Case Report: Identification of a HNF1A exons 1–10 heterozygous deletion in a Chinese MODY family
Mengyun Lei, Mei Xue, Huawei Wang, Zhe Dai, Jun Tang

TL;DR
A Chinese family with maturity-onset diabetes of the young (MODY) was found to have a rare HNF1A gene deletion, showing the importance of advanced genetic testing for accurate diagnosis and treatment.
Contribution
The study reports a novel HNF1A exon 1–10 deletion in a MODY family and emphasizes the need for CNV analysis in genetic testing.
Findings
A heterozygous HNF1A(NM_000545.8):ex1_10del was identified using MLPA after NGS failed to detect mutations.
The deletion was classified as pathogenic and associated with a typical MODY clinical phenotype.
Treatment with glimepiride improved glycemic control after switching from insulin.
Abstract
Maturity-onset diabetes of the young (MODY) is an autosomal dominant monogenic diabetes, with HNF1A-MODY (MODY3) being a common subtype. Standard genetic testing for MODY often focuses on sequencing, which can lead to the misdiagnosis of cases caused by HNF1A copy number variants (CNVs). This study investigates the diagnosis of a Chinese family with a HNF1A(NM_000545.8):ex1_10del. We evaluated a Chinese family with a clinical diagnosis of maturity-onset diabetes of the young (MODY). Clinical data and peripheral blood samples were collected from family members. A heterozygous HNF1A(NM_000545.8):ex1_10del was suspected by next-generation sequencing (NGS) using a hereditary diabetes gene panel.This finding was validated using multiplex ligation-dependent probe amplification (MLPA). We also conducted a literature review of previously reported HNF1A-MODY cases associated with heterozygous…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
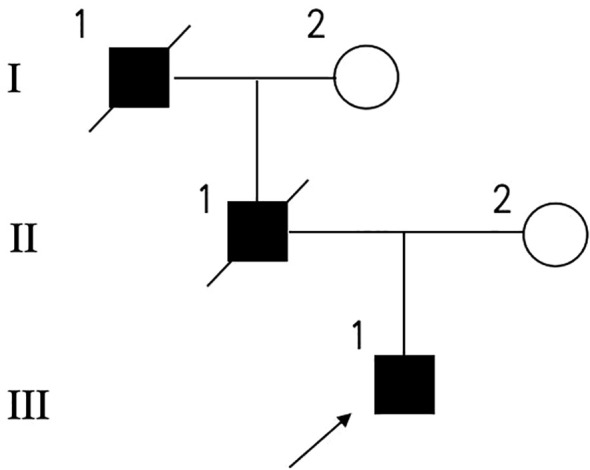 Figure 1
Figure 1 Figure 2
Figure 2| Parameter | Result | Reference value |
|---|---|---|
| Age (years) | 32 | |
| Age at diagnosis (years) | 24 | |
| BMI (kg/m2) | 18.8 | |
| HbA1C mmol/mol (%) | 72 (8.7%) | |
| GAD-Ab | Negative | Negative |
| IA-Ab | Negative | Negative |
| ICA-Ab | Negative | Negative |
| IA-2-Ab | Negative | Negative |
| Blood Urea (mmol/L) | 5.23 | 2.8-7.6 |
| Creatinine (umol/L) | 59.5 | 64-104 |
| Uric Acid (umol/L) | 205.4 | 208-428 |
| Potassium (mmol/L) | 3.81 | 3.5-5.3 |
| Sodium (mmol/L) | 140.2 | 137-147 |
| β-Hydroxybutyric Acid (mmol/L) | 0.08 | 0-0.28 |
| C-Reactive Protein (mg/L) | 1.0 | 0-10 |
| White Blood Cell Count (10^9/L) | 7.91 | 3.5-9.5 |
| Hemoglobin (g/L) | 167.3 | 130-175 |
| Platelet Count (10^9/L) | 234 | 125-350 |
| Prothrombin Time (s) | 11.6 | 9.4-12.5 |
| D-Dimer (ng/ml) | 18 | 0-500 |
| Urine glucose | ++++ | Negative |
| Urine ketone | Negative | Negative |
| Time | Blood Glucose (mmol/L) | C peptide (ng/ml) | Insulin (uIU/ml) |
|---|---|---|---|
| 0h | 6.27 | 1.18 | 2.57 |
| 0.5h | 13.67 | 2.82 | 9.09 |
| 1h | 17.97 | 4.01 | 15.3 |
| 2h | 21.94 | 3.82 | 16.1 |
| 3h | 17.38 | 2.96 | 12.5 |
| No. | Reference | Exon(s) deleted | Detection and validation method | Family segregation | Clinical and phenotypic correlation |
|---|---|---|---|---|---|
