kit v2 (Illumina), generating paired-end reads of 120 bp.
Sequence reads were analysed and mapped to the prototype TRBV isolate (HQ266581–HQ266590) using Geneious 9.1.8 software. Gaps in the genome identified after next-generation sequencing were filled by Sanger sequencing using specific primers (listed in Table S1). Tick-, mosquito-, and Culicoides-borne orbiviruses used for genetic analysis are listed in Table S2.
Nucleotide and amino acid sequence alignment and pairwise identity calculation were performed using Clustal Omega 1.2.2 within the Geneious 9.1.8 software [36]. Nucleotide phylogenetic trees were built using MEGA 7 [41]. The evolutionary history was inferred using the Maximum Likelihood method under the General Time Reversible model [42]. Initial trees for the heuristic search were obtained automatically by applying Neighbour-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood approach. The topology with superior log likelihood value was selected. A discrete Gamma distribution (+ G) with five categories was used to model evolutionary rate differences among sites, allowing for a proportion of invariable sites (+ I). All positions containing gaps and missing data were eliminated. Trees were rooted using the bluetongue virus serotype 8 (BTV-8) NET 2006/04 strain as the outgroup.
Bayesian phylogenetic analyses were performed for genome segments 5 and 10 using MrBayes v3.2.7a [43] as a complementary approach to maximum-likelihood inference. Alignments were analysed under a GTR + G model, with two independent runs of four Markov chains each. Markov chain Monte Carlo (MCMC) chains were run for 1.1–2.0 million generations, sampling every 200 generations. Convergence was assessed by monitoring the average standard deviation of split frequencies (ASDSF) and by inspecting parameter traces and effective sample sizes (ESS) in Tracer v1.7.2 [44]. All parameters showed adequate mixing (ESS > 200), and potential scale reduction factors (PSRF) were close to 1.0, indicating convergence between runs. ASDSF values below ~ 0.01 were considered indicative of convergence. The first 25% of samples were discarded as burn-in. Posterior probabilities were used to assess node support and are reported for selected nodes on the maximum-likelihood trees.
Reassortment analysis
The coding sequences of each genome segment of the TRBVs were concatenated using the Geneious software and aligned by the Clustal Omega 1.2.2. The alignment was analysed by exploratory recombination signal detection methods RDP [45], GENECONV [46], BootScan with 100 bootstrap replicates [47], MaxChi [48], Chimera [49], SiScan [50] and 3Seq [51] embedded in the Recombination Detection Program 4 [52] at significance level P ≤ 0.01, using the 16.C/2016/Dubrava/SK strain as the query sequence. Only events detected by all six methods were visualized. Recombination events supported by the exploratory methods were further analysed and visualized by the SimPlot v3.5.1 software [53].
Results
Detection and isolation of TRBV in ticks
The RT-PCR was designed for detection of segment 1 of the GIV serogroup orbiviruses. Using the designed primer set (Fig. 1a) and reaction conditions, we detected prototype TRBV strain cDNA at a dilution of 10^− 3^, corresponding to 10^3^ PFU/ml (Fig. 1b). We performed specificity testing with UUKV and TBEV, and amplification was observed only in the case of the prototype TRBV strain (Fig. 1c).
Thirty-three pooled tick samples were analysed by RT-PCR for the presence of orbivirus RNA. A homogenate prepared from an engorged female tick tested positive, yielding a DNA band of the expected size of 767 bp (Fig. 1d). The PCR product was sequenced using the Sanger method, generating a 617 bp sequence that was deposited in GenBank under accession number PP068371. Basic Local Alignment Search Tool analysis showed 97.0% pairwise nucleotide identity with segment 1 of the prototype TRBV strain (HQ266581). The same homogenate was subsequently used for virus isolation. Cytopathic effect was observed in Vero E6 cells 3 days post-infection (Fig. 1e), and the viral antigen was detected by immunofluorescence (Fig. 1f). The TRBV isolate was designated as 16.C/2016/Dubrava/SVK.
Prevalence of TRBV neutralization antibodies in grazing goats
All animals remained clinically healthy throughout the study period. None of the serum samples showed cytotoxic effects on Vero E6 cells.
