Evaluating the Diagnostic Potential of Myxovirus Resistance Protein 1 (MX1) and Myxovirus Resistance Protein 2 (MX2) As Biomarkers in Idiopathic Inflammatory Myopathies
Raghavee Neupane, Mustafa Haider, Perry Smith, Marc M Kesselman

TL;DR
This paper reviews how MX1 and MX2 proteins might help diagnose autoimmune muscle diseases like dermatomyositis by analyzing their presence in muscle tissue and blood.
Contribution
The study systematically evaluates MX1 and MX2 as potential biomarkers for idiopathic inflammatory myopathies, focusing on their diagnostic accuracy and limitations.
Findings
MX1 is consistently elevated in muscle tissue of IIM patients, especially in dermatomyositis, with high tissue specificity.
MX2 is upregulated at the transcriptomic level, but lacks sufficient protein-level data and diagnostic clarity compared to MX1.
Blood-based MX1 measurements are promising but prone to false positives due to factors like disease activity and treatment history.
Abstract
Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of autoimmune muscle diseases characterized by proximal muscle weakness, systemic involvement, and high morbidity. Current diagnosis relies on clinical assessment, serology, and muscle biopsy, but challenges remain due to overlapping features and cases lacking clear biomarkers. Identifying molecular markers that reflect underlying disease pathways could improve diagnostic accuracy and patient stratification. This systematic review evaluates the diagnostic potential of myxovirus resistance protein 1 (MX1) and myxovirus resistance protein 2 (MX2), two interferon-inducible genes, as biomarkers in IIM and related conditions. A comprehensive literature search of PubMed, Embase, Web of Science, Ovid, and CINAHL (2015-2025) identified studies examining MX1 and/or MX2 in muscle disease. Eligible studies assessed diagnostic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
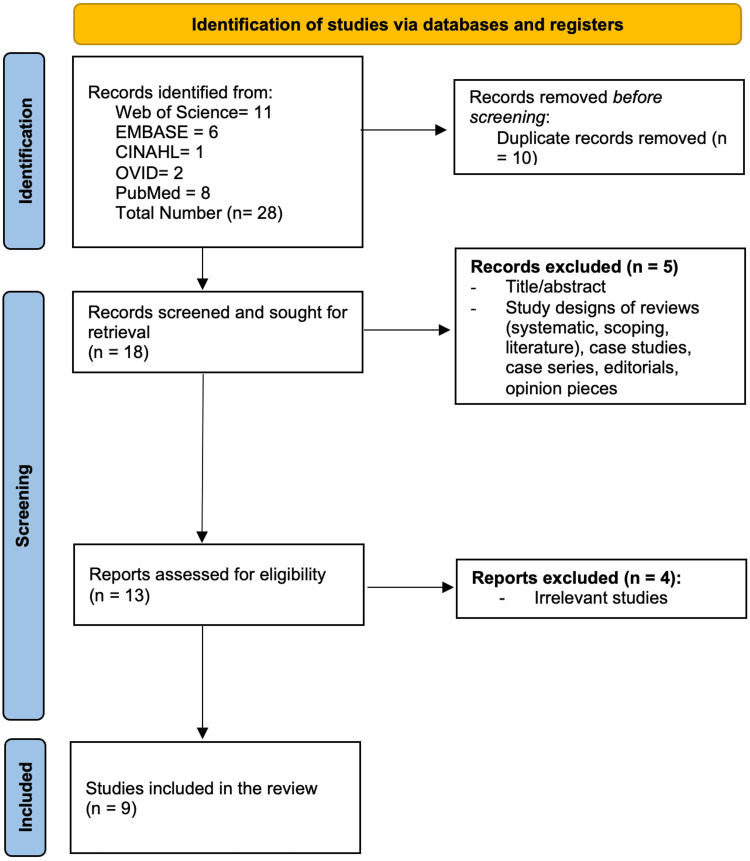 Figure 1
Figure 1| Author (Year) | Study Design | Size (M/F) | Mean Age (Years) | Medical History | Biomarker Measured (MX1/MX2) | Assay Method | Specimen Source | Expression Level Findings ↑/↓ in Disease vs. Control | Diagnostic Accuracy | Disease Activity/Severity | Conclusion |
| Tang et al., 2025 [ | Cross-sectional observational single-center study | 55 (75% F, 25% M) Subgroups: 25 MDA5(+) 19 ARS(+), 11 MSA(-) | 46 ± 12 | All had IIM. Subtypes: MDA5-positive dermatomyositis/clinically amyopathic DM, ARS-positive (anti-synthetase syndrome), or MSA-negative myositis. Many MDA5 positive has ILD | MX1 | Quantitative RT-PCR for ISG gene expression in peripheral blood, Serum cytokines/chemokines were measured through multiplex assay (34-cytokine panel), Nailfold capillaroscopy | Whole blood for molecular analyses, and nailfold capillaries for assessment of microvascular changes. | Increased | Not mentioned | MX1 levels were higher in active than in clinically stable patients, suggesting upregulation is associated with greater disease activity. | MX1/IFN-I pathway activation correlates with microvascular damage in myositis, indicating Nailfold Videocapillaroscopy may serve as a noninvasive tool to monitor disease activity and interferon status. |