| 1 | Ellard S et al. ( | ex1del, ex2_10del, ex1_10del. | Custom MLPA assay using synthetic oligonucleotide probes covering GCK, HNF1A, and HNF4A (30 exons total) | Co-segregation with early-onset diabetes confirmed within families | 4/60 (6.7%) HNF1A-negative probands had large deletions; emphasizes need for dosage analysis in MODY diagnosis |
| 2 | Willson JSB et al. ( | ex2_3del | Detected by array-CGH and confirmed by direct sequencing | Three affected family members carried same deletion (HNF1A-MODY + hepatocellular adenoma) | Coexistence of HNF1A-MODY and hepatocellular adenoma/HCC; suggests extended phenotype |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPancreatic function and diabetes · Genomics and Rare Diseases · Diabetes Treatment and Management
Introduction
1
Maturity-onset diabetes of the young (MODY) comprises a heterogeneous group of monogenic diabetes syndromes characterized by early-onset hyperglycemia, typically diagnosed before 25 years of age, and most inherited in an autosomal dominant manner (1). Although MODY accounts for only approximately 1-2% of all diabetes cases in population-based estimates (2), its true prevalence is likely underestimated due to frequent misclassification as type 1 or type 2 diabetes (3).This under-recognition is clinically important because an accurate molecular diagnosis of MODY directly determines optimal treatment, prognosis, and the need for predictive genetic testing in relatives.
MODY is caused by pathogenic variants in genes involved in pancreatic β-cell development, glucose sensing, and insulin secretion. To date, pathogenic variants in more than 11 genes have been definitively linked to MODY (4). The most common subtypes, accounting for over 90% of genetically confirmed cases, are caused by mutations in GCK (MODY2), HNF1A (MODY3), and HNF4A (MODY1) (4). GCK, located on chromosome 7p15.3, encodes glucokinase, a hexokinase that catalyzes the phosphorylation of glucose in pancreatic β-cells and hepatocytes and serves as a critical glucose sensor (5). GCK contains multiple promoters and alternative transcript isoforms, with dominant expression in liver and pancreatic islets. Pathogenic GCK variants reduce glucose-stimulated insulin secretion, resulting in lifelong, mild, stable fasting hyperglycemia (5). HNF1A, located on chromosome 12q24.31 and composed of 10 exons, encodes a homeodomain-containing transcription factor that binds DNA as a homodimer or heterodimer to regulate the expression of liver- and β-cell–specific genes, including those involved in glucose transport and insulin secretion (6). HNF4A, located on chromosome 20q13.12 and consisting of 13 exons, encodes a nuclear receptor transcription factor that directly regulates HNF1A expression and controls a broad hepatic and pancreatic transcriptional program (7). HNF1B, located on 17q12, encodes a related transcription factor that forms homo- and heterodimers with HNF1A and plays a crucial role in renal and pancreatic development (8). Other MODY-associated genes include INS (MODY10) on 11p15.5, which encodes the insulin prohormone; PDX1 (MODY4) on 13q12.2, and NEUROD1 (MODY6) on 2q31, both of which encode essential β-cell transcription factors regulating insulin gene transcription and islet differentiation (4).
