Graphene oxide/metal–organic framework composite as an effective catalyst for esterification reactions
Reza Masoudi, Ali Zarnegaryan, Zahra Dehbanipour

TL;DR
A new catalyst made from graphene oxide and a metal-organic framework was developed and shown to efficiently promote esterification reactions with high yield and reusability.
Contribution
The synthesis of a graphene oxide-impregnated MOF-801 catalyst for esterification is novel, offering high efficiency and reusability.
Findings
The MOF-801@GO catalyst achieved up to 95% esterification yield under solvent-free conditions.
The catalyst retained activity and structural integrity over multiple reuse cycles.
Characterization techniques confirmed successful integration of graphene oxide into the MOF structure.
Abstract
Herein, we report the synthesis and characterization of graphene oxide (GO) impregnated into a Zr-based metal–organic framework (MOF-801), denoted as MOF-801@GO, as a catalyst for esterification reactions. The synthesized catalyst was thoroughly characterized using various techniques, including Fourier-transform infrared (FTIR) spectroscopy, Brunauer–Emmett–Teller (BET) surface area measurements, and scanning electron microscopy (SEM), transmission electron microscopy (TEM), thermogravimetric analysis (TGA), X-ray diffraction (XRD), and inductively coupled plasma (ICP) spectroscopy. The surface acid strength was evaluated by ammonia temperature-programmed desorption (NH₃-TPD). The catalytic performance of MOF-801@GO was assessed in the esterification of carboxylic acids with alcohols using no solvent. Under optimized reaction conditions, the catalyst achieved a high esterification yield…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
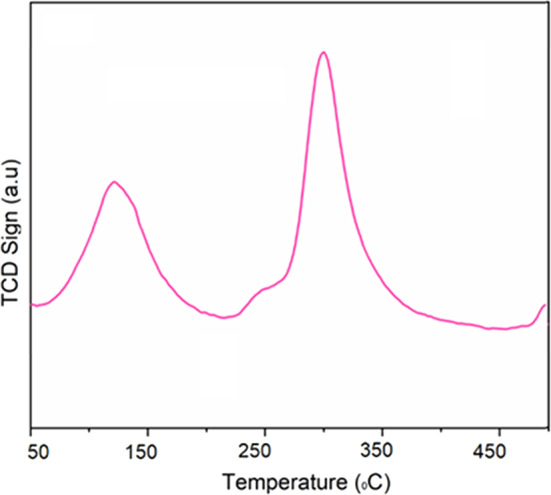 Figure 10
Figure 10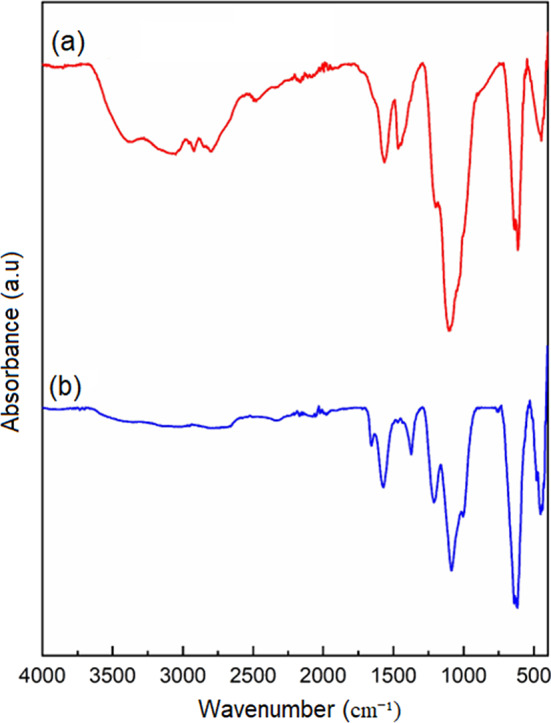 Figure 11
Figure 11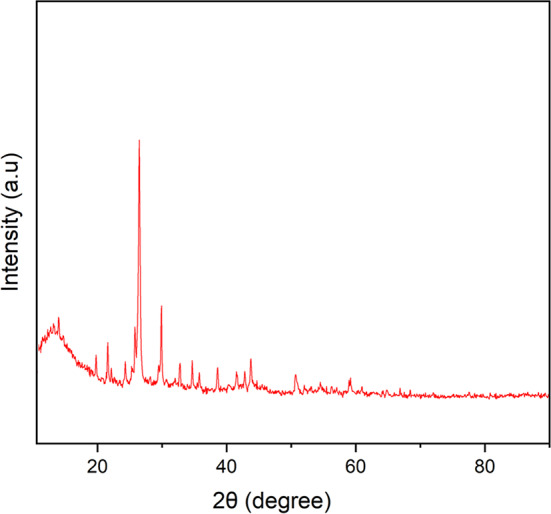 Figure 12
Figure 12 Figure 13
Figure 13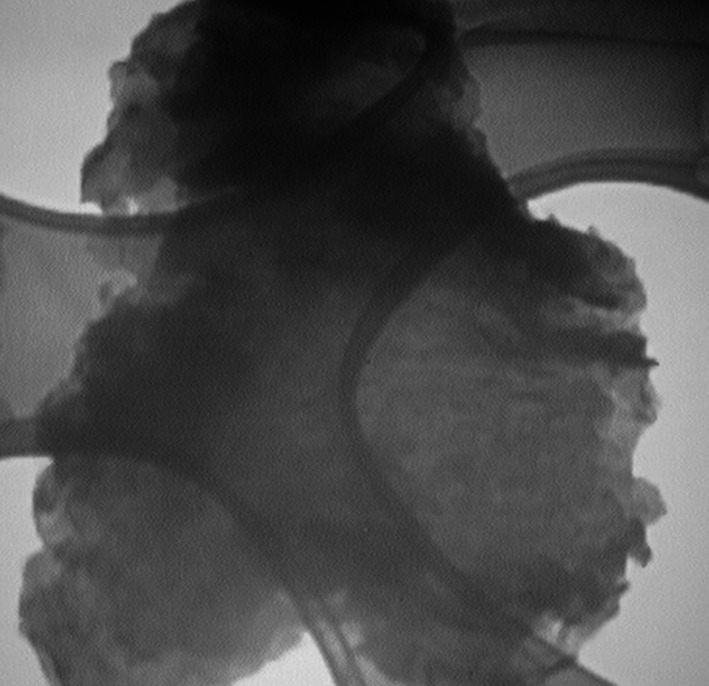 Figure 14
Figure 14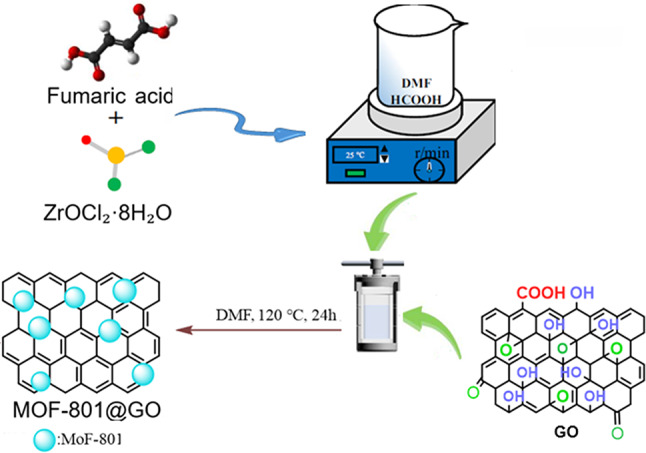 Figure 1
Figure 1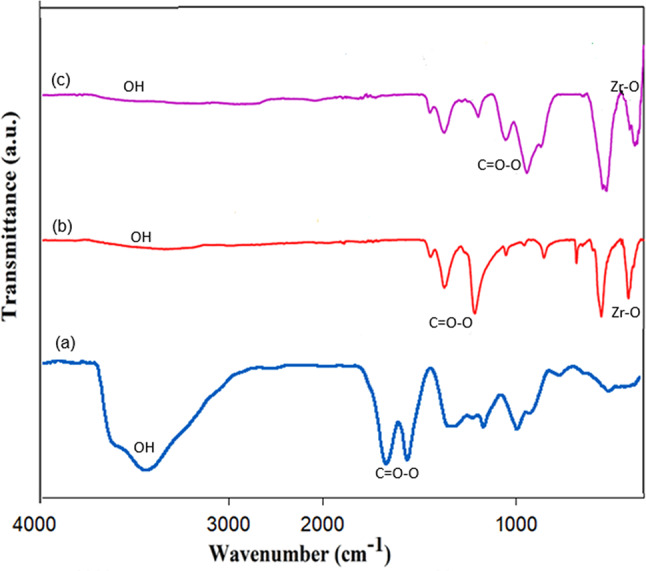 Figure 2
Figure 2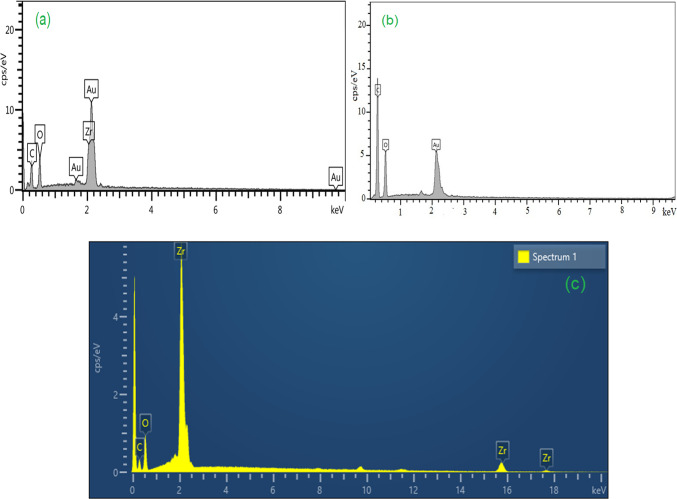 Figure 3
Figure 3 Figure 4
Figure 4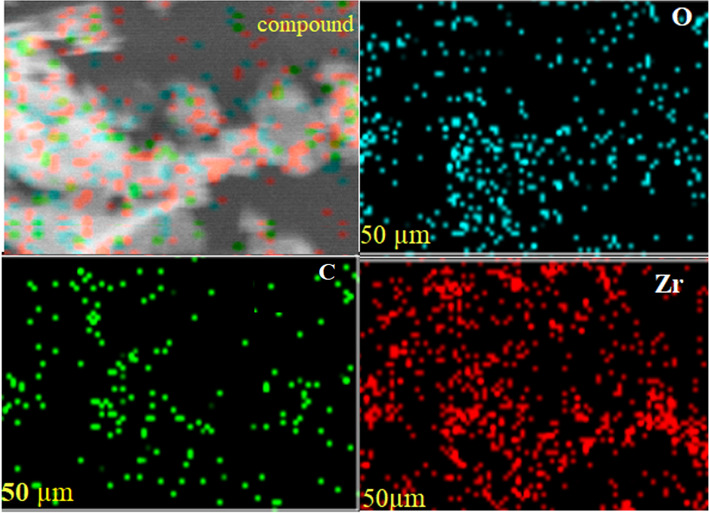 Figure 5
Figure 5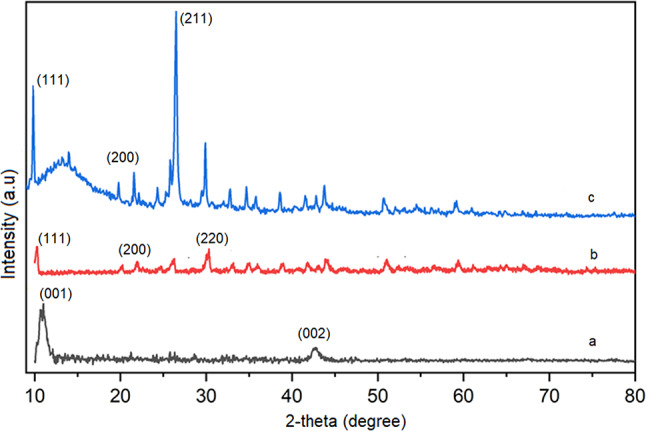 Figure 6
