Direct Fluoroformylation of the C3‐Position of Indoles with 2,4‐Dinitro(trifluoromethoxy)benzene as Fluorocarbonyl Source
Lilian Wisson, Gilles Hanquet, Fabien Toulgoat, Thierry Billard, Frédéric R. Leroux, Armen Panossian

TL;DR
A new metal-free method for adding a fluorocarbonyl group to indoles and similar compounds is developed, avoiding toxic reagents and high pressures.
Contribution
A metal-free, rapid fluoroformylation method using DNTFB as a fluorocarbonyl source is introduced for C3-position of indoles.
Findings
Fluoroformylation of indoles is achieved without metal catalysts or carboxylic acids.
The method uses 2,4-dinitro(trifluoromethoxy)benzene (DNTFB) to generate fluorophosgene in situ.
The process can be combined with amidification in a one-pot reaction.
Abstract
We herein report the direct fluoroformylation of indoles and other heteroaromatic cycles. Acyl fluorides are very useful moieties in coupling reactions with or without metal. However, they are usually obtained from the corresponding carboxylic acids or from aryl halides in pallado‐catalyzed carbonylation/fluorination reactions. Our method uses fluorophosgene generated in situ from 2,4‐dinitro(trifluoromethoxy)benzene (DNTFB) as a fluoroformylating agent without any metal and from carboxylic acid‐free heteroaromatic rings. Moreover, our method can be telescoped with amidification reactions in a one‐pot process. Indoles bearing acyl fluoride moieties are generally obtained by dehydroxyfluorination of carboxylic acids or by fluoroformylation mediated by palladium catalysts with the disadvantage of requiring toxic reagents or very high pressures. We describe herein the direct, metal‐free,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 SCHEME 1
SCHEME 1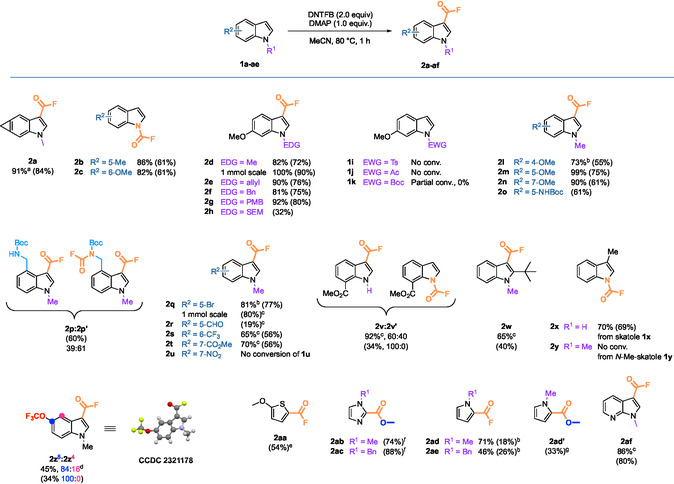 SCHEME 2
SCHEME 2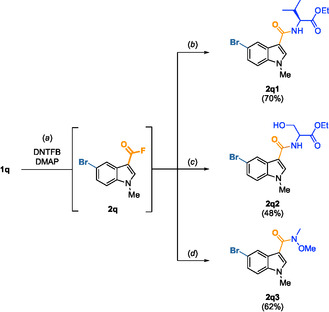 SCHEME 3
SCHEME 3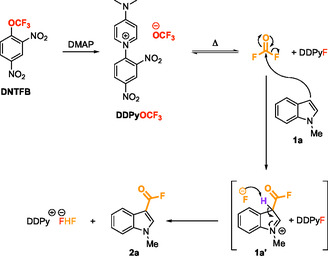 SCHEME 4
SCHEME 4|
| ||||
|---|---|---|---|---|
| Entry | X | Y | Time |
Yield ( |
| 1 | 18.1 | 9.1 | 16 h | 84% (80%) |
| 2 | 2.1 | 2.1 | 16 h | No conversion |
| 3 | 4.0 | 2.0 | 16 h | 85% |
| 4 | 4.0 | 2.0 | 1 h | 85% |
| 5 | 2.0 | 1.0 | 1 h | 91% (84%) |
| 6 | 1.0 | 0.5 | 70 min | 85% |
- —Agence Nationale de la Recherche10.13039/501100001665
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFluorine in Organic Chemistry · Synthesis and Characterization of Pyrroles · Catalytic C–H Functionalization Methods
Introduction
1