| Lee et al., 2022 [ | Retrospective cross-sectional cohort study | 129 (dermatomyositis n=66, polymyositis n=63). 66% F, 34% M; ILD + (38% of patients): 63% F ILD - (62% of patients): 68% F | Not mentioned | All patients had IIM (dermatomyositis or polymyositis). 38% had ILD confirmed by chest CT | MX1, other markers assessed included T-cell (CD3, CD4, CD8), B-cell (CD20), macrophage (CD68, CD163) markers, and MHC class I and II | IHC in formalin-fixed muscle biopsy sections | Muscle biopsy tissue (quads and delts) | Increased | Not mentioned | In univariate analysis, MX1-positive muscle cells were associated with ILD (OR ≈1.89, 95% CI 1.16–3.16, p=0.012). However, in multivariate logistic regression (adjusting for CD4 and MHC/HLA expression), MX1 did not remain an independent predictor of ILD (adjusted OR ≈1.5, p=0.15). Instead, HLA-DR expression on muscle fibers was the strongest independent predictor of ILD. No analyses correlate MX1 with muscle weakness or CK levels reported. | ILD in myositis is linked to stronger muscle immune activation, shown by more MX1 in inflammatory cells and HLA-DR on myofibers, highlighting a connection between muscle inflammation and lung involvement. |
| Ngo et al., 2024 [ | Retrospective case series | 56 (F:M ~ 3:1) | 49.7 ± 16.1 | All patients were diagnosed with IIM. The cohort included immune-mediated necrotizing myopathy (IMNM, 58.9%), dermatomyositis (23.2%), overlap myositis (8.9%), anti-synthetase syndrome (5.4%), inclusion body myositis (1.8%), and polymyositis (1.8%). | MX1/2/3 | IHC on frozen muscle biopsy tissue (quads and delts) | Frozen muscle biopsy tissue (quads and delts) | Increase in MX1/2 expression was confined to dermatomyositis | Not mentioned | Nearly all patients had either upregulated MHC-I or membrane attack complex deposition; adding Mx did not significantly improve sensitivity due to its low frequency. No sensitivity/specificity was given specifically for Mx1/2/3 | Combining multiple immunomarkers increases diagnostic yield (e.g., MHC-I + MAC detected 96% of IIMs), and IFN-induced Mx protein is a useful indicator of dermatomyositis muscle pathology. |
| Hamano et al., 2017 [ | Nested case-control study | Cohort 1 (discovery, n=48): IPF (9/1), Idiopathic nonspecific interstitial pneumonia (INSIP) (4/4), autoimmune pulmonary alveolar proteinosis (aPAP) (4/6), Sarcoidosis (1/9), HC (8/2) Total = 54.17% M, 45.83% F Cohort 2 (validation, n=114): Anti-MX1+ (9/11), Anti-MX1- (70/24) Total = 69.29% M, 30.71% F | Cohort 1 (n=48): IPF: 62.9 years, INSIP: 58.5 years, aPAP: 49.3 years, Sarcoidosis: 58.9 years, HC: 49.9 years; Cohort 2 (n=114): MX1+ 75 years, MX1- mean: 73 years | Chronic fibrosing interstitial pneumonias: IPF, INSIP, aPAP, and sarcoidosis, were included to identify disease-specific autoantibodies across the ILD spectrum. Many patients met the criteria for interstitial pneumonia with autoimmune features | Anti-MX1 autoantibody | Protein microarray (antigen array). ELISA was used in a larger cohort to quantitatively measure anti-MX1 (of IgG, IgA, and IgM isotypes) and anti-ARS autoantibodies | Serum | Increase in anti-MX1 in ~17% of fibrosing IIPs - restricted to INSIP/autoimmune ILD, 0% in IPF | Anti-MX1 is found in a minority of NSIP cases (~17% of fibrosing ILD) and never in IPF, making it very good at ruling out IPF but only catching some NSIP cases. | Anti-MX1 marks a specific NSIP/autoimmune-type ILD group and helps distinguish it from IPF. Anti-MX1 doesn't predict severity as measured by DLCO, and is a unique antibody not seen with other myositis antibodies. | Anti-MX1 may serve as a diagnostic marker for fibrosing ILD, highly specific for non-IPF, present in ~17% of cases, and could help subclassify lIPs. |