Figure 6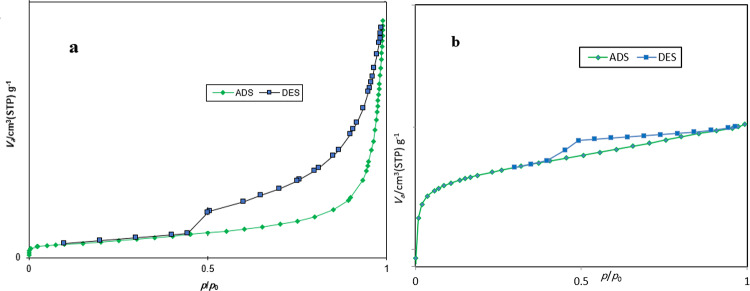 Figure 7
Figure 7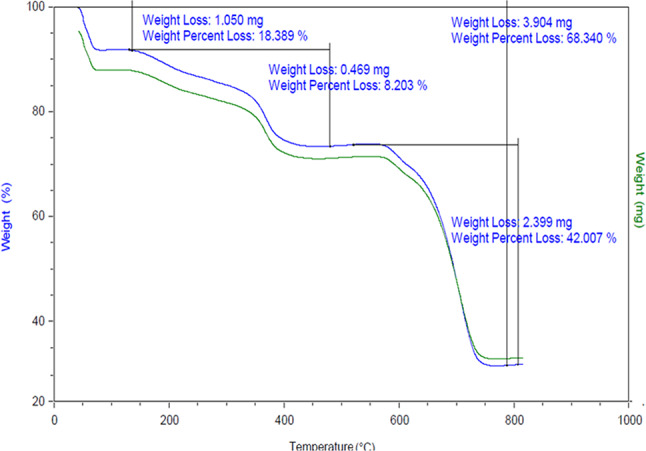 Figure 8
Figure 8 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetal-Organic Frameworks: Synthesis and Applications · Thermal and Kinetic Analysis · Biodiesel Production and Applications
Introduction
Graphene oxide (GO) is a novel carbon nanomaterial composed of monolayers of sp²-hybridized carbon atoms. It is distinguished by its exceptional chemical properties and has attracted significant attention for diverse applications, notably catalysis, sensing, supercapacitors, energy storage, photocatalysis, hydrogen storage, wastewater treatment, drug delivery systems, and biomedical applications^1^. GO possesses a unique nanostructure, typically existing as a single layer or few-layered sheets, and exhibits high thermal stability, a high surface-to-volume ratio, and a rich population of oxygenated surface functionalities^2^. GO is a two-dimensional nanomaterial with a honeycomb-like structure, consisting of a single monolayer obtained by exfoliation of GO. It features multiple hydrophilic oxygen-bearing functional moieties including hydroxyl (–OH), epoxide (–O–), carbonyl (–C=O), and carboxylic acid (–COOH) groups alongside hydrophobic aromatic regions distributed across both the edges and the basal plane. These structural features enable GO to disperse effectively in water and a range of organic solvents^3,4^. The GO sheets contain epoxy and hydroxyl groups on both basal planes, while carboxyl groups are primarily located at the edges^5^. These oxygen-containing functional groups enable GO to be readily modified into modified GO through either non-covalent or covalent interactions with small organic molecules or polymers^6–8^. Metal–organic frameworks (MOFs) are organic–inorganic hybrid crystalline materials constructed from metal ions or clusters and multitopic organic ligands via coordination-driven bonding. These materials have attracted significant attention in recent years due to their tunable pore sizes, customizable functionalities, and high surface areas. So far, MOFs have demonstrated outstanding potential in various applications, including proton conductivity, sensing, separation, gas storage, solar