An Antifungal with a Novel Mechanism of Action Discovered via Resistance Gene-Guided Genome Mining
Bruno Perlatti, Sandeep Vellanki, Yalong Zhang, Yi-Ming Chiang, Yingxia Hu, Mengdi Yuan, Kyle Dunbar, Abigail Fine, Michelle F. Grau, Sheena Li, Timothy O’Donnell, Rajani Shenoy, Hongtao Li, Hui Shi, Xia Xu, Zeyu Chen, Tara Arvedson, Yi Tang, Robert A. Cramer, Victor Cee

TL;DR
A new antifungal drug with a unique mechanism was discovered by targeting an enzyme crucial for fungal amino acid production.
Contribution
Acetolactate synthase (ALS) is validated as a novel antifungal target, with a new inhibitor discovered via resistance gene-guided genome mining.
Findings
HB-35018, a novel compound, potently inhibits ALS and outperforms existing inhibitors against pathogenic fungi.
HB-35018 forms a unique covalent bond with ALS, distinct from known inhibitors, revealed by cryo-electron microscopy.
ALS is essential for fungal virulence in a mouse model of invasive aspergillosis.
Abstract
Invasive fungal infections claim over two million lives annually, a problem exacerbated by rising resistance to current antifungal treatments and an increasing population of immunocompromised individuals. Despite this, antifungal drug development has stagnated, with few novel agents and fewer novel targets explored in recent decades. Here, we validate acetolactate synthase (ALS), an enzyme critical for branched-chain amino acid biosynthesis and absent in humans, as a promising target for new therapeutics. Using resistance gene-guided genome mining, we discovered a biosynthetic gene cluster in Aspergillus terreus encoding HB-35018 (1), a novel spiro-cis-decalin tetramic