| Kinoshita et al., 2023 [ | Case report | 1 M | 49 | Diabetes, Graves' disease, dyslipidemia, COVID-19, Hypertension, Complete atrioventricular block, and Cardiogenic shock | MX1 | IHC testing | Myocardial biopsy | Increase | Not mentioned | Testing for dermatomyositis autoantibodies, anti-SAE1, anti-MDA5, anti-TIF gamma, and anti-NXP-2, predicts COVID severity. | Myocardial biopsy shows a perifascicular staining pattern of MxA, which suggests a strong pathological association with dermatomyositis |
| Liang et al., 2021 [ | Cross-sectional and longitudinal studies | 103 F/51 M/30 controls | 49.0/47.0 (only median provided) | Dermatomyositis, Immune-mediated necrotizing myopathy | Gal-9 | ELISA, IHC staining | Subject blood serum, MRC-5 fibroblasts from American Type Culture Collection, lung biopsy | Increase in Gal-9 | Not mentioned | Gal-9 levels tend to increase with disease activity in anti-MDA5 DM and RP-ILD. | However, individual Gal-9 values cannot precisely quantify disease severity. |
| Ghang et al., 2024 [ | Case control | 129 IIM/73 controls | Not mentioned | Idiopathic inflammatory myopathies | CD3, CD4, CD8, CD20, CD68, CD163, MX1, MHC class I, MHC class II, HLA-DR | IHC staining | Muscle biopsy | Increase in CD163 and MHC Class I | AUC = 0.953, 0.961 (first is algorithms w/o IIM-histopathological features, second is with) | Both algorithms were able to identify 94.1% of patients who were diagnosed with IIM but did not meet the 2017 EULAR/ACR criteria for IIMs | Compared to histopathologic examination alone, the developed algorithms using CD163 and MHC Class I muscular expression had improved diagnostic accuracy |
| Lerkvaleekul et al., 2022 [ | Prospective multicentre study | 21 JDM/15 Healthy controls/7 Duchenne Muscular Dystrophy (controls for type I ISG expression and Siglec-1 expression on monocytes only) | 8.1/11.4 | Juvenile dermatomyositis (JDM) | Siglec-1, type 1 IFN | Multiplex immunoassay | Blood | Increase in Siglec-1 | AUC = 0.87 | Increased Siglec-1 expression "correlates with clinical disease activity", but without a quantitative relationship | Siglec-1 is a highly sensitive biomarker for risk of requiring treatment intensification |
| Shiota et al., 2022 [ | Case report | 1 F | 46 | Myelodysplastic syndrome, Acute GVHD with skin involvement | C5b9, MxA, MHC-II | IHC staining | Muscle biopsy | Increase in MxA and MHC-II | Not mentioned | Biopsy showed necrotic and regenerative fibers, interstitial fibrosis, and lymphocytic infiltrate, predominately CD8+ T cells. Expression of C5b9, MHC-II, and MxA has potential for diagnostic criteria of cGVHD-related IM. | This is the first reported case of cGVHD-related IM, showing increased MxA and MHC-I| on biopsy, consistent with the known immune pattern of cGVHD. |
| Author (Year) | JBI Tool Used | Risk of Bias |
| Tang et al., 2025 [ | Checklist for Analytical Cross Sectional Studies | Low |
| Lee et al., 2022 [ | Checklist for Cohort Studies | Low |
| Ngo et al., 2024 [ | Checklist for Case Series | Low |
| Hamano et al., 2017 [ | Checklist for Case Control Studies | Low |
| Kinoshita et al., 2023 [ | Checklist for Case Reports | Low |
| Liang et al., 2021 [ | Checklist for Analytical Cross Sectional Studies | Low |
| Ghang et al., 2024 [ | Checklist for Case Control Studies | Moderate |
| Lerkvaleekul et al., 2022 [ | Checklist for Cohort Studies | Low |
| Shiota et al., 2022 [ | Checklist for Case Reports | Low |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInflammatory Myopathies and Dermatomyositis · Viral Infections and Immunology Research · Rheumatoid Arthritis Research and Therapies
