Partial mitochondrial genome of the enigmatic Bermuda fireworm Odontosyllis enopla Verrill, 1900 (Annelida, Syllidae, Eusyllinae) and its phylogenetic implications
Lynette D. Wyant, Brendan A. Cruz, Aydanni D. Gonzalez, Joshua M. Kovalcik, Maria A. Carolus, Lauren C. Hutto, Hope Chutjian, Jude C. Roman, Anneau Cappelmann, John J. Ankney, Aidan Popp, James B. Wood, D. Tye Pettay, Mercer R. Brugler

TL;DR
This paper reports the partial mitochondrial genome of the Bermuda fireworm and explores its evolutionary relationships.
Contribution
The study provides the first extensive partial mitogenome of Odontosyllis enopla and analyzes its phylogenetic placement.
Findings
A 10,172 bp partial mitochondrial genome was assembled, including nine protein-coding genes and two rRNAs.
Intraspecific variation was analyzed among three female O. enopla mitogenomes.
A putative location for the mitochondrial origin of replication was proposed using DNA Walker analysis.
Abstract
The Bermuda fireworm, Odontosyllis enopla Verrill, 1900, is a marine polychaete that displays a unique bioluminescent mating ritual. Despite the first sighting of O. enopla more than 534 years ago, molecular data have been limited. Several syllid mitogenomes are currently available; however, there are only three published genes for O. enopla: two partial mitochondrial genes (16S [508 bp] and cox1 [653 bp]; 1,161 bp total) and one partial nuclear gene (18S [1,339 bp]). This study bioinformatically mined previously published transcriptomes of O. enopla for mitochondrial reads and subsequently assembled and annotated a partial mitochondrial genome (10,172 bp). The partial mitogenome includes nine (of 13) protein-coding genes, two ribosomal RNAs, and seven (of 22) complete tRNAs. We place the Bermuda fireworm in phylogenetic context using all available syllid mitogenomes, analyze…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
