Abnormal Newborn Screening Resembling Carnitine Palmitoyltransferase 1a Deficiency in Three Patients With COASY Protein Associated Neurodegeneration
Matthew Lynch, Sophie Manoy, Claire Murray, Geoff Wallace, Nolette Pereira, Ricky Price, Anita Inwood, Jim McGill, David Coman

TL;DR
Three patients with a rare neurodegenerative disease were identified through newborn screening due to a metabolic profile resembling a different condition, suggesting earlier diagnosis is possible.
Contribution
This study shows that COASY protein associated neurodegeneration can be detected via newborn screening patterns typically linked to another metabolic disorder.
Findings
Three patients with COASY protein associated neurodegeneration showed a metabolic profile resembling CPT1 deficiency on newborn screening.
Early detection via newborn screening can occur before neurological symptoms appear.
Inclusion of COASY-related disease in the differential diagnosis of CPT1-like profiles may improve early diagnosis.
Abstract
COASY protein associated neurodegeneration is a rare, progressive autosomal recessive neuroferritinopathy due to pathogenic mutations in the COASY gene, coding for the mitochondrial located coenzyme A synthase. Clinical manifestations include seizures, progressive spasticity, dystonia, neuropathy, cognitive decline and neuropsychiatric abnormalities. Both foetal and childhood onset phenotypes are described. We report three patients with COASY protein associated neurodegeneration who were identified on newborn screening with a dried bloodspot acylcarnitine pattern consistent with carnitine palmitoyltransferase 1a deficiency, that is, an elevated ratio of free carnitine (C0) to the sum of palmitoylcarnitine (C16) and octanoylcarnitine (C18):[C0/(C16+C18)]. Two siblings, who died in infancy, displayed neurological features from birth, with magnetic resonance imaging of the brain displaying…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
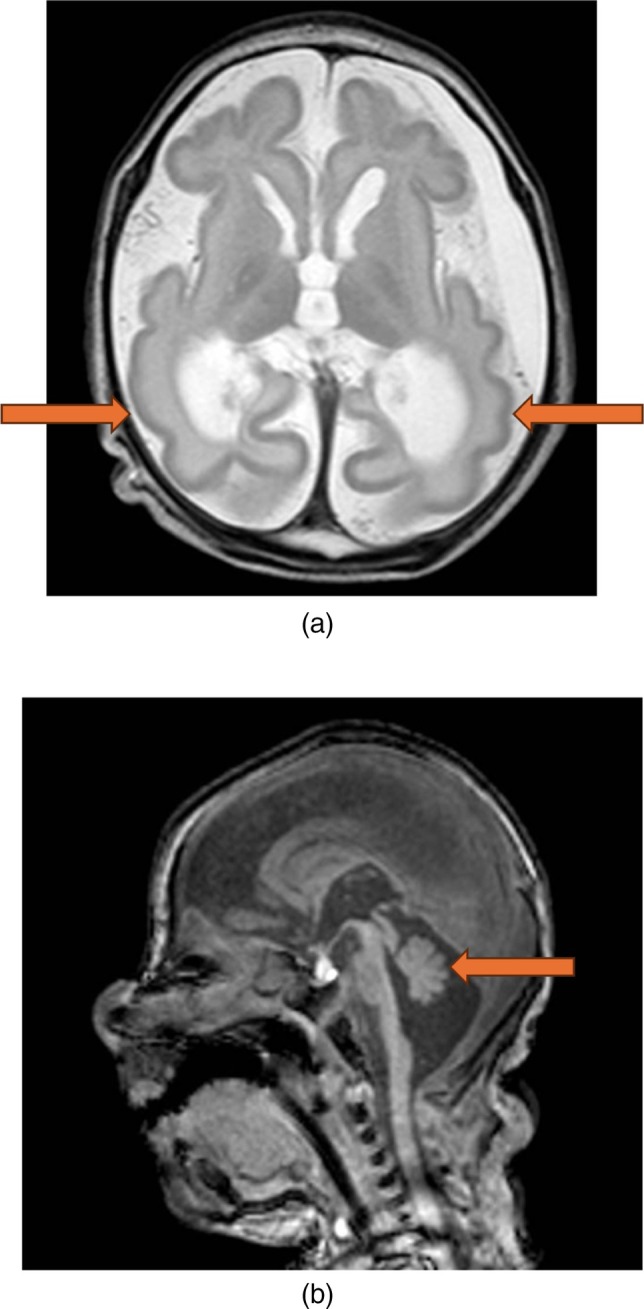 FIGURE 1
FIGURE 1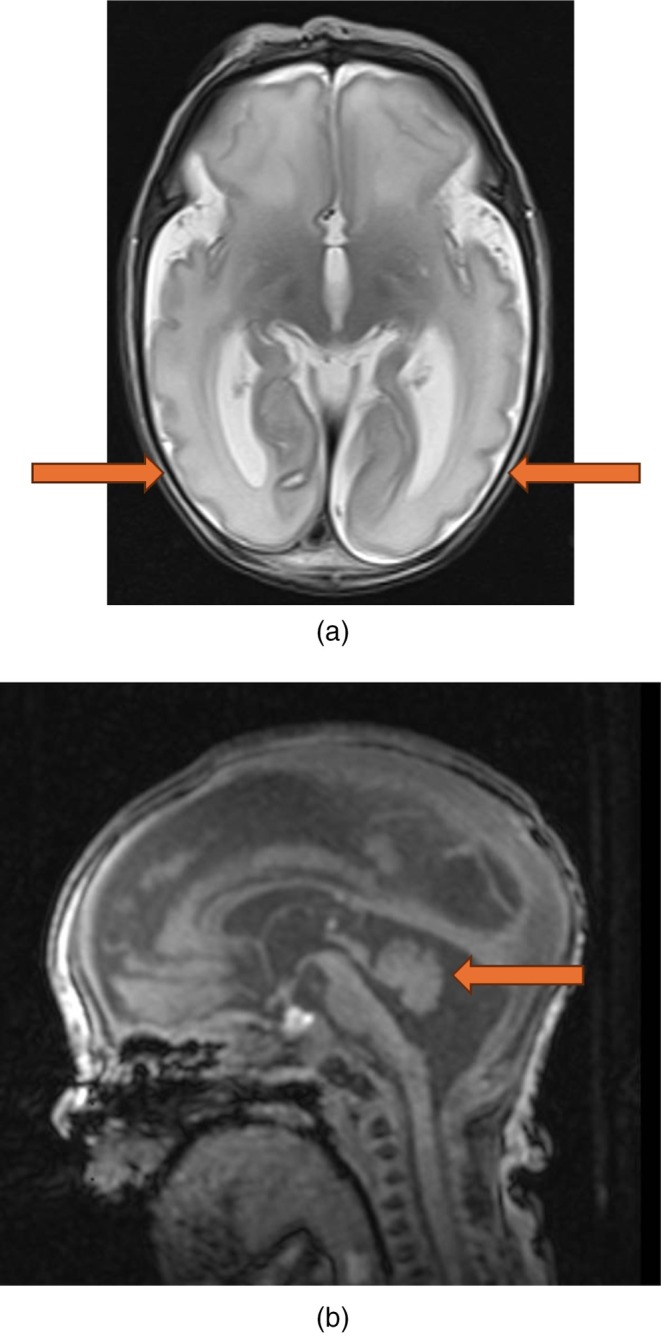 FIGURE 2
FIGURE 2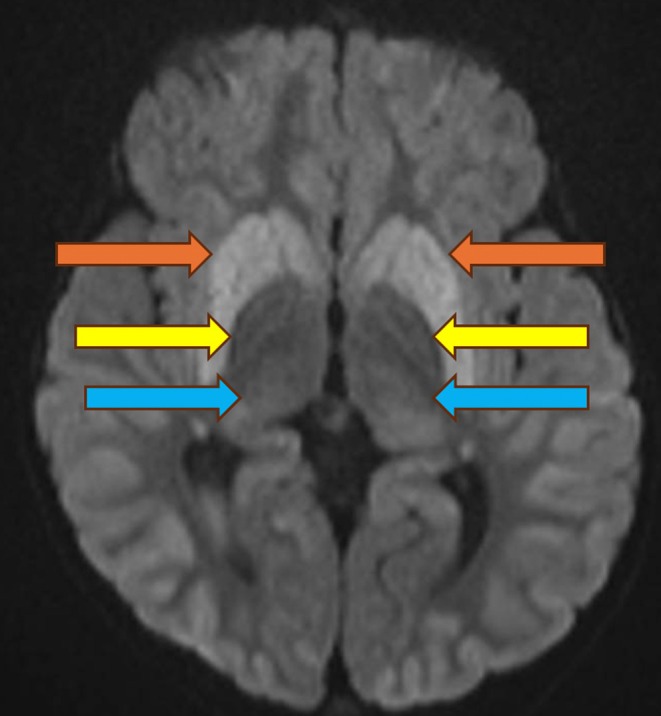 FIGURE 3