cells, drug delivery, catalysis, biomedicine, and supercapacitors^9^. Furthermore MOFs are considered a leading platform for hosting guest molecules due to their highly accessible, long-range ordered channels and large internal surface areas. To date, various active species have been successfully encapsulated within the cages or pores of MOFs, including metal oxides, noble metals, polyoxometalates (POMs), and enzymes^10–13^. MOFs can be considered ideal candidates for heterogeneous catalysis in various reactions, owing to (i) their hybrid inorganic/organic structures, (ii) the presence of accessible organic linkers and uncoordinated metal sites, (iii) favorable physicochemical properties, (iv) well-defined porous architectures, and (v) their compatibility with drug delivery system designs. MOFs have been successfully employed as catalysts due to their structural and chemical diversity, which allows them to serve as highly effective alternatives for various reactions compared to conventional catalysts^14–21^. Esters are essential building blocks in the synthesis of fine chemicals and electronic materials, and represent one of the most prevalent structural motifs found in natural products and pharmaceutical compounds^22^. Consequently, numerous methods have been developed for their efficient synthesis. Various techniques have been developed for ester synthesis, with classical approaches typically involving the condensation of carboxylic acids and alcohols. However, some of these synthetic methods require the use of corrosive reagents, often leading to low product yields and limited tolerance toward functional groups^23^. Under these harsh conditions, an efficient esterification reaction of carboxylic acids with alcohols is demonstrated, catalyzed by GO–MOF composites^24–28^. Some recently developed systems of this type include heteropolyacids, MOFs, metal oxides, etc. Recently, the integration of MOFs with GO has emerged as a promising research area due to their well-defined porosity, high surface area, and facile functionalization^29–39^. In view of the above, a metal–organic framework (MOF-801) was immobilized on GO to yield a highly active, robust, and readily recoverable heterogeneous catalyst for esterification reactions (Fig. 1).