acid that potently inhibits ALS. Biochemical and antifungal assays demonstrate that 1 surpasses existing ALS inhibitors in efficacy against Aspergillus fumigatus and other pathogenic fungi. Structural studies via…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1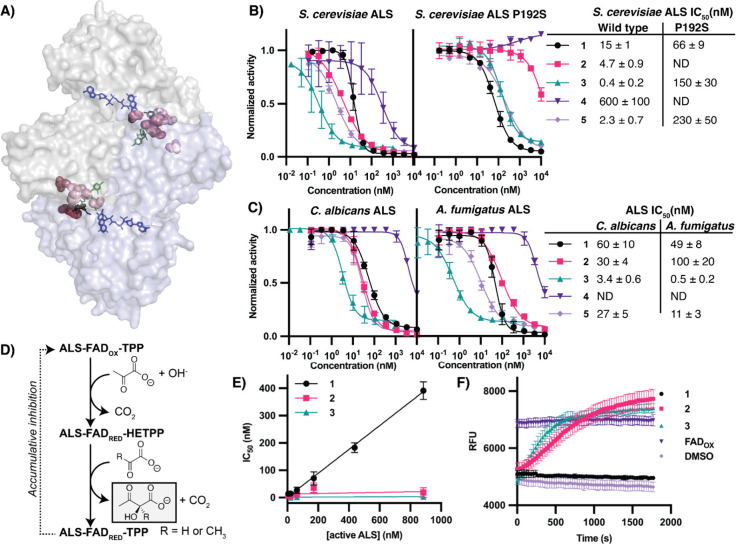 2
2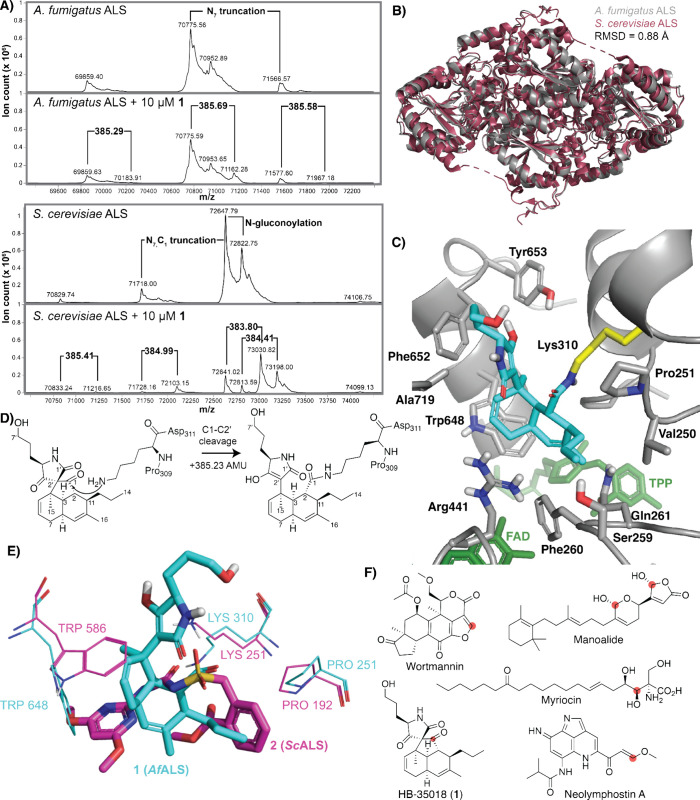 3
3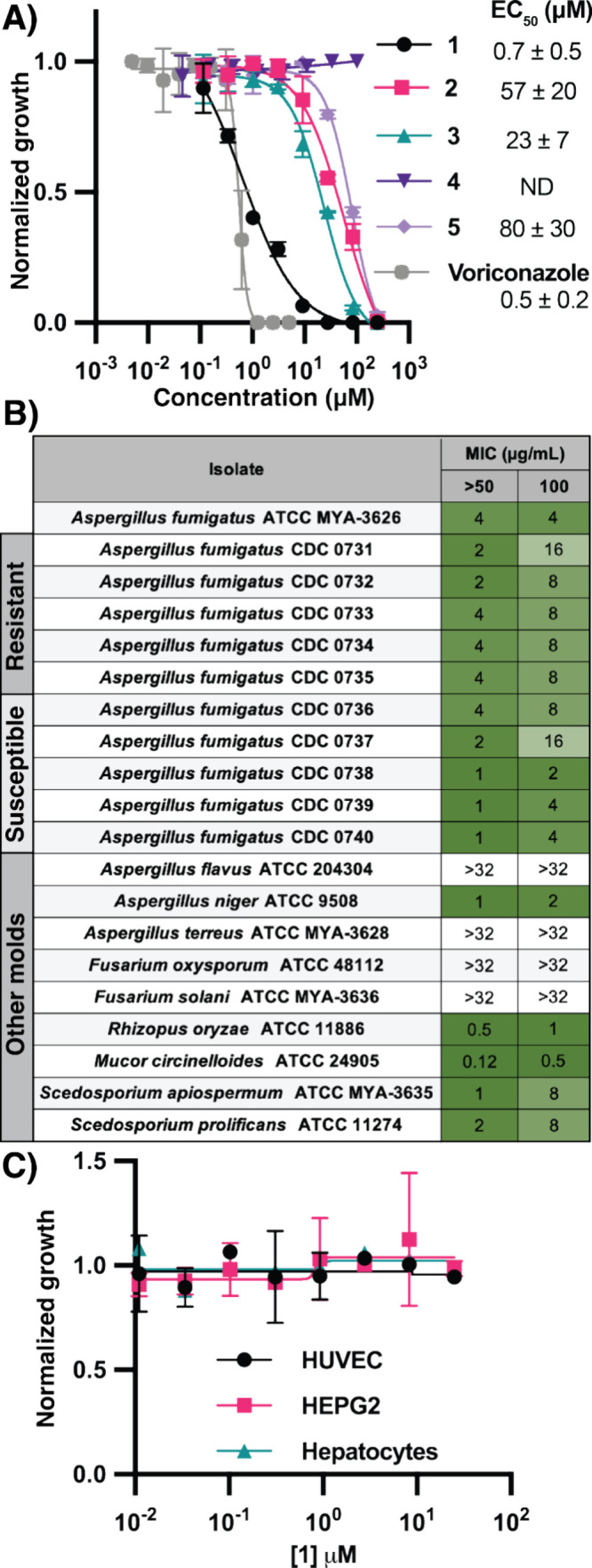 4
4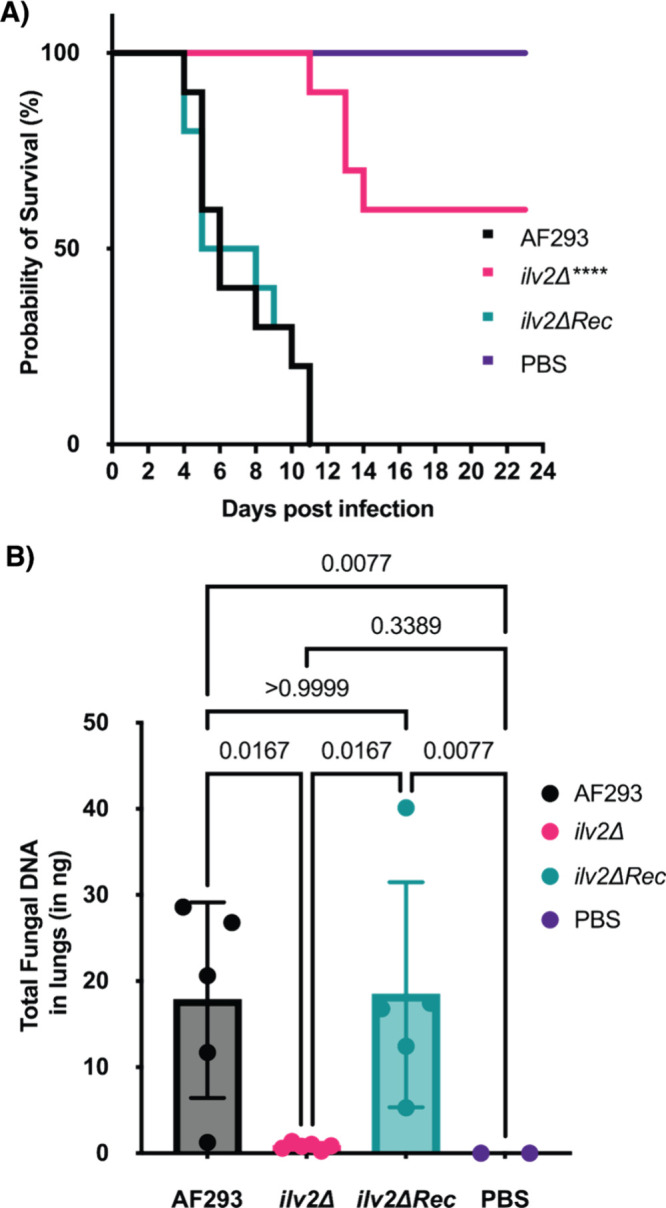 5
5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Natural Products and Biosynthesis · Antifungal resistance and susceptibility · Peptidase Inhibition and Analysis
Introduction
The need for novel antifungal therapies is urgent. Fungal infections cause an estimated 2.5 million deaths annually,? a number that is expected to rise as the number of immunocompromised patients increases and a warming climate extends the reach of pathogenic fungi. ?,? At least 1.3 million of these deaths are directly attributable to invasive aspergillosis, leading to Aspergillus fumigatus, the fungus primarily responsible for this disease, being included in the “critical priority group” of the “WHO fungal priority pathogens list to guide research, development and public health action”.? Current antifungal drugsincluding the azoles, echinocandins, and polyenesare decades old, with the efficacy of azoles and echinocandins diminishing as resistance spreads and the toxicity liabilities of the polyenes well-known.? Despite the severe impact of fungal infections and the demonstrated need for alternative therapies, development of new antifungals, particularly those with novel mechanisms of action, has been limited in recent years. ?,?