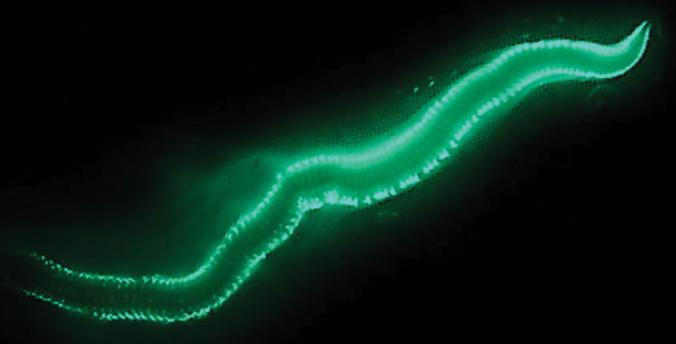 Figure 1
Figure 1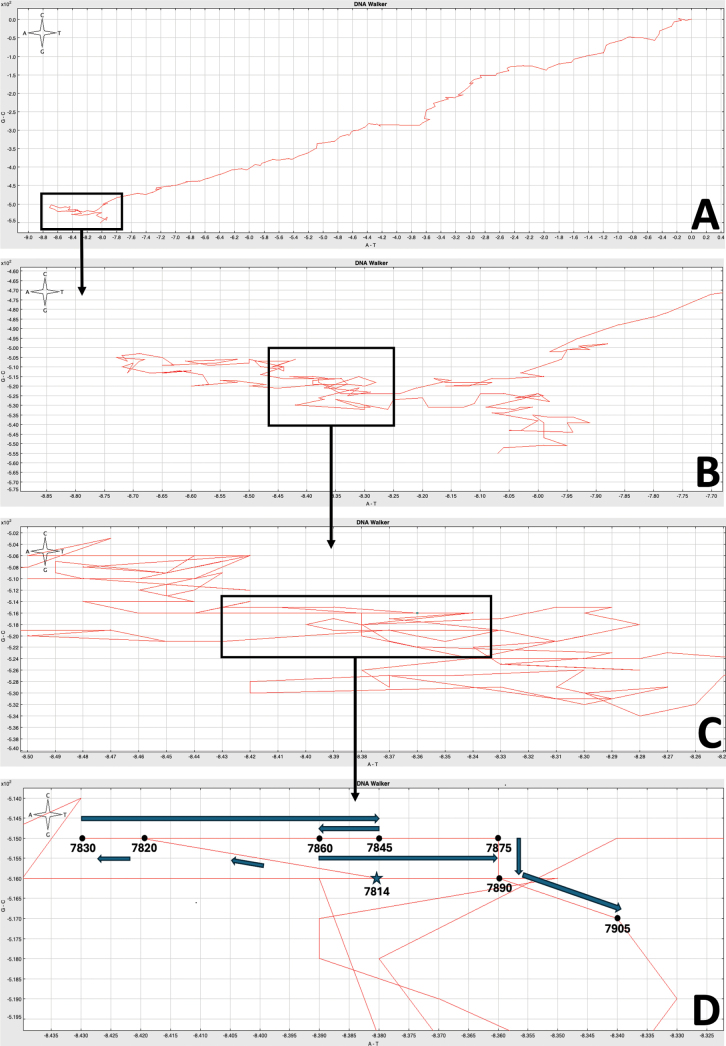 Figure 2
Figure 2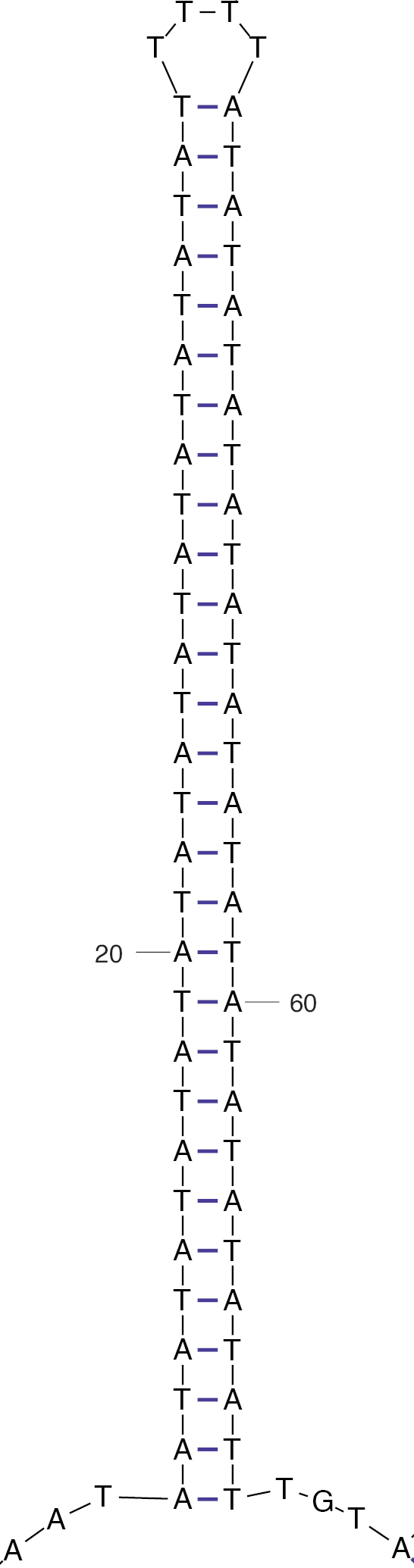 Figure 3
Figure 3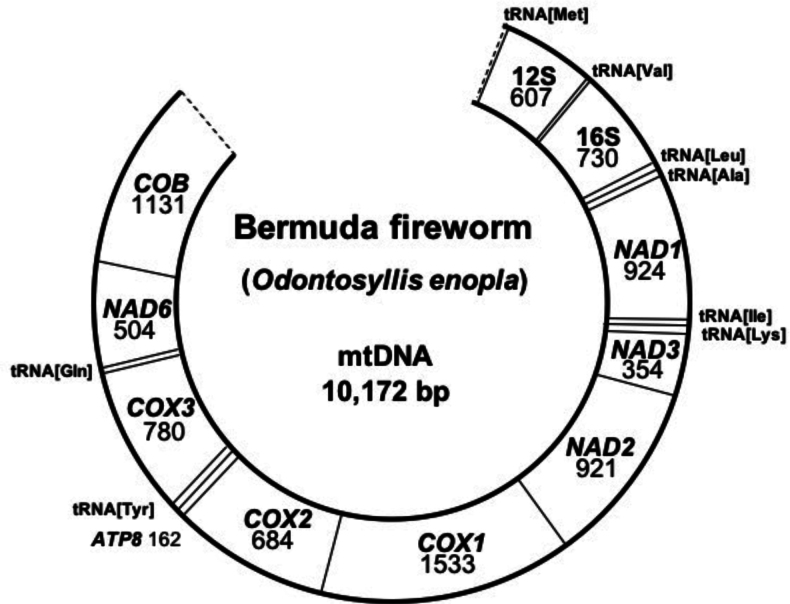 Figure 4
Figure 4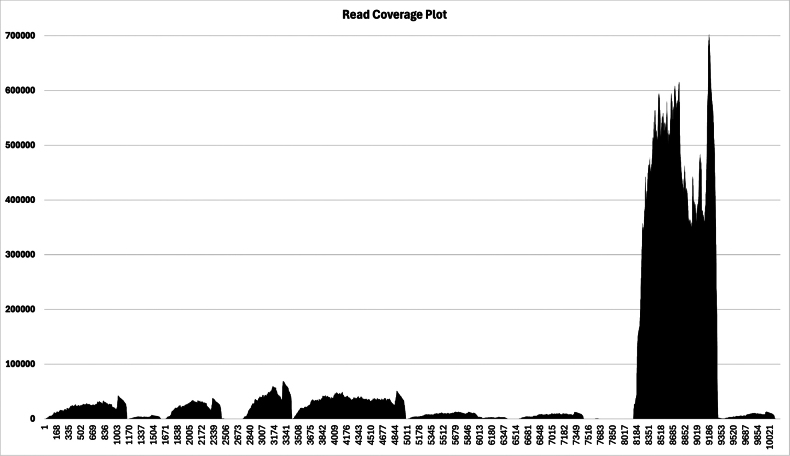 Figure 5
Figure 5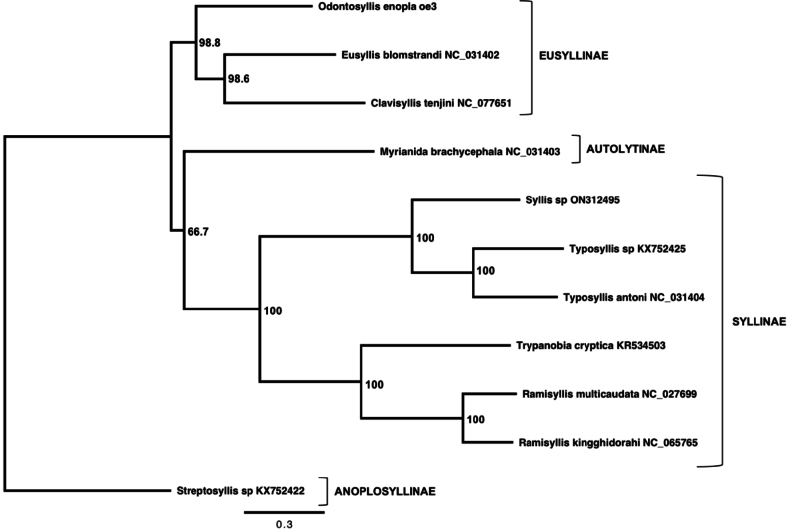 Figure 6
Figure 6| Gene | Length ( |
|---|---|
| 12S | 607 |
| tRNA-Val | 62 |
| IGR | 221 |
| 16S | 730 |
| IGR | 31 |
| tRNA-Leu | 62 |
| IGR | 123 |
| tRNA-Ala | 62 |
| IGR | 442 |
|
| 924 |
| tRNA-Ile | 61 |
| IGR | 3 |
| tRNA-Lys | 63 |
| IGR | 2 |
|
| 353 |
| IGR | 60 |
|
| 921 |
| IGR | 81 |
|
| 1533 |
| IGR | 45 |
|
| 684 |
| IGR | 64 |
|
| 162 |
| tRNA-Tyr | 62 |
| 780 | |
| tRNA-Gln** | 61 |
| IGR | 2 |
| 504 | |
| 1131 | |
| IGR | 28 |
| oe1 | oe2 | |
|
| — | — |
|
| 0.00114 (= 0.114%) | — |
|
| 0.000817 (= 0.0817%) | 0.000654 (= 0.0654%) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMarine Biology and Ecology Research · Invertebrate Taxonomy and Ecology · bioluminescence and chemiluminescence research
Introduction
Odontosyllis enopla Verrill, 1900, more commonly known as the Bermuda fireworm, is a remarkable annelid belonging to the family Syllidae (Fig. 1). These tube-dwelling worms, found on sandy coral substrates in benthic habitats (Fischer and Fischer 1995), display an incredible bioluminescent mating ritual that was first seen and recorded by Christopher Columbus in 1492 (Brugler et al. 2018) and first described by Galloway and Welch (1911).