Clinically, MODY exhibits a broad phenotypic spectrum. GCK-MODY typically presents as lifelong, mild, non-progressive fasting hyperglycemia that is often incidentally discovered, including during pregnancy screening. These patients rarely develop microvascular complications and usually do not require pharmacologic glucose-lowering therapy outside of pregnancy (4, 9). In contrast, HNF1A-MODY and HNF4A-MODY are characterized by progressive β-cell dysfunction. Patients typically present in adolescence or early adulthood with increasing hyperglycemia, marked postprandial glucose excursions, and a low renal threshold for glucose, resulting in prominent glycosuria (4, 9). Endogenous insulin secretion is usually preserved early in the disease course but declines progressively over time. HNF4A-MODY additionally shows distinctive neonatal features, including transient hyperinsulinemic hypoglycemia and large-for-gestational-age birth weight (4, 9).
The diagnosis of MODY requires a high index of clinical suspicion and integration of phenotypic, biochemical, and family history data. Typical features include early-onset diabetes, autosomal dominant inheritance across successive generations, absence of pancreatic autoantibodies, preserved C-peptide levels indicating endogenous insulin production, lack of obesity or severe insulin resistance (4, 9). Clinical tools such as MODY probability calculators may aid in selecting candidates for genetic testing, but definitive diagnosis relies on molecular genetic testing, preferably with NGS-based monogenic diabetes panels followed by segregation analysis (4). MODY represents a classical model of precision medicine because treatment can be directly guided by genotype. GCK-MODY generally requires no glucose-lowering therapy outside pregnancy. In contrast, patients with HNF1A-MODY and HNF4A-MODY are characteristically highly sensitive to low-dose sulfonylureas, which are effective as first-line therapy in most cases, particularly early in the disease course. Many patients can successfully transition from insulin to sulfonylureas without increased risk of ketoacidosis. With progressive β-cell failure, some individuals eventually require insulin therapy (4, 9).
HNF1A-MODY is caused by pathogenic variants in the HNF1A gene encoding a key transcription factor involved in pancreatic β-cell function and glucose metabolism. These variants impair downstream transcriptional programs regulating insulin secretion, leading to progressive β-cell failure (6, 10). HNF1A-MODY demonstrates high but age-dependent penetrance, with approximately 60–65% of carriers developing diabetes by 25 years of age and over 90% by mid-adulthood (11). Previous studies indicate that HNF1A-MODY is present but relatively uncommon in Chinese populations. Early family-based investigations identified HNF1A mutations in approximately 9% of MODY-like pedigrees, including several novel variants. More recent cohorts report a frequency of about 15–16% among genetically confirmed MODY cases. However, in broader young-onset diabetes populations, the prevalence is substantially lower (~0.2–0.3%). Overall, the mutation spectrum appears heterogeneous—with both inherited and de novo variants—but the overall frequency remains lower than that reported in European populations (12–14).
Molecular testing for MODY typically involves sequencing of known MODY genes. However, copy‐number variants (CNVs), such as large multi−exon deletions, can pose a diagnostic challenge. Large deletions in HNF1A or GCK can cause MODY but are not detected by standard PCR−based sequencing methods; they require gene−dosage assays such as multiplex ligation−dependent probe amplification (MLPA) (15). Here we report a 32−year−old Chinese male with early−onset, antibody−negative diabetes who was found to have a heterozygous HNF1A(NM_000545.8): ex1_10del. We detail his clinical presentation, laboratory and biochemical findings, and genetic results, and compare this case with previously reported HNF1A deletion cases. We discuss the diagnostic and therapeutic implications, highlighting lessons on precision medicine for monogenic diabetes.