Introduction and background
Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of autoimmune disorders characterized by proximal muscle weakness and variable extramuscular involvement [1-3]. Up to 85% of patients with IIM have myositis-specific antibodies (MSAs), which strongly correlate with disease phenotypes and prognosis [4]. Major clinical subtypes include dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM), amyopathic dermatomyositis (ADM), antisynthetase syndrome (ASyS), and immune-mediated necrotizing myopathy (IMNM) [1]. IMNM, often triggered by statins, is associated with anti-3-hydroxy-3-methylglutarylcoenzyme A reductase (anti-HMGCR) antibodies and persistent muscle weakness despite statin cessation [5]. IIM arises from genetic, medication-related, and environmental risk factors, involving both innate and adaptive immune responses [1,6,7]. Adult IIM subgroups, except ASyS and IBM, have a two to sevenfold increased risk of malignancies, particularly lung and breast, compared to the general population, with the risk highest one year before and after diagnosis [1].
First-line treatment includes high-dose glucocorticoids in combination with immunosuppressive drugs and immunoglobulin therapy, though standardized protocols for dosage and duration are lacking [1,4,8]. Ten-year survival rates range from 20% to 90%, with the main causes of death including malignancies, cardiovascular disease, and lung disease [1]. In North America, IIM affects an estimated 2.9 to 34 individuals per 100,000, with risk factors including viruses, smoking, and human leukocyte antigen (HLA) susceptibility [1].
Diagnosis remains challenging due to clinical heterogeneity and the absence of formal criteria. Clinicians rely on the European Alliance of Associations for Rheumatology/American College of Rheumatology (EULAR/ACR) classification criteria, combining clinical features, muscle biopsy, MRI patterns, and serology [1]. Standard laboratory markers, such as creatine kinase, aldolase, lactate dehydrogenase, and transaminases, are nonspecific, and approximately 20-30% of patients are seronegative [1,4,9]. While muscle biopsy is the gold standard for the diagnosis of IIM, MRI is increasingly used to guide sampling [4,10].
IIM subgroups differ in tissue involvement: IMNM and IBM primarily affect skeletal muscle, whereas DM and ASyS are multi-organ diseases [1]. Interstitial lung disease (ILD) occurs in up to 78% of IIM patients, especially in ASyS and DM. Anti-MDA5 antibodies are associated with an increased risk of severe respiratory complications [1]. Arthritis is common in ASyS, often mimicking rheumatoid arthritis, while dysphagia affects up to 60% of IBM patients [1]. MSAs are most reliably detected by immunoprecipitation, though it is a time consuming and not routine [1]. Indirect immunofluorescence has limited sensitivity due to weak staining of cytoplasmic antigens [1]. Conventional laboratory markers and MSAs have been previously studied. However, their sensitivity and specificity are limited, highlighting the need for novel interferon (IFN)-inducible biomarkers such as myxovirus resistance protein 1 (MX1) and myxovirus resistance protein 2 (MX2). Validated high-risk biomarkers, such as anti-MDA5 antibodies, can guide early aggressive therapy in patients at risk of ILD [6]. Early diagnosis and treatment guided by validated biomarkers are imperative to maintain improved health outcomes, as IIM is a progressive disease that causes irreversible tissue damage [1].