Natural products have long played a pivotal role in the discovery of antifungal therapies,? and resistance-gene guided genome mining has emerged as a promising method for targeted discovery of natural products with specific activities. In fungi, the phenomenon of biosynthetic gene clusters (BGCs) encoding resistance genes homologous to their product’s target has been appreciated for a range of targets, including inhibitors of glycolysis,? the proteasome,? ergosterol biosynthesis, ?−? ? ? and cyclin dependent kinases. ?,? Owing to the breadth of biology addressable by this approach, it presents a significant opportunity for the discovery of antifungal agents with novel mechanisms of action.
The ideal antifungal target is essential for the pathogen’s survival but absent in humans, reducing the risk of host toxicity. This constraint is crucial when treating the immunocompromised individuals that most commonly suffer from fungal infections.? Given the high level of conservation between humans and fungi at both the protein and pathway levels, potential targets meeting this criterion are rare.? The biosynthesis of essential amino acids, including branched-chain amino acids (BCAAs, FigureA), represents a set of such targets. Acetolactate synthase (ALS), the first dedicated enzyme in BCAA biosynthesis has been targeted extensively for agricultural applications with >50 inhibitors developed as commercial herbicides, including the widely used bensulfuron methyl (2), penoxsulam (3), bispyribac (4), and chlormuron ethyl (5, FigureB).? Despite this effort, to date, the potential for ALS inhibitors as human antifungals remains largely untapped. ?,?
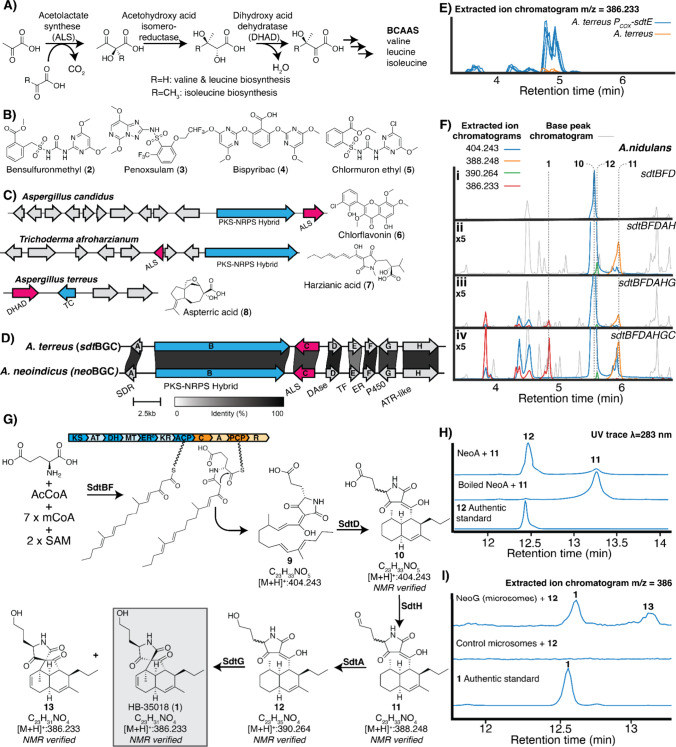
Discovery and biosynthetic characterization of 1. (A) Overview of the biosynthesis of branched chain amino acids (BCAAs). (B) Structure of select known inhibitors of ALS. (C) BGCs that produce BCAA inhibitors 6–8 containing putative resistance genes homologous to genes involved in BCAA biosynthesis. (D) sdtBGC from the genome of Aspergillus terreus and a related cluster from the genome of Aspergillus neoindicus. All proteins share >60% amino acid identity. Abbreviations: short chain reductase (SDR), polyketide synthase/nonribosomal peptide synthetase hybrid (PKS/NRPS hybrid), Diels–Alderase (DAse), transcription factor (TF), enoyl-reductase (ER), cytochrome P450 (P450), adenylation-thiolation-reductase-like (ATR-like). (E) Extracted ion chromatograms for m/z = 386.233 in wild-type A. terreus and A. terreus overexpressing the transcription for sdtE (A. terreus PCOX-sdtE). n = 4 clones per strain. (F) Base peak chromatograms and extracted ion chromatograms of cultures of A. nidulans strains expressing subsets of genes from the sdtBGC. (G) Proposed biosynthesis of 1. Key products and intermediates shown. Domain abbreviations: ketosynthase (KS), acyltransferase (AT), dehydratase (DH), methyltransferase (MT), ketoreductase (KR), acyl carrier protein (ACP), condensation (C), adenylation (A), peptidyl carrier protein (PCP), and reductase (R). See Figure S1 for a more detailed scheme inclusive of all identified shunt and side products. (H) Conversion of 11 to 12 by recombinant NeoA reductase. (I) Conversion of 12 to 1 by microsomes from S. cerevisiae strains expressing cytochrome P450 NeoG.