Odontosyllis enopla (image credit: coauthor James B. Wood). Photo taken near the shore at the north point of Ferry Island, St. George’s Parrish, Bermuda.
The Bermuda fireworm is a unique annelid due to its distinct mating behaviors and physiology. In preparation for breeding, Bermuda fireworms of both sexes undergo morphological changes, including enlargement and pigmentation of their four eyes to enhance visual sensitivity. This is particularly pronounced in males for detecting the females’ bioluminescence (Wolken and Florida 1984; Franke 1999). This sets the stage for a highly synchronized mating swarm, where selective pressures from predation favor precise timing to minimize vulnerability of isolated or early individuals (Huntsman 1948). Females initiate the ritual by rising to the surface and swimming in slow circles while emitting a continuous bluish-green glow from secreted luminous mucus and releasing their gametes. Triggered by this display, males swim towards the glowing females, producing quick, consecutive bioluminescent flashes while releasing their own gametes into the surrounding water (Fischer and Fischer 1995; Verdes et al. 2022). The bioluminescence peaks in the green portion of the visible spectrum, with wavelengths between 504–507 nm. Their visual system is most sensitive to the aforementioned wavelength, as shown by electroretinogram (ERG) recordings in response to light at multiple wavelengths (Wilkens and Wolken 1981). The similarities between the light detected and the light emitted suggest that the Bermuda fireworms are visually tuned to detect mating signals from bioluminescence.