In samples collected in 2016, no neutralizing antibodies were detected. In 2017, five of nine goats (55.6%; animals no. 1, 2, 4, 6, and 9) showed low titre seroconversion, ranging from 1:10 to 1:20 (Table 1). In 2018, three goats (animals no. 1, 4, and 9) remained seropositive with no increase in titre, while two (animals no. 8 and 12) showed low (1:10) antibody titre seroconversion. One goat (animal no. 5) exhibited no seroconversion during the study period.
The replication of TRBV isolate in rodent, human and bovine cell lines
Replication of TRBV strain 16.C/2016/Dubrava/SVK was studied in rodent (BHK-21), human (HFF-1), and bovine (BT) cell lines. Viral titres were quantified using plaque formation test performed on Vero E6 cells. The virus harvested from all three cell lines exhibited uniform plaque morphology (Fig. 2a).Fig. 2. The plaque morphology, the replication kinetic and the CPE of the TRBV isolate 16.C/2016/Dubrava/SVK. (a) plaque morphology of the new TRBV isolate in Vero-E6 cells 3 days after infection (GEL LOGIC 100); (b) virus progeny was quantified by PFT in three wells per timepoint. The mean PFU/ml and the standard deviation was plotted on logarithmic scale of the graph; (c) Representative images of TRBV isolate 16.C/2016/Dubrava/SVK induced CPE in BHK-21 (rodent), HFF-1 (human) and BT (bovine) cells 72 h after infection with infectious dose of MOI = 1 at 40× magnification: upper left – BHK-21 negative control; lower left – infected BHK-21; upper middle – BT negative control; lower middle – infected BT; upper right – HFF-1 negative control; lower right – infected HFF-1 (LD Plan-Neofluar 40×/0.6 Korr Ph 2 M27, Zeiss)
In HFF-1 and BT cells, the eclipse phase lasted approximately 6 h (Fig. 2b). In HFF-1 cells, viral titre peaked 12 h after infection, reaching 6.83 × 10^3^ PFU/ml. In BT cells, the highest titre, 5.00 × 10^4^ PFU/ml, was observed at 48 h post-infection. In contrast, replication in BHK-21 cells showed a longer eclipse phase that lasted approximately 8 h, followed by a gradual increase in viral load until throughout the observation period, reaching 5.75 × 10^5^ PFU/ml.
CPE developed in all cell lines; however, its progression differed (Fig. 2c). After 24 h, HFF-1 cells showed only minor morphological changes with preserved monolayer integrity compared with BT and BHK-21 cell lines, where numerous shrunken cells were observed. At later time points, however, prominent CPE was evident in all cell lines.
Genetic and phylogenetic analysis
Complete genome sequences of the TRBV isolate 16.C/2016/Dubrava/SVK were deposited in GenBank under accession numbers PP237014–PP237023. The nucleotide and amino acid identities of each genome segment and corresponding proteins of the TRBV, LIPV, MUKV, KEMV and GIV were compared (Table 2 and Table S3–S12). Pairwise nucleotide identities for segments 1 and 2 were below the established within-species thresholds [1]. The average amino acid identity of VP1(RdRp) among TRBV, LIPV, MUKV, and KEMV, when compared with that in GIV, was 73.1%. For VP2(T2), the nucleotide and amino acid sequences identities relative to GIV averaged 72.8% and 82.7%, respectively (Table 1 and Table S3 and S4).Table 2. Nucleotide (nt) and amino acid (aa) sequence identity comparison of TRBV strains among each other and with other GIV group orbivirusesSegmentTRBVTRBV vs. LIPVTRBV vs. MUKVTRBV vs. KEMVTRBV