In this study by leveraging resistance gene-guided genome mining we discover the sdtBGC, a BGC that encodes within it a resistance gene homologous to ALS. Using heterologous expression coupled with in situ BGC activation we identify HB-35018 (1), as the product of the sdtBGC and a novel ALS inhibitor. Through detailed biosynthetic characterization of this unique spiro-cis-decalin tetramic acid, we uncovered the activities of several notable enzymes. These include the first lipocalin-type Diels–Alderase mediating cis-decalin formation via a normal electron demand intramolecular Diels–Alder reaction and a cytochrome P450 responsible for establishing the highly strained spiro center. We further demonstrate that 1 is a strong inhibitor of recombinant ALS from multiple fungal species and employ structural analyses using intact protein mass spectrometry and cryo-electron microscopy to reveal 1 to be a covalent inhibitor of ALS, a binding mode distinct from previously known inhibitors. The activity observed in biochemical assays translates to whole fungal cells with 1 demonstrating potent antifungal activity against a panel of A. fumigatus isolates and several other fungal pathogens. Finally, using a mouse model of invasive aspergillosis, we validate ALS as essential to the pathogenicity of A. fumigatus. These findings both validate ALS as a promising antifungal target and highlight the power of genome mining to uncover innovative chemical scaffolds with potential to be developed as new antifungals.
Results
Genome Mining and Biosynthesis of 1
Resistance gene-guided genome mining has emerged as a powerful approach for identifying inhibitors of the essential fungal BCAA biosynthesis pathway. ALS performs the first dedicated step in this pathway and is the sole target to date with two distinct characterized BGC familiesthose of chlorflavonin (6)? and harzianic acid (7)?containing resistance genes encoding homologous proteins discovered in fungi. Additionally, aspterric acid (8) has been identified as an inhibitor of dihydroxy acid dehydratase (DHAD), the third protein in the BCAA pathway (FigureC).? Guided by these precedents, we initiated our discovery of novel BCAA inhibitors by mining our database of annotated fungal genomes for BGCs containing homologues of ILV2, the gene for ALS in Saccharomyces cerevisiae. This search yielded the sdtBGC in Aspergillus terreus, along with a closely related BGC in Aspergillus neoindicus (neoBGC, FigureD).
To determine the product of sdtBGC, we used two complementary approaches: in situ transcription factor activation and heterologous expression with complete refactoring in A. nidulans. In the first approach, A. terreus was transformed with a plasmid encoding sdtE, the gene for a transcription factor encoded within the BGC, under a constitutive promoter (P_COX_). Overexpression of such BGC-specific transcription factors has been shown to lead to significant upregulation of BGC expression and increased metabolite production.? Consistent with this precedent, LCMS analysis of sdtE overexpression strains led to the identification of multiple features that were upregulated as compared to the wild-type strain, the most prominent of which was a peak with m/z = 386.233 (FigureE, Figure S1). In parallel, the sdtBGC was refactored for heterologous expression in A. nidulans, resulting in the observation of multiple differential features, including the m/z = 386.233 feature identified through in situ BGC activation (FigureFiv). Upon isolation, the metabolite responsible for this feature was determined to be HB-35018 (1, Table S7, Figures S13–S19), a unique spiro-cis-decalin containing tetramic acid. While the cis-stereochemistry of the decalin was determined by NMR using J-coupling constants and 2D-NOE correlations, X-ray crystallography was required to confirm the absolute stereochemistry (Figure S4, Supporting CIF file).
Given the unique structure of 1, we sought to understand key steps in its biosynthesis using a combination of heterologous expression strains containing subsets of genes from sdtBGC and in vitro enzyme characterization. Coexpression of sdtB and sdtF in A. nidulans demonstrated that, similar to other fungal tetramic acids,? biosynthesis is 1 is initiated by the production of an octaketide-glutamic acid hybrid through the activity of polyketide synthase/nonribosomal peptide synthetase (PKS/NRPS) hybrid SdtB and in trans enoyl reductase (ER) SdtF. Release of this hybrid from SdtB via a Dieckmann cyclization catalyzed by the terminal reductase domain yielded compound 9 (FigureG, Figure S2 Ci). Upon formation of 9, a spontaneous intramolecular Diels–Alder reaction, analogous to that observed for intermediates in varicidin biosynthesis, results in partial conversion to a decalin (Figure S2Ci).? Addition of sdtD, a gene encoding a lipocalin-type Diels–Alderase (DAase), to A. nidulans sdtBF yielded cis-decalin 10 (Table S8, Figures S20–S26) as the dominant product (FigureFi, Figure S2Civ, FigureG). The stereospecific formation of cis-decalins is not commonly observed as they are thermodynamically disfavored relative to the trans- isomer. Characterized examples of cis-decalin formation in fungi include catalysis by an S-adenosylmethionine (SAM)-dependent DAase in fischerin biosynthesis? and a two-step mechanism involving an oxidation for electron-demand inversion in the biosynthesis of varicidins A and B.? To our knowledge, SdtD represents the first lipocalin-type DAase known to catalyze cis-decalin formation directly via a normal electron demand intramolecular Diels–Alder reaction. Intriguingly, CghA, the DAase from the BGC for trans-decalin Sch210971? has been successfully engineered to favor formation of the cis- product through the installation of three mutations, one of which, A242S, is present in SdtD (Figure S3), perhaps explaining this inverted stereospecificity.?