Acyl fluorides (R‐COF) are a topic of interest in synthetic chemistry. Indeed, these carboxylic derivatives show a better stability toward hydrolysis [1, 2, 3], alcoholysis [2, 3], or aminolysis [4] than other acyl halides, making them easier to handle. Several syntheses of R‐COF have been developed from carboxylic acids by dehydroxylative fluorination, from acyl halides or carboxylic anhydrides by exchange with fluoride, by metal‐catalyzed fluorination of aryl halides with carbonyl insertion or from aldehydes by hypervalent bromine‐mediated or radical‐based oxidative fluorination [5, 6, 7, 8, 9, 10, 11]. Although acyl fluorides are more stable than other halogenated equivalents, they remain reactive enough to be used as substrates, such as in peptide synthesis [12, 13, 14, 15, 16] and transition metal‐catalyzed coupling reactions [6, 16, 17, 18, 19, 20]. Indoles, on the other hand, are essential scaffolds in pharmaceuticals [21, 22] and agrochemicals [23, 24]. Many active molecules contain indole scaffolds that are functionalized in the C3 position. The introduction of an acyl fluoride at this position could therefore be of great interest from a synthesis perspective. In the literature, only two examples of C3‐fluoroformylated indoles have been reported; however, they required the use of harmful reagents, expensive and exotic palladium catalysts or very high pressures which limit their applicability [25, 26]. We present here a new, inexpensive, and easy‐to‐use method for the direct fluoroformylation of indoles to access indole‐3‐carbonyl fluoride scaffolds.
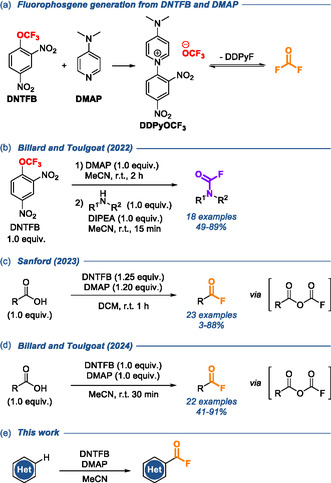
Recently, after using the combination of 2,4‐dinitro(trifluoromethoxy)benzene (DNTFB) and 4‐(dimethylamino)pyridine (DMAP) as trifluoromethoxide anion source [27, 28, 29, 30], our groups reported its use as a source of fluorophosgene (COF_2_) for the synthesis of carbamoyl fluorides with good to excellent yields (Scheme 1b) [31–33], This method is based on the degradation of the trifluoromethoxide anion (CF_3_O^–^) into COF_2_ and fluoride (Scheme 1a) and thus avoids the direct handling of toxic fluorophosgene to introduce the COF moiety on amines. Additionally, DNTFB and DMAP are inexpensive and easy‐to‐use reagents [28, 29]. During the latter project, we observed that the carboxylic acid part of ciprofloxacin underwent conversion into the corresponding acyl fluoride [31]. Therefore, a similar method was applied concomitantly by Sanford et al. [34] (Scheme 1c) and our groups [35] (Scheme 1d) to convert carboxylic acids into acyl fluorides [36] with excellent yields, without using polyfluorosulfur‐based reagents nor hydrofluoric acid. After employing DNTFB and DMAP in N‐ and O‐fluoroformylation, we were naturally interested in C‐fluoroformylation. We chose indoles as substrates due to the reasons mentioned above and due to their good C‐nucleophilicity.