Fig. 1. Schematic representation of the synthetic protocol for the MOF-801/GO composite.
Experimental section
Methods and materials
All chemicals and solvents were obtained from commercial suppliers and employed as received, without additional purification. Potassium persulfate (K_2_S_2_O_8_, Sigma, 99%), phosphorus pentoxide (P_2_O_5_, Sigma, 99%), ethanol (C_2_H_5_OH, ACS reagent, 99.8%), N,N-dimethylformamide (DMF, Sigma-Aldrich, 99.99%), sulfuric acid (H_2_SO_4_, Sigma-Aldrich, 98%), potassium hydroxide (KOH, Sigma-Aldrich, 85%), potassium permanganate (KMnO_4_, Sigma-Aldrich, 98%), hydrogen peroxide (H_2_O_2_, Sigma-Aldrich, 30%), zirconyl chloride octahydrate (ZrOCl_2_·8 H_2_O, Sigma-Aldrich, 98%), fumaric acid (C_4_H_4_O_4_, Sigma-Aldrich, 99%), formic acid (HCOOH, Sigma-Aldrich, 98%), and deionized (DI) water were used as received. N_2_ adsorption-desorption isotherm at 77 K of MOF-801/GO powder was measured on Autosorb-iQ-MP (Quantachrome Instruments) with BET surface area analysis. Fourier-transform infrared (FTIR) spectra were acquired on a Bruker Vector 22 spectrometer over the wavenumber range of 4000–400 cm^–1^. Thermogravimetric analysis (TGA) was performed on a Netzsch STA 1500 instrument under a nitrogen atmosphere (flow rate: 20 mL min^–1^) at a heating rate of 5 °C min^–1^ from 25 to 800 °C. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) analyses were carried out using a Zeiss SIGMA VP field-emission scanning electron microscope operated at 15 kV. Powder X-ray diffraction (XRD) patterns were collected using a PANalytical X’Pert^3^ PRO MPD diffractometer with Cu Kα radiation (λ = 1.5406 Å) at 40 kV and 40 mA, over a 2θ range of 5–50° with a step size of 0.02°. Transmission electron microscopy (TEM) imaging was performed using a Philips CM10 instrument operated at an accelerating voltage of 100 kV.
Catalytic yields were quantified via gas chromatography (GC) using a Shimadzu GC-16 A instrument equipped with a flame ionization detector (FID) and an HP-5 capillary column (30 m × 0.32 mm × 0.25 μm); helium was used as the carrier gas at a flow rate of 1.0 mL min^−1^. The metal loading in the catalyst was quantified via inductively coupled plasma optical emission spectrometry (ICP-OES) employing a PerkinElmer Optima 7300 DV instrument; before measurement, samples were subjected to acid digestion using a concentrated HNO₃/H_2_O_2_ mixture (3:1, v/v).
Synthesis of GO
GO was synthesized according to a published method^40^. A mixture of 3 g graphite powder, 12 mL H_2_SO_4_, 2.5 g K_2_S_2_O_8_, and 2.5 g P_2_O_5_ was heated at 80 °C under stirring for 4.5 h. Upon cooling to room temperature, the resulting solid was collected and rinsed repeatedly with a total of 500 mL of deionized water. The mixture was then stirred at room temperature for a duration of 24 h., followed by filtration. The resulting solid was sequentially washed with deionized water and ethanol, and finally dried under vacuum. The dried powder was incrementally introduced into 120 mL of concentrated H_2_SO_4_ containing potassium permanganate (15 g) in a two-neck round-bottom flask immersed in an ice bath, followed by gentle stirring until complete dissolution of the mixture was achieved. Subsequently, an additional 250 mL of deionized water and 20 mL of H_2_O_2_ (30% w/w) were added, and the reaction was quenched by stirring for 30 min in an ice bath. The supernatant was carefully decanted, and the solid residue was \ filtration. The precipitate was then subjected to repeated washing with a 1% (v/v) aqueous HCl solution until the washings yielded no turbidity upon addition of BaCl_2_ solution confirming the absence of residual sulfate ions and thus complete removal of sulfate-based impurities. Finally, the synthesized GO was further purified by repeated washing with deionized water until the washings reached a neutral pH, after which the product was dried in an oven at 35 °C. The synthesized graphene oxide was purified by repeated washing with deionized water until the pH of the supernatant approached neutrality, confirming the effective removal of residual acidic species. The purified product was then freeze-dried or oven-dried at 35 °C to yield a dry, brownish solid.