Type I IFN (IFN-I) pathway activation is a key molecular signature in DM, where it is most prominently expressed compared to other IIM subtypes. Early microarray studies in juvenile DM muscle showed upregulation of interferon-stimulated genes (ISGs) by over 100-fold compared to the controls [11], a finding later confirmed in adult DM, establishing the “interferon signature” as a distinguishing molecular feature [12]. This aligns with pathology, as perifascicular atrophy, the hallmark lesion of DM, is enriched with ISG expression, such as MX1, MX2, and ISG15 [13]. Innate immune activation, via toll-like receptors and plasmacytoid dendritic cells, drives IFN-I expression in muscle and skin [14,15]. IFN activity varies across IIM subsets, with DM largely driven by IFN-I, ASyS, and IBM by IFN-γ signatures, and IMNM showing little IFN activation [16-19]. Anti-MDA5-positive DM, prone to rapidly progressive ILD, exhibits the highest IFN-I scores [20,21]. These findings support IFN-I signaling as a key mechanism in DM and provide a rationale for IFN-inducible biomarkers.
MX1 (encoding MxA) and MX2 (encoding MxB) are IFN-inducible GTPases consistently upregulated in DM [22]. Integrated analyses show their expression alone distinguishes DM from controls with high accuracy (area under the curve (AUC) >0.90) [23]. MxA has the strongest protein level evidence as a diagnostic biomarker. Sarcoplasmic MxA staining demonstrates approximately 77% sensitivity and 100% specificity for DM, even without perifascicular atrophy [24,25]. Other IIMs, such as ASyS, IMNM, and IBM, rarely stain positive, highlighting their specificity [24]. By contrast, evidence for MX2 is primarily transcriptomic, co-upregulated with MX1 and may complement diagnostic panels [22,23,26]. Its nuclear localization has hindered assay development, but reproducible transcriptomic signals suggest potential utility once protein-level detection is standardized, e.g., via fluorescence in situ hybridization (FISH). Peripheral blood assays measuring MX1 are emerging, particularly in anti-MDA5 DM, where transcript levels correlate with disease activity, offering potential for non-invasive monitoring [21].
Despite the evidence, limitations exist. MxA sensitivity varies across studies with the biopsy timing, site, disease activity, and prior treatment [24,27]. MX2 lacks standardized protein-level assays, and its added diagnostic value over MX1 is unclear. MX1 is also elevated in other systemic autoimmune diseases, including lupus and systemic sclerosis, as well as during viral infections, potentially leading to false positives [28,29]. Comparisons of MX1 and MX2 with other IFN-inducible biomarkers, such as ISG15 or OAS family members, or clinical tools, such as autoantibody testing, MRI, or EMG, underscore current limitations in comparative and prospective data [30]. Therefore, prospective studies are needed to determine whether MX1 and MX2 can reduce diagnostic delays or misclassification.
This systematic review evaluates MX1 and MX2 as diagnostic biomarkers in IIM, focusing on their sensitivity, specificity, and clinical utility for early detection and disease differentiation.
Review
Methods
A comprehensive literature search was conducted across Embase, Web of Science, Ovid (MEDLINE), PubMed, and CINAHL using keyword terms related to MX1 and MX2 (e.g., "MX1", "MX2", "myxovirus resistance protein 1", "myxovirus resistance protein 2"), inflammatory myopathies (e.g., "dermatomyositis", "polymyositis", "myositis", "muscle inflammation"), and diagnostic relevance (e.g., "biomarker", "diagnostic accuracy", "sensitivity", "specificity"). Boolean operators ("AND", "OR") were used to combine search terms appropriately. To ensure the recency of the articles, only English-language articles published between January 1, 2015, and July 1, 2025, were assessed. Titles and abstracts were screened using Rayyan (Rayyan Systems Inc., Cambridge, MA, USA), which supported duplicate removal and two-tier review (initial title/abstract screening followed by full-text evaluation) [31]. The search was limited to peer-reviewed, published studies. The Nova Southeastern University (NSU) library database was utilized to access databases and full-text articles.