The use of DNTFB and DMAP for (a) the generation of fluorophosgene and the fluoroformylation of (b) amines, (c,d) carboxylic acids, and (e) heteroaromatic rings.
Results and Discussion
2
On the basis of these methods, we investigated the direct fluoroformylation of 1‐methylindole 1a using the conditions we developed for the trifluoromethoxylation of arynes [37] employing a large excess of DMAP and even larger excess of DNTFB (Table 1, entry 1). A complete conversion of the starting indole 1a to the desired 1‐methyl‐1H‐indole‐3‐carbonyl fluoride 2a was obtained, with a good ^1^H NMR yield of 84%. The importance of the 2:1 ratio between DNTFB and DMAP was highlighted by the attempt to reduce it to 1:1, in order to increase the rate of degradation of trifluoromethoxide anion into fluorophosgene [28], which surprisingly resulted in the absence of conversion of 1a (entry 2). Increasing the amount of DNTFB and DMAP used in a 1:1 ratio led to the full conversion of the starting material, but no fluoroformylated indole was observed (see Table S1). An intermediate 1.3:1 ratio gave a complete conversion but a lower yield (see Table S1). Going back to a 2:1 ratio and after reducing the amount of reagents and the reaction time (Table 1, entries 3–5), and after further optimization (full optimization available in Table S1), we obtained 2a with a NMR yield of 91% using 2.0 equivalents of DNTFB and 1.0 equivalent of DMAP in 1.5 mL of reaction grade acetonitrile (MeCN) at 80°C for 1 h, affording a 84% yield after purification (Table 1, entry 5). Noteworthily, an additional decrease of the amount of DNTFB and DMAP, but still maintaining a 2:1 ratio, led to incomplete conversion but a good 85% yield (entry 6).
With these conditions in hand, we conducted a study on the scope of the reaction (Scheme 2). We first investigated the influence of the protecting group of the indole nitrogen atom. When indoles were unprotected, the corresponding carbamoyl fluorides were obtained: 2b with 86% ^1^H NMR yield and 61% isolated yield, and 2c with 82% ^1^H NMR yield and 61% isolated yield. With the electron‐poor unprotected indole 1v, 92% of a 60:40 mixture of acyl fluoride 2v and carbamoyl fluoride 2v′ was observed by ^1^H NMR analysis. After purification, 2v was isolated as the unique product with 34% yield. 6‐Methoxyindoles *N‐*protected with electron‐donating groups (EDG) gave moderate to excellent yields. Indeed, whereas the N‐SEM protected product 2h was obtained in 32% yield only, the N‐methyl derivative 2d was isolated with a more satisfying 72% yield which could be improved to 90% on larger scale, while the N‐allyl, N‐benzyl, and N‐*para‐*methoxybenzyl protected derivatives (2e, 2f and 2g respectively) were obtained with yields of 75%–80% after purification. However, unsurprisingly, indoles that were N‐protected with electron‐withdrawing groups (EWG) 1i and 1j remained unconverted due to the strong decrease of nucleophilicity. N‐Boc‐protected substrate 1k did not lead to any product 2k despite a partial conversion observed by ^1^H NMR (Scheme 3). This could be explained by a degradation of the starting indole in the acidic medium formed during the reaction (Scheme 4).
Substrate scope of the fluoroformylation with DNTFB/DMAP. Yields were calculated by 1H NMR analysis with bromochloromethane as internal standard. Isolated yields are given in parentheses. aToluene was used as internal standard. b2 h reaction time. cDNTFB (6.0 equiv.), DMAP (3.0 equiv.), 80°C, 2 h. dSynthesized from the corresponding aryne precursor following the previously reported aryne trifluoromethoxylation procedure[37] eDNTFB (6.0 equiv.), DMAP (3.0 equiv.), 80°C, 16 h. fIn situ esterification by addition of methanol on the concentrated crude mixture. gIn situ esterification by addition of methanol and triethylamine on the concentrated crude mixture.