The Bermuda fireworm synchronizes its mating ritual with the lunar cycle, with swarming episodes coinciding with the first day after full moons during the summer and early autumn months (Brugler et al. 2018). Utilizing specialized chaetae, this benthic organism will swim to the surface 57 ± 1 minutes after the astronomical sunset to begin its mating ritual (Fischer and Fischer 1995). This mating swarm has been observed up to five nights following the full moon, after which both male and female Bermuda fireworms return to their benthic habitats (Fischer and Fischer 1995). The fertilized zygotes undergo cell division and, after 14 hours, become trochophores that can swim freely through the water column (Fischer and Fischer 1995).
Evidence suggests that the common ancestor of all bioluminescent syllid species was not bioluminescent itself; in fact, bioluminescence in Odontosyllis Claparède, 1863 evolved independently twice (Verdes et al. 2022). While the bioluminescence of this species is of great interest to researchers, the reversible epitoky metamorphosis in both males and females is of even greater importance. The Bermuda fireworm undergoes different physiological changes to prepare for mating, including enlargement of the eyes in males, the growth of chaetae used for rapid swimming to reach the surface, and tissues protruding from the body of the females holding the oocytes (Fischer and Fischer 1995; Brugler et al. 2018). After the mating swarm, the swimming chaetae are shed, although there appears to be no specific timeline for this reversal. Some Bermuda fireworms shed the chaetae in as little as five days after swarming, while others had at least some remaining chaetae after 35 days. Additional research needs to be conducted regarding the reversal of the size of the male eyes. Research thus far indicates that there was no reduction in male eye size two months after swarming and therefore may simply reflect sexual maturity (Fischer and Fischer 1995).
To date, only three genes have been published for the Bermuda fireworm, totaling 2,500 base pairs (bp). Of the three genes published, two are mitochondrial genes: the large subunit ribosomal RNA (16S) and cytochrome c oxidase subunit I (cox1). The third gene is the nuclear small subunit ribosomal RNA (18S). The majority of available mapped mitochondrial genomes for annelids are from the clades Errantia and Sedentaria. The family Syllidae, of which the Bermuda fireworm is a member, is known to have a highly variable mitochondrial genome in terms of gene order (Aguado et al. 2016; Schwarze et al. 2026). This manuscript presents the partial mitochondrial genome for the Bermuda fireworm and places it in phylogenetic context amongst its relatives. To our knowledge, the only study that has included Odontosyllis enopla in a phylogenetic context was a three-gene phylogeny by Verdes et al. (2022) where the authors listed O. enopla under the species ID ‘Odontosyllis sp. 9’ and specimen code ‘OenoTR.’ We also analyze intraspecific variation among three female O. enopla partial mitogenomes and propose a putative location for the mitochondrial origin of replication, which, to date, has not been definitively identified in the Bermuda fireworm or its closest relatives.
Material and methods
Background on transcriptome acquisition
Per Brugler et al. (2018), total RNA was isolated from the whole body of three female Odontosyllis enopla worms using a modified RNeasy Tissue Kit (Qiagen) protocol (voucher material not available as worms were completely macerated). Isolates were prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA) with a 350 bp insert size and run at the NY Genome Center on an Illumina HiSeq 2500 (2 × 125 bp), allocating 1/8 of a lane for each isolate. The run generated 37,063,191 (Individual #1), 39,513,743 (Individual #2), and 34,329,885 (Individual #3) raw reads. After trimming adaptors and low-quality regions, assembly with Trinity yielded 176,598 (Individual #1), 207,006 (Individual #2) and 283,041 (Individual #3) contigs (including splice variants). These represented 44,426 (Individual #1), 49,458 (Individual #2) and 61,002 (Individual #3) open reading frames (>100 amino acids) predicted by Transdecoder and included >99.0% of the 2,748 core KOGs.