Subsequent to formation of the cis-decalin, addition of putative ATR (adenylation-thiolation-reduction) gene sdtH and the sdtA short-chain dehydrogenase/reductase (SDR) gene led to production of aldehyde 11 (FigureG, FigureFii, Table S9, Figures S27–S33) and alcohol 12 (FigureG, FigureFii, Table S10, Figures S34–S40). That SdtA alone leads to conversion of 11 to 12 was confirmed by incubation of 11 with recombinant NeoA, a close homologue of SdtA from the neoBGC (FigureH). The transformations mediated by SdtH and SdtA were specific for decalin-containing substrates, with the addition of either sdtH or sdtA to strains lacking sdtD showing no impact on the metabolomic profile (Figure S2ii,iii). The final key transformation, installation of the spiro center at C2’, was catalyzed by the cytochrome P450 (P450) SdtG. Addition of sdtG to the heterologous expression system produced a complex mixture, as it could catalyze spiro center formation from both the C7’ acid and C7’ alcohol regardless of C4’ stereochemistry, resulting in a variety of shunt products (Figure S2vi, 14–19, Tables S12–S16, Figures S48–S82). To confirm SdtG alone catalyzed spiro center formation, we incubated 12 with microsomes from S. cerevisiae expressing neoG, an sdtG homologue from the neoBGC (FigureD). This reaction yielded conversion of 12 to spiro compounds 1 and its C2’ diastereomer 13 (FigureI, Table S11, Figures S41–S47).
1 Exhibits Broad Antifungal Activity via Covalent,
Nonaccumulative Inhibition of ALS
While the presence of a resistance gene homologous to ILV2 within the sdtBGC suggested that ALS is the target of 1, we undertook a genetic and biochemical approach to confirm the target. Initially we performed a forward genetic screen in which a mutagenized library of S. cerevisiae was exposed to 1 at concentrations ranging from 5 μM to 80 μM. This treatment resulted in the development of resistant clones. We performed whole genome sequencing on 24 clones and 23 had mutations in ILV2, the S. cerevisiae gene for ALS (ScALS), suggesting that ALS is the target of 1. Resistance-conferring mutations localized near the substrate entry tunnel of the ALS homodimer, proximal to the established binding sites of known ALS inhibitors 2 and 5 (FigureA). Mutations in Pro192 were present in 75% of resistant clones (FigureA, Figure S4, Table S1), the same residue whose mutation imparted resistance to the triazolopyrimidine-sulfonamides, a class of ALS inhibitors previously explored as antifungals.? To further confirm that ALS is the target of 1, we evaluated its ability to inhibit ALS-mediated formation of acetolactate from pyruvate using recombinant ScALS. We observed potent inhibition, in line with known ALS inhibitors 2–5. However, while 2–5 were significantly less potent against P192S mutant ScALS compared to wild-type ScALS, with potencies diminished by at least 100-fold in all cases, there was only a 4-fold shift in potency for 1 (FigureB). 1 is also significantly more potent against ScALS than its diastereomer 13, further supporting the assignment of 1 as the final product of the sdtBGC (Figure S6).
Confirmation that ALS is the target of 1. (A) Mutations in ALS observed in S. cerevisiae clones resistant to 1. Resistance mutations (shown in pink) are mapped onto the crystal structure of 2 (black) bound to S. cerevisiae ALS (PDB 5FEM, α-chain in gray, β-chain in blue). Essential thiamine pyrophosphate (TPP, green) and FAD (blue) cofactors shown. (B) Inhibition of wildtype (WT) and P192S mutants of ALS from S. cerevisiae by 1–5. (C) Biochemical inhibition by 1–5 of ALS from S. cerevisiae, Candida albicans, and A. fumigatus. (D) Reaction catalyzed by ALS with the oxidation state of FAD at each step indicated (FADOX = Oxidized, FADRED = reduced). HETPP = hydroxyethyl-TPP. (E) IC50 vs concentration of active S. cerevisiae ALS protein suggests tight-binding kinetics for the binding of 1 and not for 2 or 3. (F) FAD cofactor reoxidation upon incubation of S. cerevisiae ALS with 1–3. Oxidized FAD (FADOX) included as a positive control.
To assess 1 as an antifungal, recombinant ALS from the key pathogenic fungal species A. fumigatus (AfALS) and C. albicans (CaALS) was prepared. In both species, 1 demonstrated activity comparable to that observed against ScALS and of known ALS inhibitors 2-5 (FigureC).
Multiple ALS inhibitors, including both 2 and 3, have been shown to function through accumulative inhibition, a mechanism by which inhibitors function not simply by occluding the substrate binding tunnel, but by trapping reactive oxygen species in the active site, leading to sustained inactivation of the enzyme through oxidation of the flavin adenine dinucleotide (FAD) cofactor. FAD in its reduced form is essential to ALS function, so this cofactor oxidation allows the effect of accumulative inhibitors to persist after dissociation and imparts superstoichiometric activity (FigureD). ?,? Kinetic characterization indicates 1 does not function via this mechanism as standard tight-binding kinetics with IC_50_ equal to half the concentration of active ALS is observed (FigureE). Furthermore, we see no indication of FAD reoxidation upon treatment with 1 in contrast to known accumulative inhibitors 2 and 3 (FigureF).