Synthesis of MOF-801@GO
MOF-801 was synthesized following the protocol reported by Iacimi et al.^41^, with certain modifications to improve synthesis efficiency. In detail, a mixture of 1.75 g fumaric acid, 0.23 g ZrOCl_2_·8 H_2_O, and 5.3 mL formic acid was introduced into 35 mL DMF and stirred for 30 min to achieve complete dissolution. Subsequently, GO suspension (0.15 g L^–1^) was added, and the mixture was stirred thoroughly. Following this, the mixture was transferred to a 100 mL Teflon-lined autoclave and solvothermal reaction was carried out at 130 °C for 24 h to yield the final product. Upon reaction completion, the solvent was removed by filtration, and the sample was allowed to cool to 25 °C. The obtained solid residue was then immersed in ethanol and left to stand for 72 h. After the soaking step, the ethanol was eliminated through ambient-temperature drying of the material. MOF-801@GO was subsequently isolated by vacuum drying at 100 °C for 24 h in a thermostatic vacuum oven. The Zr loading, as quantified by ICP-OES, was determined to be 0.079 mmol g^–1^.
Catalytic activity tests
The esterification was performed using the synthesized MOF-801@GO as the heterogeneous catalyst. In a typical procedure, carboxylic acid (5 mmol), alcohol (3 mmol), and the catalyst (20 mg) were dissolved in ethanol (5 mL) and transferred into a reaction vessel. The reaction mixture was maintained at 100 °C under vigorous mechanical agitation for a duration of 5 h, with periodic assessment of reaction advancement conducted via thin-layer chromatography (TLC). Upon completion, the mixture was filtered through double-layer filter paper, and the filtrate was collected in a beaker. The solvent was then carefully removed by evaporation using a warm water bath. After completion of the reaction, the mixture was filtered through double-layer filter paper. The filtrate was collected in a beaker and concentrated by evaporation in a water bath to remove any excess solvent. The solid residue was then slurried with silica gel (60–120 mesh) and loaded onto a column for purification. The final product was isolated by column chromatography using a mobile phase of 2% ethyl acetate in petroleum ether.
Results and discussion
FT-IR spectra
Figure 2 shows the FT-IR spectra of GO, MOF-801, and MOF-801@GO. The FT-IR spectrum of the pristine MOF-801 aligns well with the spectral features documented previously in the literature^42^. For pure GO (Fig. 2), the intense absorption at 1721 cm^–1^ is assigned to C = O stretching in carboxylic acid functionalities. Peaks at 1059, 1228, and 1610 cm^–1^ correspond to C–O (epoxy), C–OH, and C = C vibrations, respectively. Furthermore, a broad feature centered at 3429 cm^–1^ characteristic of O–H stretching confirms abundant hydroxyl group incorporation. Additionally, a wide band at 3429 cm^–1^, assigned to O–H stretching vibrations, indicates a high concentration of hydroxyl groups. The band observed at 3423 cm^–1^ is assigned to O–H stretching vibrations, whereas the feature at 1634 cm^–1^ is attributed to C = O stretching arising from residual DMF incorporated in MOF-801. Moreover, the absorptions at 1592 and 1402 cm^–1^ are ascribed to the asymmetric and symmetric C=O stretching vibrations of carboxylate groups in MOF-801, respectively. The band at 639 cm^–1^ is assigned to the transverse optical mode of Zr–O bonds, while the one at 443 cm^–1^ corresponds to the µ-OH deformation mode^43^. Concurrently, the absorbance intensities of the bands at 639 and 443 cm^–1^ are markedly reduced, which is attributed to the complexation of -OH and GO in MOF-801^44^. The bands observed at 1081 and 1211 cm^–1^ in the FT-IR spectrum of MOF-801@GO can be assigned to the C–O and C–OH stretching vibrations of GO, respectively.
Fig. 2FT-IR spectra of (a) GO, (b) MOF-801, and (c) MOF-801@GO.
FE-SEM, EDX, and mapping analysis
Energy-dispersive X-ray spectroscopy (EDS) spectra of MOF-801, GO, and MOF-801@GO are presented in Fig. 3. EDX analysis of MOF-801 confirms the presence of zirconium (Zr), carbon (C), and oxygen (O) as the primary elemental constituents (Fig. 3a). As evident from the energy-dispersive X-ray (EDX) spectrum of GO, carbon and oxygen constitute the predominant elemental components (Fig. 3b). In the EDX spectrum of MOF-801@GO, the presence of zirconium, carbon and oxygen, a which are consistent with the expected constituents of MOF-801@GOon GO sheets (Fig. 3c). The elemental composition of the MOF-801@GO composite, determined by EDS, is summarized in Table 1.
The morphologies of GO, MOF-801, and the MOF-801@GO composite were analyzed by FE-SEM, as presented in Fig. 4. As shown in Fig. 4a, GO exhibits large, sheet-like flakes with pronounced macroscopic wrinkling. The SEM image corresponding to MOF-801 is shown in Fig. 4b small particles with sizes of approximately 1 μm and with shapes that vary between slightly deformed to small rhombic dodecahedrons are observed. In contrast to bare GO sheets and pristine MOF-801, the FE-SEM image of MOF-801@GO (Fig. 4c) reveals distinct morphological features, confirming successful composite formation.
As evidenced by the elemental mapping in Fig. 5, the uniform spatial distribution of Zr, O, and C across the MOF-801@GO composite confirms the homogeneous integration of MOF-801 with graphene oxide.
Table 1. Elemental analysis of MOF-801@GO composite from EDS.ElementLine typeWeight%Weight% SigmaAtomic %CK series41.261.6455.10OK series41.831.2341.93ZrL series16.910.502.97Total100.00100.00
Fig. 3EDX analysis of (a) MOF-801, (b) GO, and (c) MOF-801@GO.
Fig. 4FE-SEM images of (a) GO, (b) MOF-801, and (c) MOF-801@GO.
Fig. 5. Elemental mapping analysis of MOF-801@GO.