FIGURE 3| Patient | Age | C0 (μmol/L) (reference range 8–70 μmol/L) | C18:1 (μmol/L) | C18 (μmol/L) | C16 (μmol/L) | Ratio [C0/(C16+C18)] (reference range 2.5–45) |
|---|---|---|---|---|---|---|
| 1 | 4 days | 96 | 0.46 | 0.21 | 0.56 | 125 |
| 6 days | 78 | 0.53 | 0.16 | 0.62 | 96 | |
| 2 | 3 days | 100 | 0.44 | 0.16 | 0.5 | 151 |
| 9 days | 108 | 0.51 | 0.14 | 0.43 | 166 | |
| 3 | 3 days | 135 | 0.52 | 0.25 | 0.8 | 129 |
| 49 days | 76 | 0.56 | 0.21 | 0.42 | 130 | |
| 2 years | 110 | Not available | 0.1 | 0.3 | 275 |
| Patient | Clinical presentation | Genotype (COASY gene) | MRI brain findings |
|---|---|---|---|
| 1 | Seizures |
c.1403_1404dupTG c.215A>G | Microcephaly, simplified gyral pattern, parenchymal atrophy, widened extra axial CSF spaces, dilated ventricles and pontocerebellar hypoplasia. Diffusion restriction in the basal ganglia, thalami. Bilateral subdural collections. |
| 2 | Seizures |
c.1403_1404dupTG c.215A>G | Microcephaly, simplified gyral pattern, parenchymal atrophy, widened extra axial CSF spaces, dilated ventricles and pontocerebellar hypoplasia. Findings were similar to patient 1 but of a much lesser severity. |
| 3 | Global developmental delay |
c.1403_1404dupTG c.1495C>T | Thin corpus callosum, susceptibility artefact in the globus pallidi, symmetric T2 weighted imaging hyperintensity in the basal ganglia and thalami. Symmetric diffusion restriction in the cortex, basal ganglia and thalami. |
| 4 (Evers et al.) | Hypotonia, global developmental delay |
c.1495C>T; p.R499C c.C641T; p.A214V | Bilateral T2 hyperintensity and swelling of caudate nucleus, putamen and thalamus with associated restricted diffusion on DWI. |
| 5 (Evers et al.) | Broad based gait, global developmental delay |
c.1495C>T; p.R499C c.C641T; p.A214V | Bilateral T2 and FLAIR hyperintensity of caudate nucleus, putamen, thalamus and cortex. |
| 6 (Rosati et al.) | Generalised hypotonia, minimal spontaneous movements | c.1664G>A; p.R555H | T2 hyperintensity of bilateral globus pallidi, thalami and posterior limbs of the internal capsule. Progressive atrophy bilateral cerebral hemispheres, basal ganglia and brainstem. |