Bioinformatics
Mitochondrial reads were bioinformatically extracted from the transcriptomes of three female Odontosyllis enopla worms using MitoFinder v. 1.4 (Allio et al. 2020). MitoFinder employed MEGAHIT v. 3.0 (Li et al. 2015) for mitogenome assembly and tRNAscan-SE (Chan and Lowe 2019) for tRNA annotation. The following command was used to run MitoFinder on an iMac: ./mitofinder --megahit --override --new-genes -j [file name] -1 [left_reads.fastq.gz] -2 [right_reads.fastq.gz] -r [genbank_reference.gb] -o [genetic_code] -p [threads] -m [memory] -t trnascan. Eusyllis blomstrandi Malmgren, 1867 (GenBank accession no. KX752423; 14,712 bp in length) was used as the reference, and Translation Table 1 (Invertebrate Mitochondrial Code) was used as the genetic code. We would have preferred to use Odontosyllis undecimdonta Imajima & Hartman, 1964 as the reference, but only two mitochondrial genes were available on GenBank in 2025 (16S and cox1). Newly assembled partial mitogenomes were annotated using the MITOS Web Server (Bernt et al. 2013). Of the three Odontosyllis enopla worms, individual #3 (specimen ID: oe3) yielded the longest single mitochondrial contig at 10,172 bp, and thus this partial mitogenome is described herein. We used MEGA X (Kumar et al. 2018; Stecher et al. 2020) to obtain intraspecific genetic distance estimates (p-distances) among the three partial O. enopla mitogenomes.
Table 1.: Gene order and length of Odontosyllis enopla mitochondrial protein-coding genes, ribosomal RNAs, transfer RNAs, and intergenic regions (IGRs).
Phylogenetic analysis
The partial mitogenome of Odontosyllis enopla (GenBank accession no. PP998669) was combined with mitogenomes presented in Aguado et al. (2015a; Ramisyllis multicaudata Glasby, Schroeder & Aguado, 2012 and Trypanobia cryptica Aguado, Murray & Hutchings, 2015c), Aguado et al. (2016; Eusyllis blomstrandi, Myrianida brachycephala (Marenzeller, 1874), Streptosyllis sp. Webster & Benedict, 1884, Typosyllis antoni Aguado, Helm, Weidhase & Bleidorn, 2015b, and Typosyllis sp. (Langerhans, 1879)), Aguado et al. (2022; Ramisyllis kingghidorahi Aguado, Ponz-Segrelles, Glasby, Ribeiro, Jimi & Miura, 2022 [in Aguado et al. 2022]), Cejp et al. (2023; Clavisyllis tenjini Cejp, Jimi & Aguado, 2023), and Chae et al. (2023; Syllis sp. Lamarck, 1818) for a total of 11 mitogenomes. Each of the nine protein-coding genes (cob, atp8, cox1-3, nad1-3, nad6) and two ribosomal RNAs (12S and 16S) from all 11 mitogenomes were placed in individual AliView v. 1.23 (Larsson 2014) files and individually aligned using MAFFT LINS-i v7 (Katoh et al. 2019). GBlocks v. 0.91b was applied to each individual gene region to remove poorly aligned positions and divergent regions. Each individual gene region was subsequently concatenated into a single file using Seqotron v. 1.0.1 (Fourment and Holmes 2016), treating the mitogenome as a single locus. GBlocks reduced the length of the multiple sequence alignment to 7,097 bp (alignment available upon request to the corresponding author MRB).
The Akaike Information Criterion (AIC) within jModelTest v. 2.1.10 (Guindon and Gascuel 2003; Darriba et al. 2012) selected the GTR+I+G model of sequence evolution (p-inv: 0.2800; gamma: 1.0260). A maximum-likelihood-based phylogenetic tree was built using the command-line version of PhyML v. 3.1 (Guindon et al. 2010). PhyML parameters included a tree topology search consisting of the best of NNIs and SPRs, a BioNJ starting tree, and 1,000 bootstrap replicates. The resulting phylogenetic tree was visualized using FigTree v. 1.4.4 (by Andrew Rambaut; https://github.com/rambaut/figtree/releases). The tree was rooted with Streptosyllis sp. (KX752422) based on a phylogenetic analysis conducted by Aguado et al. (2016) using full mitogenomes (but see DeSalle et al. 2023).
Origin of replication
The DNA Skew Graphing tool (GraphDNA; Thomas et al. 2007), available online via the Viral Bioinformatics Research Centre (https://4virology.net/), was used to search representative mitochondrial genomes for abrupt changes in base composition bias characteristic of the origin of replication. In particular, we used the ‘DNA Walker’ graphing option (per Lobry 1996; Fig. 2). After locating a putative origin of replication, we used the default parameters in the DNA Folding Form on the UNAFold web server (Zuker 2003) to identify a stable stem-loop configuration containing a characteristic T-rich loop (Fig. 3), a common feature of origins of replication.
A DNA Walk of the partial mitochondrial genome of Odontosyllis enopla. Abrupt changes in base composition bias (switchbacks) are characteristic of the origins of replication. A. Full mitogenome walk (window size: 75); B. Zoomed-in image of switchback (window size: 20); C. Zoomed-in image of switchback (window size: 10); D Zoomed-in image of switchback with nucleotide positions and cardinal direction changes indicated (window size: 15).
The most thermodynamically stable stem-loop structure (dG = −22.02) as output by the UNAFold web server. Note the long AT-rich stem containing a characteristic T-rich loop, which is a common feature within the origin of replication.