vs. GIVProtein1nt89.2–96.388.9–95.382.2–82.968.9–70.165.8–66.8VP1(RdRp)aa97.3–98.997.6–98.994.3–95.078.0–79.172.6–73.12nt92.8–98.692.8–93.282.1–83.274.9–76.172.2–72.9VP2(T2)aa99.8–99.699.3–99.698.3–98.790.7–91.382.6–82.83nt89.2–96.2NA79.1–80.167.5–68.860.4–61.0VP3(Cap)aa97.9–99.589.8–91.673.1–77.663.4–64.14nt84.5–98.2NA79.4–81.064.0–65.858.5–59.8NS1(Tup)aa93.2–98.990.4–91.564.7–66.558.2–58.95nt82.4–98.3NA58.4–60.374.4–76.147.0–47.5VP4(OC1)aa90.4–98.659.2–60.280.9–83.236.7–37.86nt83.8–93.981.5–83.780.8–81.574.0–77.064.6–65.5VP5(OC2)aa96.8–99.494.6–95.693.0–94.086.3–87.667.2–67.67nt93.7–96.8NA80.7–82.162.3–64.557.1–58.2NS2(Vib)aa96.7–99.288.3–90.862.7–65.152.9–54.88nt87.4–93.0NA80.8–83.071.9–74.769.6–70.8VP7(T13)aa97.2–98.693.2–94.484.7–86.776.6–78.29nt86.3–95.2NA82.7–85.370.1–71.959.9–61.8VP6(Hel)aa79.8–92.376.0–80.163.7–59.345.5–46.710nt72.4–97.3NA74.0–86.571.2–75.960.5–63.2NS3aa76.1–97.177.0–78.575.6–79.456.1–57.9
Values are shown as ranges (min–max) where multiple reference sequences were available for a given virus. For each segment, nucleotide (nt) and amino acid (aa) identities are indicated separately in the table. Legend: NA – sequence not available in the GenBank; VP1 – RNA-dependent RNA-polymerase (RdRp); VP2 – major sub-core protein (T2); VP3 – capping enzyme (Cap); NS1 – tubule forming protein (Tup); VP4 – outer capsid protein (OC1); VP5 – outer capsid protein (OC2); NS2 – virus inclusion body protein (Vib); VP7 – major core-surface protein (T13); VP6 – helicase (Hel); NS3 glycoprotein.
Phylogenetic analysis of the new TRBV isolate 16.C/2016/Dubrava/SVK revealed the mosaic nature of its genome (Fig. 3, Figures S1–S9). In most genome segments, TRBV, LIPV, MUKV, and KEMV formed monophyletic groups. The new isolate shared a common ancestor with the prototype TRBV strain isolated in Slovakia in 1963 for VP2(T2), VP3(Cap), NS1(Tup), and VP5(OC2). In contrast, for VP1(RdRp), VP4(OC1; Fig. 3), and NS2(Vib), the isolate formed a separate branch within the TRBV clade. For VP7(T13), VP6(Hel), and NS3, the isolate shared a common ancestor with strains originating from geographically remote locations in Romania and Ukraine.Fig. 3. Maximum likelihood trees constructed for nucleotide sequences of orbivirus genomic segments encoding the VP4(OC1) and the NS3 proteins using MEGA 7. For better visualization of the relationship between KEMV, TRBV and MUKV, only the subtree of the VP4(OC1; Figure S5) is depicted. The reliability of each tree was estimated by bootstrap analysis of 1000 replicates using GTR substitution model with Gamma distributed evolutionary rate with invariable sites (G + I). Trees with the highest log likelihood are shown. The percentage above 75% of trees in which the associated taxa clustered together is shown above the branches. Posterior probabilities obtained from parallel Bayesian analyses are shown below the branches. Each tree was rooted on the outgroup BTV NET 2006/04 strain
In most segments, MUKV showed greater similarity to TRBV than to KEMV and shared a common ancestor. However, in segment 5 (VP4, OC1), TBRV exhibited higher nucleotide (74.4–76.1%) and amino acid identity (80.9–83.2%) with KEMV than that with MUKVs (approximately 60% at both levels; Table 2 and Table S6). These differences were also reflected in the topology of the phylogenetic tree, where the TRBVs and the KEMVs formed sister clades (Fig. 3).