Eligibility Criteria
The PICO framework guided our search strategy. The Population (P) included patients with confirmed or suspected muscle disease, including IIM. The Intervention/Index Test (I) was an assessment of MX1 or MX2 expression. Comparators (C), when available, included healthy controls or individuals with other neuromuscular or inflammatory conditions. The Outcomes (O) of interest were diagnostic performance metrics, including sensitivity, specificity, and overall diagnostic accuracy.
Studies were considered eligible if they investigated MX1 or MX2 in the context of muscle disease and assessed their diagnostic value, including sensitivity, specificity, diagnostic accuracy, or role in disease identification. Eligible study types included clinical trials, observational studies (cohort, case-control, or cross-sectional), case reports or case series, and relevant translational studies involving humans. Only studies involving patients with confirmed or suspected muscle disease were included.
Studies were excluded if they focused on MX1 or MX2 in contexts unrelated to muscle disease diagnosis, such as viral infections, oncology, or non-muscle autoimmune diseases. Articles were also excluded if they focused solely on treatment response, prognosis, or mechanistic pathways without assessing diagnostic performance. Reviews, editorials, commentaries, conference abstracts, animal-only studies, in vitro experiments, non-English publications, and articles without full-text access were also excluded.
Study Selection and Critical Appraisal of the Evidence
The initial search yielded 28 studies. After removing 10 duplicates, 18 unique articles remained. Of these, nine were excluded - five were conference abstracts and reviews, and four were unrelated to the topic. The remaining nine studies were screened through a blinded, two-tiered review process conducted by two independent reviewers, with a third reviewer resolving any discrepancies. Quality assessment was performed using the Joanna Briggs Institute Critical Appraisal Tools, which categorized risk of bias as low (>70%), moderate (50-70%), or high (<50%). Following this assessment, all nine articles that met the inclusion criteria were included in the final analysis.
The review process adhered to Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, and a PRISMA flow diagram was developed to illustrate the article selection process (Figure 1).
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram illustrating the study selection process
Results
Sample sizes ranged from individual patients to cohorts of up to 129 participants [32]. Most studies focused on patients with IIM, including DM, PM, and anti-synthetase syndrome. Four studies evaluated patients with fibrosing ILD, and one case report described MX1 expression in a patient with COVID-19-related myocarditis. Across cohorts, participants were predominantly female, representing as high as 75% of the study populations, and the mean age of participants generally ranged from the late 40s to early 60s.
MX1 was the most frequently investigated biomarker, examined in seven of the nine studies, either alone or in combination with MX2 or other IFN-stimulated genes [32-37]. Methods of assessment included quantitative reverse transcriptase PCR (qRT-PCR) of whole blood RNA (n=1) [33], immunohistochemistry (IHC) of muscle or myocardial biopsies (n=6) [32,34,36-38], protein microarrays (n=1) [35], and enzyme-linked immunosorbent assays (ELISA) for anti-MX1 autoantibodies (n=2) [35,38].
Across studies, MX1 expression was consistently upregulated in IIM compared with healthy and disease controls (n=9). DM cases demonstrated the strongest muscle staining, whereas non-IIM muscle tissues showed little to no expression. Elevated MX1 levels were correlated with markers of disease activity, including higher creatine kinase concentrations (n=1) and clinical muscle weakness (n=1). In the myocarditis case report, MX1 was markedly expressed in myocardial tissue, suggesting a broader role as a marker of IFN-mediated injury outside of skeletal muscle [36]. Anti-MX1 autoantibodies were identified in approximately 17% of patients with fibrosing ILD [35], particularly in those with non-specific interstitial pneumonia (NSIP). In these cohorts, the antibodies distinguished NSIP from idiopathic pulmonary fibrosis (IPF) with relatively high specificity but limited sensitivity.