XRD analysis
Figure 6 presents the powder X-ray diffraction (PXRD) patterns of GO, MOF-801, and the MOF-801@GO composite catalyst, which were used to assess structural features and phase purity. For GO (Fig. 6a), two distinct diffraction peaks are observed at 2θ = 11.3° and 24.2°, corresponding to the (001) reflection of GO and the (002) reflection of graphite, respectively^45^. The shift of the (001) peak to a lower angle compared to graphite reflects an increased interlayer spacing (d-spacing), which is attributed to the presence of oxygen-containing functional groups on the basal planes and intercalated water molecules between the GO layers^46^. The PXRD pattern of the as-synthesized MOF-801 (Fig. 6b) exhibits sharp, well-defined peaks that are fully consistent with the reported pattern for MOF-801, confirming its formation with a cubic crystal structure and space group^47^. Furthermore, the PXRD pattern confirms that the synthesized MOF-801 exhibits excellent phase purity and is in good agreement with previously reported data^48^. The pattern displays three prominent diffraction peaks at 2θ = 10.9°, 16.7°, and 30.4° assigned to the (111), (200), and (220) crystallographic planes, respectively^49,50^. Figure 6c shows the PXRD pattern of the MOF-801@GO composite. The diffraction peaks observed at 2θ = 11.1°, 21.2°, and 26.9° are assigned to the (111), (200), and (211) crystallographic planes of MOF-801, respectively^51–53^. The absence of the GO (001) peak in the MOF-801@GO composite does not indicate degradation or absence of GO, but rather reflects effective exfoliation and intercalation of MOF-801 nanoparticles between GO layers, leading to loss of long-range stacking order and increased, heterogeneous interlayer spacing consistent with successful composite formation.
Fig. 6PXRD patterns of (a) GO (b) MOF-801and (c) MOF-801@GO composite.
BET analysis
The N_2_ adsorption–desorption isotherms of GO and the MOF-801@GO composite are shown in Fig. 7. According to the experimental results, GO exhibits a specific surface area of 87.6 m²/g, consistent with literature-reported values^54^. Additionally, the adsorption isotherm of MOF-801@GO belongs to type IV and exhibits an obvious hysteresis loop, which may be attributed to capillary condensation in the mesopore. The low specific surface area of MOF-801@GO (49.41 m^2^/g) can be attributed to the blockage of a large number of micropores.
Fig. 7N_2_ sorption isotherms for (a) GO and (b) MOF-801@GO composite.
TGA analysis
Thermogravimetric analysis (TGA) is a widely used technique for qualitatively assessing moisture, residual solvents, and organic–inorganic composition in materials. It measures mass change as a function of temperature under a controlled atmosphere and heating program. Typically, mass loss below 200 °C corresponds to the evaporation of physiosorbed water and volatile solvents, whereas decomposition of organic moieties occurs between 200 °C and 800 °C. Additionally, TGA provides insight into the thermal stability of the material. Accordingly, TGA of MOF-801@GO was carried out under air, heating from 25 °C to 800 °C at 10 °C·min^–1^ (Fig. 8). The thermogram exhibits an initial mass loss of 18% below 180 °C, which is assigned to the desorption of surface-adsorbed solvents and moisture^55^. Subsequent weight losses at higher temperatures reflect the thermal decomposition of organic components, enabling estimation of the inorganic residue and overall thermal stability. Upon heating from 180 to 460 °C, a mass loss of 8% is observed, attributed to the partial decomposition of the MOF framework. A significant weight loss of 68% occurs between 460 and 780 °C, corresponding to the thermal decomposition of the organic linkers incorporated within the material structure. The retention of structural integrity up to 460 °C well beyond typical operating temperatures for heterogeneous catalysis demonstrates the exceptional thermal stability of the MOF-801@GO composite.
Fig. 8TGA analysis of MOF-801@GO composite.
Figure 9 presents a transmission electron microscopy (TEM) image of the MOF-801@GO nanocomposite, revealing a low-contrast, sheet-like morphology characteristic of GO. The GO sheets exhibit mild wrinkling and folding, consistent with their flexible two-dimensional (2D) structure. MOF-801 nanocrystals up to approximately 150 nm in size are homogeneously distributed and effectively intercalated between the GO layers, indicating strong interfacial interaction and successful integration of the MOF with the support.
Fig. 9TEM image of MOF-801@GO.
The acid properties of the MOF-801@GO catalyst were investigated by NH₃ temperature-programmed desorption (NH₃-TPD), and the corresponding profile is shown in Fig. 10. Two desorption peaks are observed, consistent with those reported for related materials^56,57^. The low-temperature peak in the range of 50–200 °C is attributed to weak acid sites, while the high-temperature peak between 250 and 400 °C corresponds to medium-strength acid sites. The weak acid sites are plausibly attributed to two origins: (i) residual µ₃–OH and terminal –OH groups, functioning as Brønsted acid sites, and (ii) oxygenated functional moieties present on graphene oxide (GO), including phenolic hydroxyl and carboxylic (–COOH) groups both widely recognized as sources of weak acidity. The low temperature adsorption MOF-801 is constructed from Zr₆O_4_(OH)4 octahedral secondary building units interconnected by fumarate linkers. Following thermal activation, labile terminal hydroxyl and aqua ligands are eliminated, yielding coordinatively unsaturated Zr⁴⁺ centers classified as Lewis acid sites, which account for the medium-strength NH₃ desorption observed between 250 and 400 °C. Significantly, the MOF/GO heterointerface may promote the stabilization of structural defects and induce polarization of Zr–O bonds, consequently augmenting the population and/or strength of medium-acidity sites a characteristic substantially less evident in the unmodified MOF-801. This interfacial synergy constitutes a pivotal merit of the composite architecture. MOF-801@GO exhibited weak acid sites quantified at 0.08 mmol g^–1^ and medium-strength acid sites at 1.14 mmol g^–1^.