| 7 (Rosati et al.) | Generalised hypotonia, minimal spontaneous movements | c.1664G>A; p.R555H | T2 hyperintensity of posterior limbs of the internal capsule. Persistent symmetric abnormalities in diffusion restriction. |
| 8 (Stander et al.) | Hypotonia, hyperglycaemia | Not available | Bilateral diffusion restriction in hippocampi, globus pallida, thalami, posterior limbs of internal capsule. Progressive atrophy consistent with pontocerebellar hypoplasia. |
| 9 (Stander et al.) | Hypotonia, hyperglycaemia | Not available | Bilateral diffusion restriction in hippocampi, globus pallida, thalami, posterior limbs of internal capsule. |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetabolism and Genetic Disorders · Neurological diseases and metabolism · Genomics and Rare Diseases
Introduction
1
COASY protein associated neurodegeneration (CoPAN) is an autosomal recessive disorder caused by biallelic pathogenic mutations in the COASY gene (COASY, OMIM 609855) [1]. COASY encodes the mitochondrial bifunctional enzyme coenzyme A (CoA) synthase, which catalyses the final two steps in the pathway of intracellular CoA synthesis from pantothenic acid [2, 3]. Two broad COASY phenotypes have been described on a spectrum to date. Pontocerebellar hypoplasia type 12 (PCH12, OMIM 618266) is the severe foetal onset phenotype associated with microcephaly, pontocerebellar hypoplasia and parenchymal atrophy without evidence of excessive iron deposition on brain imaging [4]. The second phenotype, CoPAN, also known as neurodegeneration with brain iron accumulation type 6 (NBIA6, OMIM 615643), is characterised by variable childhood onset of progressive spasticity, dystonia, neuropathy, cognitive decline and neuropsychiatric features [1, 5] resulting from basal ganglia iron accumulation without a clear underlying pathogenic mechanism [1]. Brain magnetic resonance imaging (MRI) demonstrates features including thin corpus callosum, symmetric diffusion weighted imaging (DWI) abnormalities and T2 hyperintensity of the corpus striatum with patchy bilateral thalamic abnormalities [1, 5].