We next characterized the structural basis for binding of 1 to ALS. Given the strained structure and the observed tight binding kinetics, we hypothesized that 1 may be binding covalently. Analysis of ALS from both AfALS and ScALS by intact protein mass spectrometry yielded traces with multiple proteoforms for both proteins. Similar analysis after incubation of both proteins with saturating concentrations of 1 (10 μM) for 30 min produced mass shifts consistent with covalent addition of 1 (FigureA) supporting this hypothesis. Limited proteolysis followed by peptide mapping localized the covalent attachment to the 239_SGRPGPVLVDLPKDVTAAIL_258 peptide of ScALS with Lys251 and Asp252 (equivalent to Lys310 and Asp311 in AfALS) serving as the most likely attachment sites (Figure S7). The ability to detect modified peptide fragments following proteolysis suggests that 1 had irreversibly modified the ALS protein.
1 is a covalent inhibitor of ALS. (A) Intact mass spectrometry traces of AfALS and ScALS both alone and treated with 10 μM 1. Observed proteoforms are annotated in the untreated trace, and m/z shifts in treated samples are highlighted. (B) Cryo-EM structure of apo-AfALS overlaid with ScALS (PDB 5FEM). (C) Cryo-EM structure of A. fumigatus ALS with 1 (aqua) covalently bound to Lys310 (yellow) blocking the substrate channel (TPP is the cofactor thiamine pyrophosphate, and FAD is the cofactor flavin adenine dinucleotide). (D) Model for covalent addition of the side chain of Lys310 into the C1 ketone of 1 with subsequent C1–C2′ bond fragmentation. (E) Overlay of 1 (aqua) bound to A. fumigatus ALS with sulfonylurea herbicide bensulfuronmethyl 2 (pink) bound to S. cerevisiae ALS (PDB 5FEM). (F) Examples of natural products that covalently modify lysine residues of target proteins. Electrophilic attachment points are highlighted in red.
Structures of multiple plant and bacterial ALS proteins exist, but fungal structures are limited to those of S. cerevisiae and C. albicans. ?,? To more fully understand the interaction of 1 with ALS from a clinically relevant fungus, as well as facilitate optimization of this scaffold and development of future antifungals, we determined the structure of AfALS. Using cryo-electron microscopy, we solved the structures of both the apo-ALS homodimer at a resolution of 2.76 Å and the 1-bound form with a resolution of 2.36 Å (FigureB,C, Figure S8, Table S2). Electron density related to compound 1 was observed in the narrow substrate channel at the homodimer interface, which is consistent with the established binding site for multiple herbicide ALS inhibitors, including 3-5. In agreement with our proteomic data, the electron density was continuous with the side chain of Lys310, consistent with covalent modification of the protein and inconsistent with the spiro center of 1 being intact (as would be expected for noncovalent binding) (FigureC, Figure S8A). The structure achieving the best fit to the observed density contained an amide between C1 and the ε-amine of Lys310 with fragmentation of the C1–C2’ bond. This could arise through the reversible reaction of Lys310 with the C1 ketone, followed by irreversible fragmentation of the C1–C2’ bond in a retro-Dieckmann-like reaction (FigureD). The proposed fragmentation is similar to the amine-induced fragmentation of tricarbonyl C-acyl β-ketoesters first described by C. Kitsiou et al.? The driving force for this fragmentation is likely the relief of ring strain in the spiro center. In the covalently bound, postfragmentation state, the decalin portion of the ligand is within 4.5 Å of hydrophobic amino acid side chains of Trp648, Phe260, Pro251, and Val250, as well as the π-faces of polar side chains of Arg441 and Gln261, and the oxygen of the Ser259 side chain. The tetramic acid portion of the ligand, modeled for convenience as the enol tautomer, is within 4.5 Å of Tyr653, Phe652, and Ala719 (FigureC). Compared to the structure of apo AfALS, binding of 1 triggered major conformational changes in the side chains of Lys310, Arg441, Trp648, and Phe652 (Figure S8B). The side chain of Trp649 changed orientation due to spatial clash with the decalin ring of 1, the aromatic ring of Phe652 shifted closer to interact with 1 via π-stacking interactions, while the side chain of Arg441 also changed orientation to interact with 1 through electronic interactions. Due to the proximity of the Tyr653 oxygen and the C3′ oxygen (O–O distance 4.1 Å), it is possible that the OH of Tyr653 assists in C1–C2’ fragmentation by providing a hydrogen-bond to the C3′ ketone, activating it to accept the electrons from the C1–C2’ bond. The ligand-protein interactions of inhibitor 1 and AfALS are largely consistent with the S. cerevisiae forward genetics experiments, with 5/6 mutated residues observed within 5 Å of ligand atoms in the AfALS structure (Supporting Table S1). Comparison of the binding of 1 and 2 to AfALS provides a potential justification for the differential resistance to P192 mutants as P192 (P251 in AfALS) contacts a flexible propyl group in 1, likely resulting in a weaker interaction, while P192 contacts a much more rigid benzyl group in 2, likely resulting in a stronger interaction.
While 1 binds the same region as synthetic herbicides, the binding mode is distinct as shown in the overlay in FigureE. In the 1-AfALS (aqua) and 2-ScALS (pink) structures the side chains of Trp648 (Af numbering) and Trp586 (Sc numbering) are observed in different conformations and there is little overlap between the atoms of 1 and 2.
The side-chain thiol of cysteine is by far the most frequent point of attachment to proteins by covalent inhibitors, but lysine represents an attractive target, partially due to lysine’s significantly higher abundance in the proteome (5.8% lysine vs 1.9% cysteine).? Despite this promise, design of lysine targeting covalent inhibitors is often challenging due to the high pK a of the Lys ε-amino group.? Several natural products are among those compounds known to covalently modify lysine, including manoalide, which irreversibly inhibits phospholipase A2,? wortmannin and neolymphostin A, both of which target phosphoinositide 3-kinases (PI3K), ?,? and myriocin, which covalently binds to serine palmitoyltransferase (FigureF).? The mechanism of 1 is unique among these examples with strain release via C–C bond fragmentation serving to irreversibly trap a lysine side chain in an amide linkage.