The highest genetic diversity was observed in segment 10, which encodes the NS3 glycoprotein (Table 2 and Table S12). Within this segment, the monophyletic clade of TRBVs was divided. Strains from Slovakia and Romania shared 84.4–90.7% nucleotide and 91.4–97.1% amino acid identity (Table S12). The Ukrainian strains displayed higher identity among themselves (nucleotide, 97.3%; amino acid, 97.1%), but lower identity with Slovak and Romanian strains, comparable to that observed for KEMVs (nucleotide, 72.4–76.2%; amino acid, 76.1–81.8%). Interestingly, MUKVs shared 83.9–86.5% nucleotide and 91.4–94.3% amino acid identity with Slovak and Romanian TRBV strains, sharing a common ancestor in the phylogenetic tree, whereas the Ukrainian strains formed sister clades with the KEMVs (Fig. 3).
Reassortment analysis
RDP4 software, using a significance threshold of P ≤ 0.01, predicted one reassortment event between TRBV strain 16.C/2016/Dubrava/SVK and the prototype SVK strain (Fig. 4a). This event was supported by six out of seven methods: BootScan (P = 9.361 × 10^− 35^; average bootstrap support 95.25%), RDP (average P = 3.711 × 10^− 28^), MaxChi (P = 1.805 × 10^− 22^), Chimera (P = 3.003 × 10^− 18^), SiScan (P = 5.428 × 10^− 15^), and 3Seq (P = 5.164 × 10^− 09^). GENECOV did not show support at this significance threshold. The predicted recombination breakpoints were located at positions 8191 and 10,049, which correspond to segment 4 in the concatenated sequence. SimPlot analysis further supported a possible reassortment event at this genomic region (Fig. 4b).Fig. 4. BootScan (RDP4) and SimPlot analysis for detection of genomic segment reassortment events between TRBVs genomes based on concatenated segments of the Slovak TRBV strains, Romanian TRBV strain and the MUKV MUV-hay strain. (a) The relative bootstrap support plot for nearest neighbour groupings of the recombinant sequence. Colour coding: dark grey zone – 95% breakpoint confidence interval; pink zone – tract of sequence with recombinant origin; BootScan plot of TRBV strains: yellow line – Romania (major parent) with prototype (minor parent); green line – Romania (major parent) with 16.C/2016/Dubrava/SVK (recombinant); purple line – prototype (minor parent) with 16.C/2016Dubrava/SVK (recombinant). The dashed line marks the bootstrap cut-off (70%). (b) In the SimPlot analysis the TRBV 16.C/2016/Dubrava/SVK strain was the query sequence. Colour coding of TRBV strains: red line – prototype TRBV, yellow line – TRBV Romania, blue line – MUKV MUV-hay. Dashed horizontal lines express the similarity and the vertical lines indicates the genome segments. Similarity plot was generated based on the following setting: window: 200 bp, step: 20 bp, gap strip: on, Kimura (2-parameter), T/t: 2,0
Discussion
The significance of tick-borne arboviruses lies in their severe clinical impact, their emergence as global health threats, and the diagnostic challenges they pose to modern medicine. TBEV is considered the most significant human tick-borne arbovirus pathogen in Eurasia, especially in Slovakia, with increasing incidence over the past 50 years [54, 55]. TBEV, TRBV, LIPV, and KEMV share similar biotopes and transmission cycles, and infections caused by the GIV group orbiviruses can clinically resemble tick-borne encephalitis infections [2, 5, 56]. Despite evidence of their neurotropic potential and the absence of pathognomonic clinical symptoms, TRBV, LIPV, and KEMV are often neglected in the differential diagnosis of viral central nervous system (CNS) infections. The extent of this diagnostic challenge is evident in Slovakia, where between 2016 and 2018, 40% of all viral CNS infections were diagnosed as unspecified viral encephalitis, meningitis, or unspecified viral CNS infection [57].
GIV group orbiviruses have been isolated in Russia, Kazakhstan, Egypt, Slovakia (formerly Czechoslovakia), Moldavia, Ukraine, Japan, and Romania, from questing or engorged ticks collected from mammal and avian hosts [2, 8, 10, 11, 18, 58–61]. In the present study, we detected a new TRBV strain using newly designed oligonucleotides. Under the applied conditions, the analytical sensitivity of the RT-PCR reaction was sufficient to isolate the virus from a PCR-positive tick homogenate. Nevertheless, the assay could be further improved through detailed optimization of the reaction conditions, including template and/or primer concentration and cycling conditions.