A previous report describes two siblings with CoPAN identified with newborn screening dried blood spot (DBS) acylcarnitine profiles resembling carnitine palmitoyltransferase 1a (CPT1) deficiency (CPT1, OMIM 255120) [5]. CPT1A is a mitochondrial outer membrane protein that reversibly converts long chain fatty acyl‐CoA species to their corresponding acylcarnitine moieties [6]. CPT1 deficiency is suggested on extended newborn screening via a characteristic pattern of elevated free carnitine (C0) and an elevated molar ratio of free carnitine to the sum of palmitoylcarnitine and octanoylcarnitine [C0/(C16+C18)] [6, 7]. This pattern is often reproducible on both DBS and plasma acylcarnitine profiles, although discrepancies between plasma and DBS free carnitine levels are recognised in CPT1 deficiency [8]. Whilst certain DBS acylcarnitine profile abnormalities require evaluation for several potential aetiologies, there are recent reports where DBS patterns of CPT1 deficiency had an alternative molecular diagnosis, with this being CoPAN [5].
We describe three patients who demonstrated a pattern of CPT1 deficiency on newborn screening DBS with subsequent normal plasma acylcarnitine profiles. All presented with or subsequently developed neurological abnormalities, and genetic studies identified pathogenic mutations in the COASY gene. This case series confirms that both COASY phenotypes can present with this DBS acylcarnitine pattern and CoPAN should be considered in the differential diagnosis of an abnormal extended newborn screening result suggestive of CPT1 deficiency.
Case Series
2
Patient 1 was the first child to non‐consanguineous Caucasian parents. She was born at 37 weeks gestation via vaginal delivery in good condition, with APGAR scores of 9^1^ and 9^5^. Severe microcephaly was evident with a head circumference of 28 cm (< 1st centile). Birth weight was 2200 g (< 1st centile), and length 43 cm (< 1st centile). Physical examination demonstrated jitteriness, increased tone in limbs and brisk deep tendon reflexes. Nasogastric feeding was required from the first day of life due to a poor suck. Seizures presented on day one of life and remained refractory to anti‐epileptic drugs.
A DBS newborn screening card was collected on day four of life, and a subsequent repeat sample on day 6 of life, with the acylcarnitine profile demonstrating an elevated C0 and [C0/(C16+C18)] ratio, consistent with CPT1 deficiency (see Table 1). A plasma acylcarnitine profile collected on day 12 of life demonstrated a mild elevation in C0 but normal [C0/(C16+C18)], excluding CPT1 deficiency.
An electroencephalogram (EEG) on day 12 of life identified frequent subtle clinical and subclinical seizures. An MRI brain on day 15 of life demonstrated diffuse structural brain abnormalities including microcephaly, simplified gyral pattern, parenchymal atrophy, widened extra axial CSF spaces, dilated ventricles and pontocerebellar hypoplasia (see Figure 1a,b). Diffusion restriction was seen in the basal ganglia, thalami and there were bilateral subdural collections.