Case presentation
2
The proband (III-1, Figure 1), a 32-year-old man, was diagnosed with diabetes at the age of 24 due to a two-year history of polydipsia and polyuria. At the time of evaluation, the body mass index (BMI) was 18.8 kg/m², indicating a lean body type. The HbA1c level was 8.7%, suggesting poor long-term glycemic control. Pancreatic autoantibodies were negative, indicating the absence of autoimmune diabetes. Renal function showed normal blood urea but slightly decreased creatinine and uric acid; electrolytes, β-hydroxybutyric acid, inflammatory markers, and hematological parameters were within reference ranges. Urine glucose was strongly positive (++++), while urine ketones were negative (Table 1). The results of the oral glucose tolerance test (OGTT) are summarized in Table 2. The fasting blood glucose was 6.27 mmol/L, which increased sharply to 21.94 mmol/L at 2 hours and remained elevated (17.38 mmol/L) at 3 hours, indicating impaired glucose tolerance and poor glycemic regulation. C-peptide and insulin levels both showed an initial increase following glucose load, peaking at 1 hour (C-peptide: 4.01 ng/mL; insulin: 15.3 μIU/mL) and gradually declining thereafter (C-peptide: 2.96 ng/mL; insulin: 12.5 μIU/mL at 3 hours). Although insulin and C-peptide secretion increased in response to glucose stimulation, the levels were relatively low in comparison to the marked hyperglycemia, suggesting an inadequate β-cell secretory response.
Pedigree of the family. Circles represent females, and squares represent males, and a diagonal line through a symbol indicates that the individual is deceased. Filled symbols indicate diabetic individuals and proband is shown by an arrow.
The patient’s family history was significant for diabetes on the paternal side (Figure 1). His father (II-1) was diagnosed with diabetes at age 42 and was reportedly managed as type 2 diabetes; he later developed diabetic nephropathy and ultimately died from end-stage renal disease. The patient’s paternal grandfather also had diabetes and died from related complications. The patient’s mother (II-2) has no history of diabetes, and no other relatives were available for genetic testing. Given the young age at onset, negative autoantibodies, preserved BMI, low C-peptide, and a suggestive family history (father and grandfather with diabetes), monogenic diabetes was suspected (1).
Genetic testing by NGS identified no pathogenic single nucleotide variants (SNVs) or insertions/deletions (indels) in any known MODY genes, suggesting the possible presence of a large deletion in the HNF1A gene. We then conducted MLPA analysis targeting the HNF1A gene, which identified a heterozygous HNF1A(NM_000545.8): ex1_10del. (Figure 2). According to ACMG criteria, this deletion is considered pathogenic, confirming the diagnosis of HNF1A-MODY.
Detection of a heterozygous HNF1A(NM_000545.8):ex1_10del by MLPA.
Based on this molecular diagnosis, the patient’s treatment was changed from insulin to an oral sulfonylurea. Insulin (mealtime aspart 8 units) was gradually withdrawn, and glimepiride was initiated at 1 mg daily. Within three months of switching to glimepiride alone, the patient’s blood glucose levels normalized. At follow-up one year later, his HbA1c had improved to 6.7% on glimepiride monotherapy, with no hypoglycemic episodes reported. This excellent response to low-dose sulfonylurea is consistent with the known pharmacogenetic signature of HNF1A-MODY (6, 10).
We summarized two representative reports of HNF1A exon or whole-gene deletions associated with HNF1A-MODY (Table 3). The first report by Ellard et al (15). described a cohort of probands screened using a custom MLPA assay covering GCK, HNF1A, and HNF4A (30 exons total). Multiple deletion patterns were identified in different probands, including HNF1A(NM_000545.8):ex1del, ex2_10del, and ex1_10del. Familial segregation analysis confirmed co-segregation of the deletions with early-onset diabetes. In this cohort, 4 out of 60 (6.7%) HNF1A-negative probands carried large deletions, highlighting the importance of gene dosage analysis in MODY diagnosis.Willson et al. (16) reported an in-frame HNF1A(NM_000545.8):ex2_3del in a HNF1A-MODY family, initially detected by array-CGH and confirmed by direct sequencing. The deletion co-segregated in three affected family members, all of whom developed both HNF1A-MODY and hepatocellular adenomas, indicating an extended phenotype beyond diabetes. These studies collectively demonstrate that HNF1A deletions may present with HNF1A-MODY and, in some cases, hepatic adenomas, underscoring the clinical relevance of CNV testing when sequencing alone is negative.
Materials and methods
3
Clinical evaluation
3.1
We obtained a detailed history and conducted a physical examination. Height, weight, and body mass index (BMI) were recorded. The family history of diabetes was documented through interviews with the patient and relatives.