With the mechanism of ALS inhibition by 1 established and understood, we next tested the translation of this biochemical potency to antifungal activity. While the potency of 1 was approximately equivalent to that of 2-5 in biochemical screens, 1 inhibited A. fumigatus growth similarly to voriconazole (IC_50_ = 0.7 μM and 0.5 μM, respectively) and was more potent than the all the known ALS inhibitors (IC_50_= 200 μM (2), 25 μM (3), ND (4), 90 μM (5))(FigureA). The antifungal activity of 1 extends beyond lab strains and persists in clinically relevant isolates of A. fumigatus from the antimicrobial resistance cell bank from the Center for Disease Control and Prevention (CDC)? including multiple isolates resistant to azoles, the standard of care for aspergillosis? (FigureB). 1 also inhibits growth of a collection of rarer pathogenic fungi including Aspergillus niger, Rhizopus oryzae, Mucor circinelloides, Scedosporium apiospermum, and Scedosporium prolificans (FigureB).
1 is a potent inhibitor of the pathogenic fungi. (A) Inhibition of A. fumigatus growth by ALS inhibitors 1–5. (B) Minimum inhibitory concentrations (MICs) for >50% and 100% growth inhibition of a panel of fungi by 1 at 24 h. Isolates of A. fumigatus are labeled as either “resistant” or susceptible, depending on azole susceptibility per the CDC ARIsolate bank. (C) Cytotoxicity of 1 against primary human cells HUVECs (human umbilical vein endothelial cells) and hepatocytes and the liver cancer cell line HEPG2.
Consistent with its observed antifungal activity arising from specific inhibition of ALS, a target with no close homologue in humans, 1 shows no cytotoxicity when tested against a panel of primary human cells and a cell line at concentrations up to 100 μM, suggesting that off-target toxicity is unlikely to be a liability (FigureC).
ALS Is Required for Full Virulence of A. fumigatus
Essential amino acids, including BCAAs, are so named because humans must acquire them through their diet. This is in contrast to fungi and bacteria, which can synthesize these nutrients de novo. This distinction makes proteins involved in BCAA biosynthesis attractive targets for antifungal development. However, a potential limitation of targeting these pathways is the ability of pathogenic organisms to scavenge BCAAs from their environment, rendering these targets conditionally essential. Despite this concern, ALS has been shown to be essential for the pathogenicity of both Cryptococcus neoformans ? and Candida albicans ? in mouse models of infection. These findings suggest that host-derived BCAA scavenging is insufficient to compensate for ALS loss from these species in the context of an infection. We aimed to determine whether the same holds true for invasive aspergillosis caused by A. fumigatus.
To investigate this, an A. fumigatus AF293 strain lacking the ilv2 gene was constructed. In vitro growth assays revealed that this ilv2Δ strain required exceptionally high levels of BCAAs in the medium to proliferate (Figure S10A–C), indicating limited BCAA import capacity. In vivo infection studies in a mouse model of invasive aspergillosis showed a significant increase in the survival of mice infected with an ilv2Δ strain (FigureA, n = 10 for infected groups, n = 6 for PBS), demonstrating reduced virulence. Furthermore, restoration of ilv2 expression in the mutant background fully recapitulated the pathogenicity of the wild-type strain (ilv2ΔRec, FigureA). The inability of the ilv2Δ strain to sufficiently grow in the respiratory tract at day 3 postinfection was supported by a 20-fold reduction in fungal DNA levels in the lungs of infected mice, suggesting significantly reduced fungal burden as compared to those infected with wild-type AF293 or ilv2ΔRec strains (FigureB, n = 5 for AF293 and ilv2ΔRec groups, n = 6 for ilv2Δ, n = 2 for PBS).
*ALS is required for full virulence in invasive aspergillosis. (A) Survival curves for a triamcinolone mouse model of invasive aspergillosis using Aspergillus fumigatus AF293 (wild-type), a mutant of AF293 with ilv2 (the gene for ALS) deleted (ilv2Δ), and the deletion mutant with ilv2 expression reconstituted at aft4 safe haven site (ilv2ΔRec). Log-rank (Mantel-Cox) test was significant (****P < 0.0001; n = 10 for infected groups; PBS n = 6). (B) Total fungal DNA quantified in the lungs of mice in each group. Kruskal–Wallis nonparametric test was significant (***P = 0.0002; AF293/ilv2ΔRec: n = 5; ilv2Δ: n = 6; PBS n = 2). Dunn’s posthoc test was used for pairwise comparisons. The P-values are indicated on the graph.
These results demonstrate that ALS is required for full virulence of A. fumigatus in the context of invasive aspergillosis and establish it as a promising therapeutic target for antifungal treatment. Next we assessed the potential of 1 for in vivo efficacy through detailed characterization of its physicochemical and pharmacokinetic properties (Table S3). We found that 1 is unstable in multiple matrices, including pH 7.4 PBS buffer (t 1/2 = 141 min), mouse plasma (t 1/2 = 88 min), and mouse liver microsomes (t 1/2 = 3 min). It is likely that the instability in these matrices is due to fragmentation chemistry similar to that described between 1 and ALS (FigureD), with water (or another biorelevant nucleophile) reacting with the C1 ketone followed by fragmentation of the C1–C2’ bond. From these data, we reasoned that even though 1 is a potent, irreversible covalent ALS inhibitor, it would be unlikely to achieve the plasma concentrations needed for 100% inhibition of pathogenic A. fumigatus strains at feasible dose levels. Consistent with these assumptions, we see a significant decrease in the potency of antifungal activity after 48 h incubations (Table S4). Taken together, these data lead us to believe 1 will require further optimization to achieve success in in vivo efficacy studies.