MX2 was evaluated in only one of the included studies, all of which assessed its expression in the context of IFN-stimulated gene signatures [35]. Compared with MX1, MX2 expression was less consistently reported and generally appeared at lower levels across tissues. In DM cohorts, MX2 was modestly upregulated in both blood and muscle but did not show as strong an association with clinical disease activity as MX1. One study incorporating MX2 into a multi-gene panel found that it contributed to overall IFN-I scores but added limited discriminatory power when analyzed independently [34]. No studies examined anti-MX2 autoantibodies, and none specifically investigated their diagnostic or prognostic utility in IIM or related conditions.
Only three studies formally evaluated diagnostic or prognostic performance [35,37,39]. Among these, the presence of anti-MX1 antibodies showed potential in differentiating fibrosing ILD subtypes, and elevated MX1 expression was proposed as a biomarker of IFN-I pathway activation in IIM. Despite these findings, validation in larger, multi-center cohorts remains limited (Table 1).
The methodological quality and risk-of-bias assessment of the included studies, conducted using the Joanna Briggs Institute Critical Appraisal Tools, is summarized in Table 2.
Discussion
In this systematic review, we synthesized evidence on the role of MX1 and MX2 in IIM. The findings reinforce the central role of the IFN-I pathway in IIM and highlight MX1 as a reproducible and biologically grounded biomarker, particularly in DM [31-33,36]. Across heterogeneous study designs and assay platforms, MX1 was consistently upregulated in affected muscle and blood, correlating with markers of disease activity, including creatine kinase, muscle weakness, and inflammation [32]. These findings highlight MX1 as a clinically relevant biomarker for assessing disease activity and guiding treatment, particularly in IIM cases lacking serologic markers.
Sensitivity of MX1 as a biomarker may be influenced by disease phase and timing of tissue sampling. Activation of the IFN pathway is the most pronounced early in DM, prior to the development of extensive fibrosis or fatty replacement. As a result, MX1 expression may be most informative during early or preclinical diagnosis, when conventional histopathologic findings are subtle or absent. If confirmed in prospective studies, MX1 could enable early diagnosis and support the timely initiation of immunomodulatory therapy, thereby reducing the risk of irreversible muscle damage.
MX1 may also help bridge between tissue-based and blood-based diagnostics. Although muscle biopsy remains the diagnostic gold standard, it is invasive and may be nondiagnostic depending on sampling site and disease stage. Similarly, serologic testing for MSAs is clinically useful but remains negative in up to 30% of patients. In this context, MxA IHC provides a direct tissue-based measure of IFN activation and demonstrates high specificity, even in cases lacking classic histologic features. This suggests that MX1 could serve as a complementary diagnostic marker, particularly in seronegative or diagnostically ambiguous cases.
Beyond skeletal muscle, MX1 expression has been implicated in systemic manifestations. Anti-MX1 autoantibodies have been detected in patients with fibrosing ILD, and MX1 expression has been reported in COVID-19-associated myocarditis, suggesting broader relevance as a marker of IFN-driven injury [34,35]. By contrast, MX2 has been less well studied. Evidence suggests modest upregulation in DM, with weaker associations with disease activity than MX1 and limited independent diagnostic or prognostic value [33]. No studies evaluated anti-MX2 antibodies, leaving their clinical significance uncertain.
Limitations of the current literature include small, single-center studies and heterogeneity in assay methods, ranging from quantitative reverse transcription polymerase chain reaction (qRT-PCR) to IHC and serologic testing [31-36]. Few studies systematically evaluated diagnostic accuracy (e.g., sensitivity, specificity, or area under the curve (AUC)) or prognostic value [34,36,38], but none examined the cost-effectiveness or feasibility of integrating MX1 or MX2 testing into clinical practice. Importantly, longitudinal studies are lacking, limiting conclusions about whether MX1 can track disease progression or predict therapeutic response over time. Standardization remains a major barrier to the widespread adoption of MX1 testing. Variability in immunohistochemical protocols, antibody clones, scoring thresholds, and biopsy site selection likely contributes to inter-study heterogeneity. Establishing consensus guidelines for MX1 assessment, similar to those developed for myositis-specific autoantibodies, will be critical before MX1 can be incorporated into routine diagnostic workflows. Multi-center validation using harmonized protocols should therefore be a priority for future research [24,25,27].