Laboratory testing
3.2
OGTT and glycated hemoglobin (HbA_1_c) were measured. Pancreatic autoantibodies – including anti-human Insulin Antibody, anti-Glutamic Acid Decarboxylase Antibody, anti-Pancreatic Islet Cell Antibody, anti-Tyrosine Phosphatase Antibody. Additional workup included renal function tests, liver enzymes, lipid profile, and screening for microvascular complications (fundoscopic exam, neuropathy, and albuminuria).
Genetic analysis
3.3
Genomic DNA was extracted from peripheral blood leukocytes using standard protocols. For the identification of candidate variants, we first performed next-generation sequencing (NGS) using a targeted monogenic diabetes gene panel, which included HNF1A, GCK, HNF4A, HNF1B, PDX1, and other known MODY-associated genes. Sequencing libraries were prepared following the manufacturer’s instructions, and the NGS workflow included rigorous quality control metrics, with rigorous evaluation of sequencing quality metrics, including minimum read depth per targeted region, base-calling accuracy scores, and evenness of coverage across all exons, to guarantee robust and reproducible variant identification. Variants were called using validated bioinformatics pipelines and interpreted according to the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines for pathogenicity.
We performed multiplex ligation-dependent probe amplification (MLPA) to detect potential large deletions or duplications. Custom MLPA probes were designed to cover all 10 coding exons of HNF1A. MLPA assays were performed following standard protocols, and data quality was assessed based on probe signal intensity, inter-probe consistency, and reference sample comparison to ensure accurate detection of copy number variations.
All participants provided written informed consent, and the study was approved by the Medical Ethics Committee of Zhongnan Hospital of Wuhan University (No: 2023029).
Discussion
4
This case demonstrates that a heterozygous HNF1A(NM_000545.8):ex1_10del can cause a classic HNF1A-MODY phenotype. The clinical features – early-onset diabetes, low BMI, negative autoantibodies, low C-peptide indicating β-cell failure, and a multigenerational family history – strongly resembled those of MODY cases (1). Importantly, despite the unusual mutation type (a whole-gene deletion), the clinical phenotype did not differ qualitatively from that seen with point mutations or small indels in HNF1A. Like many HNF1A-MODY patients, our proband had progressive β-cell failure yet achieved excellent glucose control on an oral sulfonylurea. Our patient’s response to low-dose glimepiride – achieving HbA1c of 6.7% is consistent with the known sulfonylurea sensitivity of HNF1A-MODY (6). Since the genetic defect impairs glucose-stimulated insulin secretion upstream of sulfonylurea action (6). Over time, patients with HNF1A-MODY typically maintain good control on sulfonylureas, although progressive β-cell decline may eventually necessitate insulin.
HNF1A encodes a homeodomain-containing transcription factor crucial for regulating genes in pancreatic β cells (as well as in the liver and kidney) (6, 10). Haploinsufficiency of HNF1A leads to reduced transcription of insulin and other β-cell genes, resulting in impaired glucose-stimulated insulin secretion (6, 10). Furthermore, HNF1A regulates renal glucose handling; loss of one allele lowers the renal threshold for glucose reabsorption, causing glycosuria at lower blood glucose levels (17). Although we did not formally measure the renal threshold, the presence of polyuria despite only modest hyperglycemia in our patient suggests the renal glycosuria typical of HNF1A-MODY. The HNF1A(NM_000545.8):ex1_10del eliminates the entire coding sequence, effectively creating a null allele. Yet the resulting phenotype in our patient (and others with this deletion) is strikingly like that of patients with missense or smaller truncating mutations in HNF1A. This underscores that the phenotype of HNF1A-MODY is driven by the loss of HNF1A function, regardless of mutation type.
Multi-exon HNF1A deletions can underlie HNF1A-MODY and are often missed by standard sequencing, highlighting the critical role of CNV analysis (15). Although whole-gene or multi-exon deletions are most commonly associated with HNF1B-MODY (18), they also occur in HNF1A and GCK (15). In our patient, MLPA identified a HNF1A exons 1–10 deletion after panel sequencing was uninformative; without gene dosage analysis, the molecular diagnosis would have been missed, potentially leading to unnecessary insulin therapy. Prior studies support this approach: Ellard et al. detected HNF1A deletions in 6.7% of sequencing-negative HNF1A -MODY cases and recommended routine MLPA (15), and current guidelines likewise advocate including dosage analysis in MODY gene testing (19).