One‐pot, two‐step fluoroformylation and amidification of 1q. (a) DNTFB (2.0 equiv.), DMAP (1.0 equiv.), MeCN, 80°C, 2 h. (b) Ethyl L‐valinate hydrochloride (3.2 equiv.), KOH (7.0 equiv.), MeCN, 80°C, 18 h. (c) Ethyl‐DL‐serinate hydrochloride (3.2 equiv.), NEt3 (7.0 equiv.), MeCN, 80°C, 18 h. (d) N‐Methoxymethylamine hydrochloride (3.2 equiv.), KOH (12.0 equiv.), DCM, 80°C, 18 h.
Mechanism of the fluoroformylation of 1a.
Substituents of the N‐methylated indole core were also varied. Indoles bearing electron‐donating substituents on various positions of the 6‐membered ring reacted to afford acyl fluorides 2l‐o with yields ranging from 55% to 75%. In the case of N‐Boc‐protected 4‐(aminomethyl) substrate 1p, a mixture of mono‐ and bis‐fluroformylated indoles 2p and 2p′ was obtained with a 39:41 ratio and 60% yield after isolation. This indicates that primary amines must be fully protected to prevent the formation of carbamoyl fluoride in addition to the fluoroformylation of the C3 position of the indole. Interestingly, such N‐Boc‐protected carbamoyl fluorides are, to the best of our knowledge, unprecedented. Pursuing the study of the scope of the fluoroformylation reaction, deactivating groups were also assessed. Longer reaction times or more DNTFB and DMAP equivalents were needed to reach full conversions and good yields for 2q,s–t. Although the aldehyde moiety was partially tolerated as 2r was obtained with a moderate yield of 19% explained by deoxyfluorination side‐reactions, the highly deactivating nitro group gave no conversion in our conditions. It is also worth‐mentioning that 5‐bromo‐1‐methyl‐1H‐indole‐3‐carbonyl fluoride 2q, displaying a functionalizable C–Br bond was also obtained with a good yield of 80% on a 1.0 g scale using 6.0 equivalents of DNTFB, 3.0 equivalents of DMAP and a reaction time of 2 h.
Steric hindrance had a negative impact on the reaction. 4‐Methoxy‐1‐methyl‐1H‐indole‐3‐carbonyl fluoride 2l was isolated with 55% yield, which was lower than for indoles substituted by methoxy groups on other positions of the carbocycle (2d, m–n). Although the yield difference was not always large, unlike its congeners, the isolated product 2l showed a poor stability at room temperature and degraded into the corresponding carboxylic acid after 24 h. A similar observation was made with the 2‐(tert‐butyl)‐substituted product 2w which not only required 6.0 equivalents of DNTFB, 3.0 equivalents of DMAP and 2 h of reaction to be formed with a 40% yield, but also proved unstable at room temperature, degrading to give back the starting product 1w. We hypothesized a hydrolysis of the acyl fluoride to the corresponding carboxylic acid followed by a decarboxylation of the latter. When the C3 position was substituted, fluoroformylation did not occur, as observed with N‐methylated skatole 1y. However, when skatole was used, the corresponding carbamoyl fluoride was obtained with 70% ^1^H NMR yield and 69% isolated yield.
Finally, we wondered if it would be possible, with the DNTFB/DMAP system, to carry out at the same time the fluoroformylation and the trifluoromethoxylation [37] of an indolyne derivative. Gratifyingly, indoles 2z ^ 5 ^ and 2z ^ 4 ^, which differ by the position of the OCF_3_ group due to the two electrophilic positions of the aryne intermediates, were formed in a moderate 45% ^19^F NMR yield and an 84:16 regioisomeric ratio of the 5‐ and 4‐OCF_3_ products, respectively. We isolated the 5‐OCF_3_ isomer 2z ^ 5 ^ with 34% yield and its structure was confirmed by X‐ray diffraction crystallography (CCDC number 2 321 178).