(a) T2 axial imaging showing diffuse parenchymal volume loss and simplified cortex. (b) T1 sagittal imaging showing pontocerebellar hypoplasia.
On day 17 of life, she developed necrotising enterocolitis. After initial successful management with antibiotics and cessation of enteral feeds, necrotising enterocolitis recurred on day 41 of life, with patient death on day 42 of life.
Patient 2 was a dichorionic, diamniotic twin and a sibling of patient 1. Delivery was performed by caesarean section at 36 weeks due to poor foetal growth, with APGAR scores of 9^1^ and 9^5^. Microcephaly was evident, with a head circumference of 31.5 cm (< 1st centile). Birth weight was 2530 g (3rd centile) and length 46 cm (1st centile). Physical examination demonstrated increased limb tone and absent deep tendon reflexes. Like patient 1, nasogastric feeding was required from the first day of life due to poor suck. Drug resistant epilepsy also presented on the first day of life.
On day 2 of life, MRI brain identified diffuse structural brain abnormalities including microcephaly, simplified gyral pattern, parenchymal atrophy, widened extra axial CSF spaces, dilated ventricles and pontocerebellar hypoplasia (see Figure 2a,b). Finding were similar to patient 1 but of a much lesser severity.
(a) T2 axial imaging showing simplified cortex. (b) T1 sagittal imaging showing pontocerebellar hypoplasia.
A DBS card was collected on day 3 of life, with the DBS acylcarnitine profile demonstrating an elevated C0 and [C0/(C16+C18)], consistent with CPT1 deficiency (see Table 1). A repeat DBS card collected on day 9 of life demonstrated the same pattern of CPT1 deficiency with elevated C0 and [C0/(C16+C18)] (see Table 1). A plasma acylcarnitine profile performed on day 2 of life was normal, excluding CPT1 deficiency.
During infancy, the clinical course was characterised by a severe developmental and epileptic encephalopathy and drug resistant epilepsy. At age 17 months, feeding intolerance and recurrent intestinal pseudo‐obstructions developed, requiring intravenous parenteral nutrition. Further clinical deterioration ensued and the patient died at age 18 months. Posthumous whole exome sequencing performed at Victorian Clinical Genetics Service (VCGS) in the sibling pair identified pathogenic COASY mutations (c.1403_1404dupTG, paternal inheritance, ACMG class 5), (c.215A>G, maternal inheritance, ACMG class 4).
Patient 3 was the first child of a nonconsanguineous Caucasian couple. Vaginal delivery occurred at 38 + 3 weeks following induction of labour for maternal gestational diabetes mellitus, with APGAR scores of 9^1^ and 9^5^. Birth weight was 2680 g (12th centile), head circumference was 33 cm (20th centile) and length was 48 cm (25th centile). On day 3 of life, admission to the special care nursery was required for phototherapy for unconjugated hyperbilirubinemia.
A DBS card was collected on day 3 of life, with the DBS acylcarnitine profile demonstrating an elevated C0 and [C0/(C16+C18)], consistent with CPT1 deficiency (see Table 1). A plasma acylcarnitine profile performed on day 12 of life demonstrated a mild elevation in C0 but normal [C0/(C16+C18)], excluding CPT1 deficiency. A repeat card was collected on day 49 of life, again demonstrating the CPT1 pattern of increased C0 and [C0/(C16+C18)].
At 5 months of age, she was diagnosed with gastroesophageal reflux and constipation. Mild motor developmental delay was apparent during infancy, with rolling at age 6 months and sitting without support achieved at 12 months.
Her first focal seizure occurred at age 15 months. A computerised tomography (CT) brain was performed, demonstrating symmetric calcification in the globus pallidus. An MRI brain was performed at age 18 months demonstrating susceptibility artefact on susceptibility weighted imaging, a thin corpus callosum, symmetric T2 weighted imaging hyperintensity in the basal ganglia and thalami, and symmetric diffusion restriction in the cortex, basal ganglia and thalami (see Figure 3). Dystonia was present in the lower limbs, with evidence of brisk deep tendon reflexes in both upper and lower limbs.
DWI demonstrating bilateral symmetric diffusion restriction in caudate nucleus and putamen (orange arrow), globus pallidus (yellow arrow) and thalami (blue arrow).