Fig. 10NH_3_-TPD profile of the prepared MOF-801@GO catalyst.
Catalytic studies
The availability of suitable precursors is crucial in synthetic organic chemistry for the efficient synthesis of organic compounds. In esterification reactions, GO and MOF-801 have been individually reported as catalysts; however, their catalytic performance often results in unsatisfactory yields. Given that both GO and zirconium-based materials particularly MOF-801 exhibit catalytic activity in esterification processes, their combination is expected to yield a synergistic effect, leading to enhanced catalytic efficiency. Based on this rationale, we designed and synthesized a nanocomposite of GO and MOF-801 as a promising heterogeneous catalyst for esterification reactions. The catalytic performance of the synthesized MOF-801@GO nanocomposite was evaluated using a carboxylic acid and aliphatic alcohols as model substrates. Initially, acetic acid and 1-Heptanol alcohol were reacted in a 1:4 molar ratio to optimize the catalyst loading for the esterification reaction. The effect of varying MOF-801@GO dosage on the product yield is summarized in Table 2. As shown in Table 2, the product yield increases with increasing catalyst loading over 4 h at 80 °C. A yield of approximately 31% was obtained with 4 mg of MOF-801@GO, which increased to 98% when the catalyst amount was raised to 20 mg (1.32 mol%). In the absence of the catalyst, only a negligible amount of ester product was detected, highlighting the essential role of MOF-801@GO in promoting the reaction. The effect of reaction temperature on catalytic efficiency is illustrated in Entries 6–8 of Table 2. Based on these results, the optimal conditions for this transformation are the use of 20 mg of MOF-801@GO under solvent-free conditions at 80 °C (Table 2). As shown in Table 2, the product yield increases with increasing catalyst loading over 4 h at 80 °C. The elimination of the water by-product from the catalytic microenvironment appears to promote improved reaction yields at higher temperatures. Motivated by the encouraging outcomes observed with diminished catalyst loadings at 80 °C, kinetic profiling was conducted to track reaction progression over time, employing reduced catalyst amounts of 20 and 14 mg under identical experimental conditions (Table 2, entries 9–13). According to this analysis, full conversion was attained within 4 h and 5 h using 20 mg and 14 mg of catalyst MOF-801@GO, respectively (Table 2, entries 5 and 13).
Table 2. Effect of different catalysts on the esterification of acetic acid with alcohol.EntryCatalystsCatalyst [mg]Time[h]Temperature[°C]Yield [%]^a^[email protected]@GO8480683MOF-801@GO12480754MOF-801@GO16480875MOF-801@GO20480986MOF-801@GO20425407MOF-801@GO20440538MOF-801@GO20460719MOF-801@GO141803410MOF-801@GO142804511MOF-801@GO143807012MOF-801@GO144809213MOF-801@GO145809814GO206803615MOF-801204802416ZrOCl_2_20108018^a^ Reaction conditions: carboxylic acid (5 mmol), alcohol (5 mmol), catalyst (20 mg), solvent-free, 80˚C.
Table 3. The esterification of carboxylic acid with alcohols in the presence of the MOF-801@GO.
EntryROHTime [h]TON^a^TOF(h^− 1^)^b^Yield [%]^c^1CH_3_(CH_2_)7_OH471.917.9952CH_3(CH_2_)2_OH472.718.1963CH_3 (CH_2_)5 OH474.218.5984CH_3_(CH_2_)3_OH569.717.4925CH_3_CH(OH)CH_2_CH_3_571.214.2946CH_3(CH_2_)3_CH(OH)CH_3_668.111.3907CH_3(CH_2_)_6_OH465.916.4878CH_3_CH_2_OH473.418.3797^a^Turnover number [calculated by this equation: Yield (%)/Cat. (mol %)], ^b^Turnover frequency [calculated by this equation: TON/time (h)], 20 mg (containing 1.32 mol% of zirconium), ^c^Isolated yield.
The effect of reaction temperature on catalytic efficiency is illustrated in Entries 6–8 of Table 2. Based on these results, the optimal conditions for this transformation are 20 mg of MOF-801@GO under solvent-free conditions at 80 °C (Table 2).
Following optimization of the reaction parameters, the catalytic performance of MOF-801@GO was evaluated in the esterification of various carboxylic acids and alcohols (Table 3). The high yields achieved for the corresponding esters demonstrate that MOF-801@GO functions as a highly effective and robust catalyst for synthesizing a range of esters.
Catalyst reusability and stability
One of the most important properties of catalyst is their recyclability and reusability without significant loss of catalytic activity or structural integrity. Therefore, the recyclability and reusability of MOF-801@GO were subsequently investigated in the esterification reaction between acetic acid and 1-octanol, selected as a model reaction. The results demonstrated that the MOF-801@GO catalyst could be recycled and reused multiple times without a significant loss of catalytic efficiency. Moreover, the heterogeneous nature of the catalyst was further confirmed by evaluating zirconium leaching via ICP. The analysis revealed negligible leaching of Zr species upon reuse: the Zr content was 0.079 mmol·g^–1^ in the fresh catalyst and decreased only slightly to 0.075 mmol·g^–1^ after six catalytic cycles. This minimal loss (< 5%) strongly supports the robust structural integrity and truly heterogeneous nature of the catalytic system. The recoverability of MOF-801@GO was evaluated in the esterification reaction. After completion of each reaction cycle, the catalyst was recovered via centrifugation, rinsed with ethanol, and reintroduced into the next reaction run. The results show that the catalyst can be recovered and reused for at least five consecutive cycles with only a minor decrease in yield from 98% in the first run to 91% in the fifth cycle (Table 4). This slight reduction in activity may be attributed to minor catalyst loss during recovery and partial deactivation of active sites due to adsorption of reactants or products.