Other heteroaromatic cycles underwent fluoroformylation. A good yield of 54% was obtained for 5‐methoxythiophene‐2‐carbonyl fluoride 2aa. However, due to its lower reactivity compared with indoles, it required more equivalents of DNTFB and DMAP and an overnight reaction time. Unfortunately, we were unable to isolate fluoroformylated imidazoles due to hydrolysis during the aqueous workup. Therefore, we derivatized them into the corresponding methyl esters by stirring concentrated crude reaction mixtures in methanol. Using this method, we were able to isolate methyl 1‐methyl‐1H‐imidazole‐2‐carboxylate 2ab and methyl 1‐benzyl‐1H‐imidazole‐2‐carboxylate 2ac with excellent yields of 74% and 88% respectively. N‐Methyl and N‐benzyl pyrroles gave corresponding acyl fluorides 2ad and 2ae with 71% and 46% NMR yields. They were isolated in modest yields of 18% and 26% respectively. The difference between NMR and isolated yields of 2ad and 2ae could be attributed to the instability of the product during workup or the column. To overcome this issue, the derivatization method was employed with N‐methylpyrrole to give a modest isolated yield of 33% of methyl 1‐methyl‐1H‐pyrrole‐2‐carboxylate 2ad′ after treatment of the concentrated crude with methanol and triethylamine. Finally, we obtained 1‐methyl‐1H‐pyrrolo[2,3‐b]pyridine‐3‐carbonyl fluoride 2af with an excellent yield of 80%. However, 3‐bromofuran, benzofuran, and benzothiophene did not undergo conversion, indicating insufficient nucleophilicity to react with fluorophosgene under our conditions (refer to Supporting Information for further details). Electronically enriched aromatic rings such as N,N‐dimethylaniline or 1,3,5‐trimethoxybenzene were not reactive either under our conditions.
Acyl fluorides are often used in peptide synthesis [9, 12, 13, 14, 15, 16], which encouraged us to merge our new method with amidification reactions. The amidification protocol was inspired by the work of Qin et al*.* [38] who used in situ generated acyl fluorides to synthesize amide bonds with primary and secondary amines as nucleophiles in the presence of KOH. To take into account the reaction between amino acids and 2,4‐dinitrofluorobenzene (DNFB, Sanger's reagent) [39], a side‐product of the fluoroformylation reaction, we had to use high excesses of amine and base. Therefore, we performed the fluoroformylation of 1q followed by an amide bond formation in a one‐pot, two‐step procedure (Scheme 3). Valine and serine derivatives produced 2q1 and 2q2 with 70% and 48% yields respectively. Triethylamine was used as the base with the serine derivative to avoid transesterification reactions occurring when KOH was employed. Similarly, we obtained the analogous Weinreb amide 2q3 with a good yield of 62%. These results along with the esterification reaction used to obtain 2ab and 2ac demonstrate the compatibility of our method with nucleophilic additions on the formed acyl fluorides in one‐pot strategies.
Finally, the plausible mechanism of the reaction is depicted in Scheme 4. After formation of the DDPyOCF_3_ salt from DNTFB and DMAP, fluorophosgene is generated in situ by the degradation of the CF_3_O^–^ ion. An aromatic electrophilic substitution (S_E_Ar) by fluorophosgene then takes place at the nucleophilic C3 carbon of the indole. The indolium intermediate formed is then deprotonated to restore the aromaticity of the molecule. Control experiments were conducted (see Supporting Information for more details) and confirmed that the fluoride anion was able to play the role of the proton trap, which concurs with the findings of Sanford et al. [34] and Zhang et al. [32, 33, 36] and that fluorophosgene alone is able to mediate the fluoroformylation reaction. However, a sufficient activation of DNTFB is required to obtain fluoroformylated heteroaromatic rings in good yields.