Based on the DBS acylcarnitine profiles, clinical presentation and MRI features, a diagnosis of CoPAN was suspected. Whole exome sequencing performed at Fulgent Diagnostics demonstrated biallelic pathogenic mutations in COASY (c.1403_1404dupTG, ACMG class 4), (c.1495C>T, ACMG class 5). Parental segregation studies have not been performed to date.
Methods
3
Dried Blood Spot Acyclcarnitine Profile
3.1
In Queensland, DBS acylcarnitine profiles are performed by flow injection analysis tandem mass spectrometry. From 2004 to 2015, this was performed on a Waters quattro micro, and since 2015 on a Waters xevo‐TQD.
Next Generation Sequencing
3.2
Whole genome sequencing was performed on patient 2 at VCGS using massively parallel sequencing (Nextera TM DNA Flex Library Prep kit, illumine sequencers). Nuclear DNA had a mean target coverage of 30× and a minimum of 90% bases sequenced to at least 10×. Mitochondrial DNA analysis had a mean coverage of 800×. Variant analysis and interpretation were conducted within the selected target region using Agilent Alissa Interpret. There was insufficient specimen available for confirmatory genetic testing in patient 1.
Patient 3 underwent genomic testing at Fulgent Genetics using next generation sequencing technology. Phenotype‐related genes had 99.37% of target regions sequenced with at least 10× coverage of coding regions. Variants were interpreted manually using locus specific databases, literature searches and other molecular biological principles.
Discussion
4
This case series demonstrates a diagnosis of CoPAN present on newborn screening with a DBS acylcarnitine profile consistent with CPT1 deficiency. In our case series, all DBS samples collected in the neonatal and infantile periods had the pattern of CPT1 deficiency, with all plasma samples collected demonstrating a normal pattern. This pattern of abnormal DBS profiles and normal plasma acylcarnitine profiles is consistent with the pattern reported by Evers et al. in 2 siblings with CoPAN [5]. However, Stander et al. report a plasma acylcarnitine profile also consistent with CPT1 deficiency in 2 other siblings. The clinical and neuroimaging features of these reported cases are similar and summarised in Table 2.
DBS acylcarnitine patterns resembling CPT1 deficiency are rare. Since 2004 in Queensland, more than 1 million infants have had DBS acylcarnitine screening, with only 4 patients identified with this profile. One patient had the mild Kiribati variant of CPT1 deficiency, with a plasma acylcarnitine profile consistent with CPT1 and identification of the pathogenic homozygous c.100C>Y mutation in CPT1A. The three remaining patients identified all had CoPAN.
CoA is an essential co‐factor in all living organisms and is required for several biochemical pathways including fatty acid metabolism and the tricarboxylic acid cycle [9]. Intracellular CoA is formed by a highly conserved five‐step pathway from pantothenic acid [10]. There are currently two known genetic disorders affecting enzymes in the CoA synthesis pathway: pantothenate kinase associated degeneration (PKAN, OMIM 606157), caused by pathogenic mutations in PANK2, and CoPAN [11]. In our case series, the c.1403_1404dupTG variant was present in all cases (although noting two patients were siblings). This is a nonsense variant predicted to lead to loss‐of‐function through truncated protein production [12].
The biochemical mechanism explaining the CPT1‐associated acylcarnitine abnormalities is not fully understood in CoPAN. Underlying mitochondrial dysfunction in these conditions may be an overlapping mechanism. A possible explanation is that defective de novo production of mitochondrial CoA results in the inability for fatty acids to be transferred from CoA to carnitine at the outer mitochondrial membrane. This theoretically could result in increased levels of free carnitine and decreased levels of long chain acylcarnitine species, resulting in the pattern of CPT1 deficiency. Evers et al. suggest this may lead to the biochemical abnormalities found on DBS [1]. However, there is currently no mechanism to explain the discrepancies between DBS and plasma acylcarnitine patterns in our cases of CoPAN. Acylcarnitine patterns are known to differ across tissue and body fluid types, which may be a contributor to this finding [13].