Results
The partial mitogenome of the Bermuda fireworm, Odontosyllis enopla, is 10,172 bp in length and contains nine of the 13 protein-coding genes (cob, atp8, cox1-3, nad1-3, nad6), two ribosomal RNAs (12S, 16S) and eight transfer RNAs (Met, Val, Leu, Ala, Ile, Lys, Tyr, Gln) (Fig. 4). We were unable to bioinformatically recover nad4L, atp6, and nad4-5. The partial mitogenome is available under GenBank accession no. PP998669. Gene order for the partial mitogenome of Odontosyllis enopla is as follows: tRNA[Met]-12S-tRNA[Val]-16S-tRNA[Leu]-tRNA[Ala]-nad1-tRNA[Ile]-tRNA[Lys]-nad3-nad2-cox1-cox2-atp8-tRNA[Tyr]-cox3-tRNA[Gln]-nad6-cob (Table 1). Of the genes recovered, the gene order of O. enopla matches that of Eusyllis blomstrandi (GenBank accession no. NC_031402; the E. blomstrandi mitogenome is 14,712 bp in length). We are missing the following gene segment: atp6-nad5-nad4L-nad4. MitoFinder found no evidence of circularization of the 10,172 bp fragment. A read coverage plot is presented in Fig. 5. The 16S ribosomal RNA (position 8182–9304) was the most transcriptionally active gene (upwards of 700K reads per position), followed by cytochrome c oxidase subunit II (cox2; position 2749–3432; maximum of ~70K reads per position) and cytochrome c oxidase subunit I (cox1; position 3458–5008; maximum of ~50K reads per position). Cells typically manufacture significant amounts of ribosomal RNA (16S and 12S) and can terminate transcription after these genes are successfully copied. Interestingly, 12S ribosomal RNA (position 9386–9984) was one of the least transcriptionally active genes. Similar to other invertebrates, the O. enopla mitogenome is AT-rich (A: 3,880, T: 3,075, G: 1,886, C: 1,331). We obtained 6,120 bp of comparable sequence data from the three O. enopla mitogenomes, yielding eight variable sites. Individual #1 (specimen ID: oe1) had four unique substitutions, individual #2 (specimen ID: oe2) had three unique substitutions, and individual #3 (specimen ID: oe3) had one unique substitution (Table 2). A maximum-likelihood-based phylogenetic tree based on nine protein-coding genes (cob, atp8, cox1-3, nad1-3, nad6) and two ribosomal RNAs (12S, 16S) showed Odontosyllis enopla grouping sister to a clade comprised of Eusyllis blomstrandi (NC_031402) and Clavisyllis tenjini (NC_077651) with 98.8 node support (Fig. 6).
Partial mitochondrial genome map of Odontosyllis enopla. We were unable to bioinformatically recover nad4L, atp6, and nad4-5.
A read coverage plot showing the number of individual reads (y-axis) that mapped to the different parts of the assembled mitogenome (x-axis; 10,172 bp in length).
A maximum-likelihood-based phylogenetic tree based on nine protein-coding genes (cob, atp8, cox1-3, nad1-3, nad6) and two ribosomal RNAs (12S, 16S). Node support is based on 1,000 bootstrap replicates. The tree was rooted with Streptosyllis sp. based on a phylogenetic analysis by Aguado et al. (2016) using complete mitogenomes. The following sequences were used: Odontosyllis enoplaPP998669 (this study), Ramisyllis multicaudataNC_027699 and Trypanobia crypticaKR534503 (Aguado et al. 2015a), Eusyllis blomstrandiNC_031402, Myrianida brachycephalaNC_031403, Streptosyllis sp. KX752422, Typosyllis antoniNC_031404, and Typosyllis sp. KX752425 (Aguado et al. 2016), Ramisyllis kingghidorahiNC_065765 (Aguado et al. 2022), Clavisyllis tenjiniNC_077651 (Cejp et al. 2023), and Syllis sp. ON312495 (Chae et al. 2023).
Table 2.: Genetic distance estimates (p-distances) among the three Odontosyllis enopla mitogenomes (based on 6,120 bp of comparable sequence data). A total of eight variable sites were identified. Individual #1 (specimen ID: oe1); Individual #2 (specimen ID: oe2); Individual #3 (specimen ID: oe3).
Discussion and conclusion
Prior to this publication, only two partial mitochondrial sequences for Odontosyllis enopla were available in GenBank: the large subunit ribosomal RNA gene (16S; 508 bp) and the cytochrome c oxidase subunit 1 gene (cox1; 653 bp). Combined, these sequences are 1,161 bp in length. Our newly obtained sequence data (10,172 bp) represent more than 8.76 times the amount of mitochondrial DNA than was previously available.