Conclusion
3
In conclusion, we have developed a new, easy‐to‐use, and cheap method for the direct fluoroformylation of indoles on the C3 position using the combination of DNTFB and DMAP as fluoroformylating system. The method is safer than the direct use of gaseous fluorophosgene, a toxic gas, which is here produced in situ in controlled amount. This method is compatible with both electron‐donating groups on the nitrogen atom or the benzene ring of indoles, and with electron‐withdrawing groups on the arene ring, resulting in fluoroformylated indoles with good to excellent yields. However, protecting the nitrogen atom of the indole core with an electron‐withdrawing group prevented conversion while leaving it unprotected resulted in the formation of the corresponding carbamoyl fluorides unless an electron‐poor ring was used. This strategy has also been successfully applied to other heteroaromatic cycles, such as pyrroles, imidazoles or thiophenes. Additionally, the mild process was tolerated by acid‐sensitive functions, and esterification and amidification reactions were carried out in a one‐pot, two‐step process starting from the C3‐H indole with good overall yields.
Supporting Information
Additional supporting information can be found online in the Supporting Information Section. The authors have cited additional references within the Supporting Information [40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70]. Supporting Table S1: Optimization of the fluoroformylation of 1‐methylindole 1a. Supporting Table S2: Specific rotation measurements for a solution of 2 q 1 in MeOH. Supporting Table S3: Atomic coordinates (× 10^4^) and equivalent isotropic displacement parameters (Å^2^ × 10^3^) for **2z^5^ **. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. Supporting Table S4: Bond lengths [Å] and angles [°] for **2z^5^ **. Supporting Table S5: Anisotropic displacement parameters (Å^2^ × 10^3^) for *2z^5^ . The anisotropic displacement factor exponent takes the form: ‐2 pi^2^ [ h^2^ a^2^ U11 + … + 2 h k a b U12 ]. Supporting Table S6: Hydrogen coordinates (× 10^4^) and isotropic displacement parameters (Å^2^ × 10^3^) for **2z^5^ **. Supporting Table S7: Torsion angles [°] for **2z^5^ **.
Funding
This study was supported by Agence Nationale de la Recherche (grant ANR‐ 20‐CE07‐0004‐02 (Ap‐PET‐I)).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1C. G. Swain and C. B. Scott , “Rates of Solvolysis of Some Alkyl Fluorides and Chlorides,” Journal of the American Chemical Society 75 (1953): 246.
- 2B. D. Song and W. P. Jencks , “Mechanism of Solvolysis of Substituted Benzoyl Halides.,” Journal of the American Chemical Society 111 (1989): 8470.
- 3D. N. Kevill and M. J. D’Souza , “Correlation of the Rates of Solvolysis of Benzoyl Fluoride and a Consi deration of Leaving‐Group Effects,” The Journal of Organic Chemistry 69 (2004): 7044.15471451 10.1021/jo 0492259 · doi ↗ · pubmed ↗
- 4M. L. Bender and J. M. Jones , “Nucleophilic Reactions of Morpholine with the Benzoyl Halides. The Presence of an Element Effect,” The Journal of Organic Chemistry 27 (1962): 3771.
- 5F. Aldabbagh , “Acid Halides.” In Comprehensive Organic Functional Group Transformations II, (Elsevier, 2005). 1.
- 6Y. Ogiwara and N. Sakai , “Acyl Fluorides in Late‐Transition‐Metal Catalysis,” Angewandte Chemie International Edition 59 (2020): 574.30969455 10.1002/anie.201902805 · doi ↗ · pubmed ↗
- 7M. Gonay , C. Batisse , and J.‐F. Paquin , “Recent Advances in the Synthesis of Acyl Fluorides,” Synthesis 53 (2021): 653.
- 8F. Pulikkottil , J. S. Burnett , J. Saiter , C. A. I. Goodall , B. Claringbold , and K. Lam , “e Fluorination for the Rapid Synthesis of Carbamoyl Fluorides from Oxamic Acids,” Organic Letters 26 (2024): 6103.39016380 10.1021/acs.orglett.4c 01605 PMC 11287745 · doi ↗ · pubmed ↗