Discussion
The rising global incidence of invasive fungal infections, now responsible for over 2 million deaths annually, underscores an urgent need for new antifungal treatments. Current therapies face increasing resistance and limited efficacy, driving a critical need for novel antifungal agents with new mechanisms of action. Here, we utilize resistance gene-guided genome mining to discover such an agent in HB-35018 (1), an inhibitor of BCAA biosynthesis with potent in vitro activity across a breadth of pathogenic fungi. This antifungal activity is in stark contrast to what we observe from known ALS inhibitors which each show potent biochemical activity, but are very poor antifungals. It is possible that, for these herbicides, their anionic nature at cellular pH is a liability. In fungi, ALS is localized to the mitochondria, meaning that ALS inhibitors must cross the cell wall, cell membrane, and mitochondrial membrane to exert activity.? The passive permeability of anionic compounds is poor and they are likely to become substrates for the organic anion transporter (OAT) family of efflux pumps, providing a possible explanation for the lack of antifungal activity among most known ALS inhibitors.?
Intriguingly, the sdtBGC, encoding 1, represents the third BGCalong with those of chlorflavonin (6) and harzianic acid (7)to contain an ALS-homologous putative resistance gene that produces a verified ALS inhibitor. Furthermore, the sdtBGC was identified in the genome of A. terreus, which also harbors the BGC for aspterric acid (8). The latter contains a homologue DHAD, the molecular target of 8 and a key enzyme downstream of ALS in the BCAA pathway. The evolution of three distinct BGC families with ALS-homologous resistance genes, coupled with the occurrence of two BGCs with resistance genes from the same pathway in a single fungal genome, demonstrates a convergence on BCAA biosynthesis as a significant target for antifungal activity.
While this convergence on BCAA biosynthesis suggests the promise of this pathway as an antifungal target in the ecologies in which these fungi evolved, here we demonstrate that this promise translates into a therapeutic context by showing that ALS is essential to pathogenicity in a mouse model of invasive aspergillosis. Whether or not this essentiality translates to biosynthesis of other essential amino acids remains to be seen, but were this to be the case, it would unlock another large set of promising antifungal targets.
Finally, in establishing that 1 acts through a covalent modification of a lysine residue, we report the cryo-EM structure of the ALS of A. fumigatus. While the inherent instability of 1 may limit its direct use in in vivo studies, this structure provides a clear path forward for optimizing the scaffold into a viable therapeutic. The detailed structural insights will not only enable the rational design of more stable analogs but will also facilitate future efforts toward the discovery and development of novel antifungals targeting this protein. While this manuscript was in the final stages of preparation, another study was published describing the biosynthesis of 1 and demonstrating that it has modest herbicidal activity, demonstrating another path forward for the development of 1.?
Overall, this study highlights the power of genome mining as an effective strategy for the discovery of unique natural product scaffolds with therapeutic potential and by demonstrating the repeated evolutionary targeting of BCAA biosynthesis by distinct fungal BGCs, reinforces the relevance of this pathway as a target in further antifungal drug development efforts.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Denning D. W.Global Incidence and Mortality of Severe Fungal Disease Lancet Infect Dis 2024247 e 428e 43810.1016/S 1473-3099(23)00692-838224705 · doi ↗ · pubmed ↗
- 2Seidel D.Wurster S.Jenks J. D.Sati H.Gangneux J.-P.Egger M.Alastruey-Izquierdo A.Ford N. P.Chowdhary A.Sprute R.Cornely O.Thompson G. R.3rd Hoenigl M.Kontoyiannis D. P.Impact of Climate Change and Natural Disasters on Fungal Infections Lancet Microbe 202456 e 594e 60510.1016/S 2666-5247(24)00039-938518791 · doi ↗ · pubmed ↗
- 3Casadevall A.Global Warming Could Drive the Emergence of New Fungal Pathogens Nat. Microbiol 20238122217221910.1038/s 41564-023-01512-w 38030896 · doi ↗ · pubmed ↗
- 4World Health Organization . WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action; World Health Organization, 2022.
- 5Cavassin F. B.Baú-Carneiro J. L.Vilas-Boas R. R.Queiroz-Telles F.Sixty Years of Amphotericin B: An Overview of the Main Antifungal Agent Used to Treat Invasive Fungal Infections Infect Dis Ther 202110111514710.1007/s 40121-020-00382-733523419 PMC 7954977 · doi ↗ · pubmed ↗
- 6Perfect J. R.The Antifungal Pipeline: A Reality Check Nat. Rev. Drug Discovery 201716960361610.1038/nrd.2017.4628496146 PMC 5760994 · doi ↗ · pubmed ↗
- 7Puumala E.Fallah S.Robbins N.Cowen L. E.Advancements and Challenges in Antifungal Therapeutic Development Clin. Microbiol. Rev.202410.1128/cmr.00142-23PMC 1093889538294218 · doi ↗ · pubmed ↗
- 8Vanreppelen G.Wuyts J.Van Dijck P.Vandecruys P.Sources of Antifungal Drugs J. Fungi (Basel)20239217110.3390/jof 902017136836286 PMC 9965926 · doi ↗ · pubmed ↗