Odontosyllis enopla was included in a three-gene phylogeny by Verdes et al. (2022), where the authors listed O. enopla under the species ID “Odontosyllis sp. 9” and specimen code “OenoTR”. In that phylogeny, Odontosyllis was recovered as paraphyletic. O. enopla and congeners (“Clade 2” per Verdes et al. (2022)) are grouped sister to Nudisyllis Knox & Cameron, 1970. The clade consisting of “Clade 2” Odontosyllis + Nudisyllis is grouped sister to “Clade 1” Odontosyllis + Eusyllis + Pionosyllis Malmgren, 1867. Mitochondrial genomes of Nudisyllis and Pionosyllis were not available at the time of this analysis; however, the complete mitogenome of Eusyllis blomstrandi (GenBank accession no. NC_031402) was available and included in our phylogenetic analysis.
The phylogenetic reconstruction placed Odontosyllis enopla as a sister (ML bootstrap support: 98.8) to a clade containing Eusyllis blomstrandi (NC_031402) and Clavisyllis tenjini (NC_077651). These three species are all classified in the subfamily Eusyllinae. Additionally, all three taxa share the same mitochondrial gene order. The topology of our phylogenetic reconstruction was also recovered by Schwarze et al. (2026), who presented a 26-taxon ML tree based on all 13 mitochondrial protein-coding genes, mitochondrial 16S and 12S, and nuclear 18S and 28S. Their phylogeny included two additional members of Autolytinae (Virchowia christophi Aguado, Springer, Oguchi, Sato, Jimi & Miura, 2025 [in Springer et al. 2025] and Myrianida sp. Milne Edwards, 1845) and nine additional members of Syllinae (Eurysyllis tuberculata Ehlers, 1864, Trypanosyllis sp. Claparède, 1864, Parasphaerosyllis ezoensis Imajima & Hartman, 1964, Syllis malaquini Ribeiro, Ponz-Segrelles, Helm, Egger & Aguado, 2020, Haplosyllis sp. Langerhans, 1879, Syllis maganda Martínez & San Martín, 2020, Syllis okadai Fauvel, 1934, Paraopisthosyllis rufa Springer, Aguado, Sato, Oguchi, Jimi & Miura, 2025, and Megasyllis nipponica (Imajima, 1966)).
We were unable to bioinformatically recover nad4L, atp6, and nad4-5. These four genes are found in tandem (atp6-nad5-nad4L-nad4) in the mitogenomes of Clavisyllis tenjini (NC_077651), Eusyllis blomstrandi (NC_031402), Myrianida brachycephala (NC_031403; Autolytinae), and Streptosyllis sp. (KX752422; Anoplosyllinae). In C. tenjini, E. blomstrandi, and M. brachycephala, these four genes are located between cob and 12S. In Streptosyllis sp., these four genes have been translocated between cox2 and cox3. In Ramisyllis multicaudata (NC_027699) and Trypanobia cryptica (KR534503), both members of Syllinae, nad5 has been split from the 4-gene segment and moved to a different location (between cox3 and 16S). In Typosyllis antoni (NC_031404) and Typosyllis sp. (KX752425), also members of Syllinae, both nad5 and atp6 have been split from the 4-gene segment and moved to different locations (nad5 is between nad3 and nad1, while atp6 is between 12S and cox1). Given the considerable variability in the structure and placement of this four-gene segment across these mitogenomes, these four genes may have been lost during a rearrangement event. That said, MitoFinder found no evidence of circularization. A more plausible explanation is that this four-gene segment (atp6-nad5-nad4L-nad4) was not transcriptionally active when the three female Odontosyllis enopla worms were collected and preserved. These results also suggest that the four-gene segment may not play a significant role in bioluminescence display, gamete formation, or sexual reproduction more broadly.
According to Patra et al. (2016), most annelid mitochondrial control regions (i.e. the origin of replication or D-Loop) are located between tRNA[Arg] and tRNA[His], but the position does indeed vary in some orders. We searched the nine (of 13) protein-coding genes, two ribosomal RNAs, and seven (of 22) complete tRNAs (totaling 10,172 bp) for evidence of a putative origin of replication. A DNA Walk analysis identified a switchback in cardinal direction in a ~440 bp non-coding region between tRNA[Ala] and nad1 (Fig. 2). After identifying this putative origin of replication, we used the default parameters in the DNA Folding Form on the UNAFold web server to locate a lengthy stem-loop configuration containing a characteristic T-rich loop (Fig. 3), which is a common feature within the origin of replication. Excluding the tRNAs Leucine and Alanine, the putative control region in O. enopla is located between 16S and nad1. This placement is notable as Cejp et al. (2022) sequenced the complete mitochondrial genome of four species within the family Chrysopetalidae (Errantia, Phyllodocida, Nereidiformia) and also located the control region between 16S and nad1 (more specifically, between tRNA[Ile] and tRNA [Leu1] or tRNA [Leu1] and tRNA [Ser2]). Aguado et al. (2016: 93) noted that “In the five mt genomes (Streptosyllis sp., Eusyllis blomstrandi, Myrianida brachycephala, Typosyllis antoni and Typosyllis sp.), the longest non-coding regions are AT-rich and are suggested to be the putative control regions.” In E. blomstrandi and M. brachycephala, these regions are located between 12S and 16S (specifically, between tRNA[Val] and 16S). As noted earlier, cells typically manufacture significant amounts of ribosomal RNA (16S and 12S) and can terminate transcription after these genes are successfully copied. Thus, any gene rearrangement that places the origin of replication immediately upstream of ribosomal 16S and 12S genes would have a selective advantage.