Table 4. Reusability and recoverability of [email protected] [%]98 ± 197 ± 195 ± 193 ± 193 ± 191 ± 1
Hot filtration test
To investigate the heterogeneous nature of the catalyst, a hot filtration test was performed during the esterification reaction under optimized reaction conditions using the MOF-801@GO catalyst. After 1 h of reaction, the reaction was temporarily halted, and the catalyst was isolated from the reaction mixture via hot filtration. At this point, a product yield of 69% was achieved. The filtrate was subsequently transferred to a new reaction vessel and maintained at 80 °C for a further 2 h. No significant increase in product yield was observed, indicating that catalytic activity ceased upon catalyst removal and confirming the heterogeneous nature of the catalyst. Furthermore, ICP analysis revealed no significant leaching of zirconium, supporting the structural stability of the catalyst under the reaction conditions.
To evaluate the chemical and structural stability of the catalyst over six reuse cycles, the recovered catalyst was characterized by FT-IR, PXRD, TEM, and SEM analyses. The FT-IR spectrum of the recovered catalyst exhibited complete correspondence with that of the as-synthesized catalyst, confirming the exceptional chemical stability of the MOF-801@GO composite under the employed reaction conditions (Fig. 11).
Fig. 11FT-IR spectra of the catalyst (a) before and (b) following the catalytic reaction.
As shown in Fig. 12, the PXRD pattern of the recovered catalyst is nearly identical to that of the fresh catalyst, confirming the high structural stability of the catalyst during multiple recovery cycles.
Fig. 12PXRD pattern of the recovered MOF-801@GO catalyst.
The SEM image of the recovered catalyst closely matches that of the as-synthesized material, corroborating the exceptional structural integrity of the catalyst under the employed reaction conditions (Fig. 13).
Fig. 13SEM image of the recovered MOF-801@GO catalyst.
The TEM image of the recovered catalyst was acquired and compared with that of the fresh catalyst (Fig. 14). As shown, the recovered catalyst retains the characteristic morphology of the fresh sample, including a few twisted nanosheets, indicating excellent structural stability under the applied reaction conditions.
Fig. 14. The TEM image of the recovered MOF-801@GO catalyst.
Finally, the catalytic performance of the developed catalyst was compared with that of several recently reported catalysts used the esterification reaction. The results demonstrate that the present catalyst outperforms previously reported systems in terms of recyclability and reaction time. Moreover, this study confirms the superior efficiency and stability of the current catalytic system compared to earlier alternatives (Table 5).
Table 5. Comparison of the performance of several catalysts with the present work for esterification.EntryCatalystConditionsYield [%]References1SHBPAO6 h, 5 run recovery, 90˚C76^58^2SO_3_H@P-E155 h, and 3 run recovery, 100 ˚C99^59^3IL@SBA-15-Pr-SO_3_H40 h, 4 run recovery, 45˚C91^60^4PAFR12 h and 3 run recovery, 50 ˚C96^61^5MOF-801@GO4 h, 6 run recovery, 80˚C98This work
Conclusion
In this study, we report the synthesis and characterization of a composite catalyst composed of GO incorporated into a zirconium-based metal–organic framework (MOF-801), designated as MOF-801@GO. GO was employed as a supporting material to enhance the catalytic properties of zirconium. Furthermore, this work demonstrates the effective esterification of carboxylic acids for the synthesis of various esters applicable as biodiesel, using the MOF-801@GO as a heterogeneous catalyst. Under optimized reaction conditions, the catalyst achieved high product yields. The MOF-801@GO composite was characterized using FTIR, BET, SEM, TEM, TGA, and XRD techniques. Significantly, the catalyst could be easily recovered and reused for multiple cycles without significant loss of catalytic efficiency or stability.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Salahshoori, I., Babapoor, A. & Seyfaee, A. Elevated performance of THZ neat, hybrid and composite membranes by the addition of nanoparticles (ZIF-67): A molecular dynamics study. Polym Bull 1–36 (2022).
- 2Zhang, Q., Lei, Y., Wang, J., Zhang, Y. & Ma, P. Zeolitic imidazolate Frameworks-Based materials for accelerating sustainable biodiesel production: A mini review. Catal Surv. Asia 1–20 (2025).
- 3Zhang, Q. et al. Facile fabrication of Bi 2O 2CO 3/MOF-801 nanocomposites for efficient photodegradation of noxious Rh B pollutants. J Inorg. Organomet. Polym. Mater 1–18 (2025).
- 4Furukawa, H. et al. M Water adsorption in porous metal–organic frameworks and related materials. J Am. Chem. Soc 4369–4381 (2014).10.1021/ja 500330 a 24588307 · doi ↗ · pubmed ↗
- 5Aghajani Hashjin, M., Zarshad, S., Motejadded Emrooz, H. B. & Sadeghzadeh, S. Enhanced atmospheric water harvesting efficiency through green-synthesized MOF-801: a comparative study with solvothermal synthesis. Sci Rep 13, 16983, (2023).10.1038/s 41598-023-44367-1PMC 1056238037813977 · doi ↗ · pubmed ↗
- 6Karimi, B. & Vafaeezadeh, M. SBA-15-functionalized sulfonic acid confined acidic ionic liquid: a powerful and water-tolerant catalyst for solvent-free esterifications. Chem Commun 483327 (2012).10.1039/c 2cc 17702 a 22361844 · doi ↗ · pubmed ↗