The virus was isolated in Vero E6 cells, and its replication dynamics were studied in rodent (BHK-21), bovine (BT), and human (HFF-1) cell lines. The replication curve in rodent cells showed efficient multiplication without inhibitory effects, whereas lower yields of infectious progeny were observed in human and bovine cells. Orbivirus genomes consist of dsRNA segments, which are potent stimulators of the interferon (IFN) response. BHK-21 cells are known to have a disrupted type I IFN response due to defects in the cytosolic RLR sensors, specifically RIG-1 and MDA5; however, HFF-1 and BT cells have this cascade intact [62–66]. A similar growth curve in BHK-21 cells has been described for MUKV in a previous study [58]. Despite reduced TRBV replication in IFN type I-competent cells, replication did occur, suggesting the presence of viral immune evasion mechanisms. At present, knowledge of such potential mechanisms in tick-borne orbiviruses remains limited. The NS4 protein, which is believed to have IFN antagonistic potential, is derived from an alternative ORF in segment 9 of GIV orbiviruses [67]. Recombinant GIV NS4 protein protects dsRNA from Dicer cleavage, indicating a dsRNA-binding domain. In addition, ectopic expression of the NS4 protein in viral dsRNA-stimulated HeLa cells significantly suppresses mRNA expression of key antiviral genes, including those encoding PKR, RIG-I, MDA5, IFN-β, and IRF-3, −7, and − 9 [26].
TRBV, LIPV, MUKV, and KEMV are currently classified as GIV serotypes [1]. Orbiviruses within the same species typically share at least 78% amino acid identity in RdRp, and at least 76% nucleotide and 83% amino acid identity in the major subcore structural protein T2. In our study, the average RdRp amino acid identity of TRBV, LIPV, MUKV, and KEMV compared to GIV was 73.1%. For the T2 protein, the average nucleotide and amino acid identities were 72.8% and 82.7%, respectively. The VP7(T13) protein, which is also used to identify and classify orbiviruses [24], showed a maximum nucleotide and amino acid identity of 71.1% and 82.2%, respectively, relative to GIV. Together with previous reports [12, 24, 25, 28, 58] our findings suggest that TRBV, LIPV, MUKV, and KEMV may represent strains of different orbiviruses with a wide geographical distribution from Japan to Central Europe. Limited reassortment between GIV and KEMV has been reported under laboratory conditions [34, 35], and these viruses are therefore classified into different subgroups within the same species [1].
Most genome segments and proteins of TRBV showed higher sequence identity to MUKVs isolated from Japan than to KEMVs circulating in Russia. However, segment 5, encoding the outer surface protein VP4(OC1), was an exception and shared higher identity and a common ancestor with KEMVs. Viral surface proteins mediate host-cell entry and may be responsible for virulence. Being exposed to the immune system, they are under positive selective pressure, which contributes to their variability. Orbiviruses have two outer capsid proteins carrying neutralizing epitopes and form the basis of orbivirus serotyping [68–70]. Accordingly, cross reactivity between TRBVs and MUKVs is plausible and is likely quantitative, as in the case of TRBV versus KEMV [71, 72].
Genome segment 10 encodes the NS3 and NS5 proteins, which localize to the smooth vesicle membrane of host cells during orbivirus infection and facilitate transport across the cytoplasm to the plasma membrane [73–75]. Differences in interaction with NS3-containing vesicles have been observed between different orbiviruses and host-cell types. Such differences may impact the efficiency of orbivirus replication in vectors, which is dependent on virion release from salivary gland cells into the salivary ducts of a given vector species [76]. NS5 supports virus replication by reducing host-cell protein synthesis and preventing ribosomal RNA degradation [26]. To date, the role of NS3 proteins in TRBVs has not been fully investigated. In our study, the highest degree of variability was observed in segment 10. Slovak and Romanian TRBV strains clustered with MUKV, whereas Ukrainian TRBV strains formed a distinct clade with KEMV. Further studies of the geographical strains are required to understand how this variability in segment 10 affects the biological properties of TRBV, including its replication ability and geographical spread.