We analyzed intraspecific variation among three female O. enopla partial mitogenomes and identified eight variable sites across 6,120 bp of comparable sequence data. Individual #1 (specimen ID: oe1) had four unique substitutions, individual #2 (oe2) had three unique substitutions, and individual #3 (oe3) had one unique substitution. Genetic distance estimates (p-distances) ranged from 0.114% (comparing oe1 and oe2) to 0.0654% (comparing oe2 and oe3). Within the molecular-based literature on annelids, intraspecific genetic divergences based on mitochondrial DNA are only available for 16S and cox1 (e.g. Nygren et al. 2010; Aguado et al. 2019; Del Olmo et al. 2024); thus, no comparable data currently exist at the partial or whole mitogenome level. Although not directly comparable, intraspecific genetic distances (uncorrected Kimura two-parameter) within the Syllis prolifera Krohn, 1852 species complex ranged from 0.2 ± 0.2 to 0.9 ± 0.2 for 16S and 0.3 ± 0.1 to 1.5 ± 0.2 for cox1 (Del Olmo et al. 2024). Intraspecific genetic divergences (p-distances) within Amblyosyllis Grube, 1857 were <1% for 16S (with one exception being 1.9%) and 0–4% for cox1 (Aguado et al. 2019). According to Kvist S (2016), the median value for intraspecific and interspecific distances within annelid mitochondrial cox1 is 3.56%, and 20.06%, respectively. We suggest that future studies determine if these thresholds also hold true at the whole mitogenome level.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aguado MT, Glasby CJ, Schroeder PC, Weigert A, Bleidorn C (2015 a) The making of a branching annelid: An analysis of complete mitochondrial genome and ribosomal data of Ramisyllis multicaudata. Scientific Reports 5(1): e 12072. 10.1038/srep 12072 PMC 450532626183383 · doi ↗ · pubmed ↗
- 2Aguado MT, Helm C, Weidhase M, Bleidorn C (2015 b) Description of a new syllid species as a model for evolutionary research of reproduction and regeneration in annelids. Organisms, Diversity & Evolution 15(1): 1–21. 10.1007/s 13127-014-0183-5 · doi ↗
- 3Aguado MT, Murray A, Hutchings P (2015 c) Syllidae (Annelida: Phyllodocida) from Lizard Island, great barrier reef, Australia. Zootaxa 4019(1): 35–60. 10.11646/zootaxa.4019.1.526624065 · doi ↗ · pubmed ↗
- 4Aguado MT, Richter S, Sontowski R, Golombek A, Struck TH, Bleidorn C (2016) Syllidae mitochondrial gene order is unusually variable for Annelida. Gene 594(1): 89–96. 10.1016/j.gene.2016.08.05027590441 · doi ↗ · pubmed ↗
- 5Aguado MT, Capa M, Lago-Barcia D, Gil J, Pleijel F, Nygren A (2019) Species delimitation in Amblyosyllis (Annelida, Syllidae). P Lo S ONE 14(4): e 0214211. 10.1371/journal.pone.0214211 PMC 645752130970025 · doi ↗ · pubmed ↗
- 6Aguado MT, Ponz-Segrelles G, Glasby CJ, Ribeiro RP, Nakamura M, Oguchi K, Omori A, Kohtsuka H, Fischer C, Ise Y, Jimi N, Miura T (2022) Ramisyllis kingghidorahi n. sp., a new branching annelid from Japan. Organisms, Diversity & Evolution 22(2): 377–405. 10.1007/s 13127-021-00538-4 · doi ↗
- 7Allio R, Schomaker‐Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F (2020) Mito Finder: Efficient automated large‐scale extraction of mitogenomic data in target enrichment phylogenomics. Molecular Ecology Resources 20(4): 892–905. 10.1111/1755-0998.13160 PMC 749704232243090 · doi ↗ · pubmed ↗
- 8Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF (2013) MITOS: Improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution 69(2): 313–319. 10.1016/j.ympev.2012.08.02322982435 · doi ↗ · pubmed ↗