Beyond diagnosis, MX1 may have important implications for precision medicine in inflammatory myopathies. As targeted therapies directed against IFN signaling pathways continue to emerge, reliable biomarkers of pathway activation are needed to guide patient selection and therapeutic response. MX1 expression could potentially identify patients most likely to benefit from IFN-targeted or Janus kinase-inhibitor therapies, thereby avoiding unnecessary exposure to broad immunosuppression in patients without active IFN signaling. This positions MX1 not only as a diagnostic biomarker, but also as a candidate predictive biomarker for treatment stratification [41].
Taken together, MX1 emerges as a promising biomarker of IFN pathway activation in IIM, with potential applications in diagnosis, disease monitoring, and stratification of systemic involvement such as ILD [31-34]. However, its clinical utility remains preliminary, and MX2 is even less characterized.
Conclusions
This review highlights MX1 as a consistent marker of IFN-I pathway activation in IIM and related conditions. Across multiple studies, MX1 expression was elevated in muscle and blood, correlated with disease activity, and showed potential for identifying systemic involvement, such as ILD. The presence of anti-MX1 autoantibodies further suggests a role in stratifying fibrosing lung disorders, although sensitivity remains limited. In contrast, evidence for MX2 is sparse, with only modest upregulation reported and little independent diagnostic or prognostic value. Overall, current findings support MX1 as a promising biomarker for diagnosis, disease monitoring, and potential therapeutic stratification in IFN-mediated diseases. However, the evidence remains preliminary, and widespread clinical adoption will require validation in larger, multicenter cohorts with standardized methodologies. Future work should also clarify the clinical significance of MX2 and assess whether combined IFN signatures can provide added value beyond MX1 alone.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Idiopathic inflammatory myopathies Nat Rev Dis Primers Lundberg IE Fujimoto M Vencovsky J 86720213485778010.1038/s 41572-021-00325-7PMC 10425161 · doi ↗ · pubmed ↗
- 2Metabolic profiling of patients with different idiopathic inflammatory myopathy subtypes reveals potential biomarkers in plasma Clin Exp Med Zhao Q Hu Q Meng S 341734292320233710365210.1007/s 10238-023-01073-6PMC 10618316 · doi ↗ · pubmed ↗
- 3Imaging biomarkers in the idiopathic inflammatory myopathies Front Neurol Zubair AS Salam S Dimachkie MM Machado PM Roy B 11460151420233718157510.3389/fneur.2023.1146015 PMC 10166883 · doi ↗ · pubmed ↗
- 4Idiopathic inflammatory myopathies: a review Intern Med J Ashton C Paramalingam S Stevenson B Brusch A Needham M 8458525120213415576010.1111/imj.15358 · doi ↗ · pubmed ↗
- 5Statin-associated autoimmune myopathy: current perspectives Ther Clin Risk Manag Tiniakou E 4834921620203258154310.2147/TCRM.S 197941 PMC 7266943 · doi ↗ · pubmed ↗
- 6Biomarkers and autoantibodies of interstitial lung disease with idiopathic inflammatory myopathies Clin Med Insights Circ Respir Pulm Med Yoshifuji H 141146920152708132210.4137/CCRPM.S 36748 PMC 4820065 · doi ↗ · pubmed ↗
- 7Epidemiology of the idiopathic inflammatory myopathies Nat Rev Rheumatol Khoo T Lilleker JB Thong BY Leclair V Lamb JA Chinoy H 6957121920233780307810.1038/s 41584-023-01033-0 · doi ↗ · pubmed ↗
- 8Trial of intravenous immune globulin in dermatomyositis N Engl J Med Aggarwal R Charles-Schoeman C Schessl J 1264127838720223619817910.1056/NEJ Moa 2117912 · doi ↗ · pubmed ↗