Genome segment reassortment in tick-borne orbiviruses has been previously reported [34, 35]. Our results indicate that segment 4 encoding the NS1 protein of TRBV 16.C/2016/Dubrava/SVK originated from the prototype strain isolated in the 1960 s in western Slovakia [77]. Tubule formation in infected cells is a characteristic feature of orbivirus infection [78, 79]. NS1 acts as a positive regulator of viral protein synthesis, including itself, which creates a positive feedback loop that leads to a rapid increase in the expression of all viral proteins [78]. A long-term study focusing on the genetic analysis of BTV strains in India indicates the potential impact of NS1 reassortment on virus fitness—likely related to the replication efficiency in the dominant local vector, Culicoides imicola [70]. Strong evidence suggests a selective sweep event, in which a single NS1 variant invaded each Indian BTV serotype through reassortment and subsequently spread over a large geographic area within a few decades, suggesting enhanced adaptation of BTV to new hosts. Currently, it is not possible to draw any conclusion regarding the reassortment event in the new TRBV isolate. However, further studies comparing replication of the prototype TRBV and the new reassortant in cells of different host origins would provide deeper insight into the impact of NS1 gene reassortment on the replication kinetics of orbiviruses.
We screened goats grazing in the locality where the novel TRBV strain was isolated. Neutralization test results showed that five goats became infected in this area. Although with low titre, three of the animals remained seropositive. Additionally, two other goats also became infected, as indicated by the seroconversion, suggesting maintenance of the virus within the natural focus. The low antibody titre in our study is in concordance with previous studies. Seroconversion in goats has been previously investigated in a natural focus [6]. Out of fourteen animals, two exhibited neutralizing antibodies from the 56th and the 135th day of pasturing, with titres of 1:16 and 1:32. Low antibody titres (1:4–1:16) have also been reported in humans with neurological signs of TRBV and LIPV (Slovakia) [4]. Notably, cross-serological reactions between TRBV and KEMV have been reported [72] and the occurrence of KEMV has been suggested in Central Europe [16]. For this reason, cross-neutralization tests are planned for goat serum samples from this study, as well as for samples from other wild and farm animals.
In conclusion, this study advances current knowledge of the replication and genetic diversity of tick-borne orbiviruses. Results from genetic analyses and exploratory recombination detection methods indicate that segment 4, which encodes the NS1 protein of the new TRBV isolate, originated from the prototype TRBV strain isolated during the 1960 s in Slovakia. Given the high variability observed in certain genomic regions and their potential implications for viral spread, pathogenicity, and virulence, further studies focusing on the ecology, host immune responses, and antiviral evasion mechanisms of zoonotic tick-borne orbiviruses are warranted.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1 (PPTX 819 KB)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Matthijnssens J, Attoui H, Bányai K et al (2022) ICTV Virus Taxonomy Profile: Sedoreoviridae 2022. J Gen Virol 103. 10.1099/jgv.0.001782
- 2Bratuleanu BE, Răileanu C, Bennouna A et al (2023) Diversity of viruses in Ixodes ricinus in Europe including novel and potential arboviruses. Transboundary and Emerging Diseases 2023:e 6661723. 10.1155/2023/6661723
- 3Csank T, Drzewnioková P, Čurlík J et al (2017) Tribeč virus (Great Island serogroup; genus Orbivirus) in ruminants in Slovakia. In: Sbrorník abstraktů z Československé virologické konference 2017. Biologické centrum AV ČR, v.v.i., České Budějovice, p 76
- 4Jakimovski D, Mateska S, Dimitrova E et al (2023) Tick-borne encephalitis virus and Borrelia burgdorferi seroprevalence in Balkan tick-infested individuals: A two-centre study. Pathogens 12. 10.3390/pathogens 12070922
- 5Libíková H, Řeháček J, Grešíková M et al Cytopathic viruses isolated from Ixodes ricinus ticks in Czechoslovakia. Acta Virol 8:96