Beyond glycemic management, HNF1A mutations can have extrapancreatic manifestations. In particular, hepatic adenomas are reported in some patients with HNF1A-MODY (16, 20, 21). Although our patient currently has normal liver imaging and function tests, periodic abdominal surveillance has been arranged given this risk. Hepatic adenomas should be monitored due to potential hemorrhage or rare malignant transformation. These findings reinforce that comprehensive genetic testing, including CNV assessment, is essential not only for accurate HNF1A-MODY diagnosis and tailored diabetes management but also for anticipating extrapancreatic complications such as hepatic adenomas.
In summary, we identified a heterozygous HNF1A(NM_000545.8):ex1_10del in a Chinese patient with early-onset, antibody-negative diabetes, confirming HNF1A-MODY. The clinical features—including strong family history, sulfonylurea responsiveness, and absence of obesity or autoimmunity—were typical of HNF1A-MODY despite the atypical genetic lesion. This case highlights the importance of comprehensive genetic testing for MODY, which should include CNV analysis (e.g., MLPA) in patients with negative sequencing results. Early molecular diagnosis allowed tailored therapy, switching from insulin to low-dose sulfonylurea, and informed family screening and periodic surveillance for hepatic adenomas, exemplifying precision medicine in diabetes care.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dzhemileva LU Zakharova EN Goncharenko AO Vorontsova MV Rumyantsev SA Mokrysheva NG . Current views on etiology, diagnosis, epidemiology and gene therapy of maturity onset diabetes in the young. Front Endocrinol (Lausanne). (2024) 15:1497298. doi: 10.3389/fendo.2024.1497298, PMID: 39902162 PMC 11788143 · doi ↗ · pubmed ↗
- 2Gardner DS Tai ES . Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabetes Metab Syndr Obes. (2012) 5:101–8. doi: 10.2147/DMSO.S 23353, PMID: 22654519 PMC 3363133 · doi ↗ · pubmed ↗
- 3Kleinberger JW Pollin TI . Undiagnosed MODY: time for action. Curr Diabetes Rep. (2015) 15:110. doi: 10.1007/s 11892-015-0681-7, PMID: 26458381 PMC 4785020 · doi ↗ · pubmed ↗
- 4Tosur M Philipson LH . Precision diabetes: Lessons learned from maturity-onset diabetes of the young (MODY). J Diabetes Investig. (2022) 13:1465–71. doi: 10.1111/jdi.13860, PMID: 35638342 PMC 9434589 · doi ↗ · pubmed ↗
- 5Abu Aqel Y Alnesf A Aigha II Islam Z Kolatkar PR Teo A . Glucokinase (GCK) in diabetes: from molecular mechanisms to disease pathogenesis. Cell Mol Biol Lett. (2024) 29:120. doi: 10.1186/s 11658-024-00640-3, PMID: 39245718 PMC 11382428 · doi ↗ · pubmed ↗
- 6Li LM Jiang BG Sun LL . HNF 1A:From monogenic diabetes to type 2 diabetes and gestational diabetes mellitus. Front Endocrinol (Lausanne). (2022) 13:829565. doi: 10.3389/fendo.2022.829565, PMID: 35299962 PMC 8921476 · doi ↗ · pubmed ↗
- 7Herrera-Pulido JA Boisvert FM Boudreau F . Hepatocyte nuclear factor 4alpha multiple isoforms, their functions, and their interactomes. Proteomics. (2023) 23:e 2200372. doi: 10.1002/pmic.202200372, PMID: 37232233 · doi ↗ · pubmed ↗
- 8Sanchez-Cazorla E Carrera N Garcia-Gonzalez MA . HNF 1B transcription factor: key regulator in renal physiology and pathogenesis. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms 251910609, PMID: 39408938 PMC 11476927 · doi ↗ · pubmed ↗