In pantothenate kinase associated degeneration (PKAN, OMIM 606157), fibroblast metabolomic studies demonstrated reduced levels of long chain acylcarnitine species. However, there have been no reports to date of patients with PKAN presenting on newborn screening with a pattern of CPT1 deficiency [14]. In a CoPAN mouse model, no significant reduction in CoA levels was measured on mass spectrometry of murine cerebrum compared with controls [15]. Fibroblast studies of two siblings with CoPAN also did not demonstrate a significant difference in measured levels of CoA compared with unaffected siblings.
Conclusion
5
In summary, this case series demonstrates that the phenotypic spectrum associated with pathogenic COASY mutations can present with the DBS acylcarnitine profile of CPT1 deficiency. A DBS pattern of CPT1 deficiency with a normal plasma acylcarnitine profile is suggestive of CoPAN and can be detected prior to the onset of neurological manifestations, identifying new potential for early diagnosis.
Author Contributions
M.L. and S.M. prepared the original manuscript. All authors provided input into the manuscript. D.C. provided oversight. All authors take responsibility for the content of the article.
Funding
The authors have nothing to report.
Ethics Statement
Local human research ethics committee protocols were followed, including obtaining informed consent with a caregiver.
Consent
Informed consent for preparation of this case report was obtained with a caregiver for each case subject.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1S. Dusi , L. Valletta , T. B. Haack , et al., “Exome Sequence Reveals Mutations in Co A Synthase as a Cause of Neurodegeneration With Brain Iron Accumulation,” American Journal of Human Genetics 94, no. 1 (2014): 11–22.24360804 10.1016/j.ajhg.2013.11.008PMC 3882905 · doi ↗ · pubmed ↗
- 2A. Zhyvoloup , I. Nemazanyy , A. Babich , et al., “Molecular Cloning of Co A Synthase. The Missing Link in Co A Biosynthesis,” Journal of Biological Chemistry 277, no. 25 (2002): 22107–22110.11980892 10.1074/jbc.C 200195200 · doi ↗ · pubmed ↗
- 3S. Aghajanian and D. M. Worrall , “Identification and Characterization of the Gene Encoding the Human Phosphopantetheine Adenylyltransferase and Dephospho‐Co A Kinase Bifunctional Enzyme (Co A Synthase),” Biochemical Journal 365, no. Pt 1 (2002): 13–18.11994049 10.1042/BJ 20020569 PMC 1222654 · doi ↗ · pubmed ↗
- 4T. van Dijk , S. Ferdinandusse , J. P. N. Ruiter , et al., “Biallelic Loss of Function Variants in COASY Cause Prenatal Onset Pontocerebellar Hypoplasia, Microcephaly, and Arthrogryposis,” European Journal of Human Genetics 26, no. 12 (2018): 1752–1758.30089828 10.1038/s 41431-018-0233-0PMC 6244412 · doi ↗ · pubmed ↗
- 5C. Evers , A. Seitz , B. Assmann , et al., “Diagnosis of Co PAN by Whole Exome Sequencing: Waking Up a Sleeping Tiger's Eye,” American Journal of Medical Genetics. Part A 173, no. 7 (2017): 1878–1886.28489334 10.1002/ajmg.a.38252 · doi ↗ · pubmed ↗
- 6C. A. Stanley , F. Sunaryo , D. E. Hale , J. P. Bonnefont , F. Demaugre , and J. M. Saudubray , “Elevated Plasma Carnitine in the Hepatic Form of Carnitine Palmitoyltransferase‐1 Deficiency,” Journal of Inherited Metabolic Disease 15, no. 5 (1992): 785–789.1434517 10.1007/BF 01800021 · doi ↗ · pubmed ↗
- 7R. Fingerhut , W. Röschinger , A. C. Muntau , et al., “Hepatic Carnitine Palmitoyltransferase I Deficiency: Acylcarnitine Profiles in Blood Spots Are Highly Specific,” Clinical Chemistry 47, no. 10 (2001): 1763–1768.11568084 · pubmed ↗
- 8S. Primassin and U. Spiekerkoetter , “ESI‐MS/MS Measurement of Free Carnitine and Its Precursor γ‐Butyrobetaine in Plasma and Dried Blood Spots From Patients With Organic Acidurias and Fatty Acid Oxidation Disorders,” Molecular Genetics and Metabolism 101, no. 2–3 (2010): 141–145.20637671 10.1016/j.ymgme.2010.06.012 · doi ↗ · pubmed ↗