Resolution of vanishing bile duct syndrome in a patient associated with refractory hodgkin lymphoma following Anti–PD-1 therapy: a case report and literature review
Yi-Ching Lin, Wan-Chen Hsieh, Yu-Hsuan Tuan, Hsu-Hua Tseng, Jia-Huei Tsai, Tung-Hung Su, Tai-Chung Huang

TL;DR
A patient with a rare liver condition caused by Hodgkin lymphoma showed full recovery after treatment with an immune checkpoint inhibitor.
Contribution
This is the first report of successful treatment of Hodgkin lymphoma-related vanishing bile duct syndrome using a PD-1 inhibitor.
Findings
Pembrolizumab induced complete metabolic remission of refractory Hodgkin lymphoma.
Liver function normalized without immune-related hepatic adverse events.
Immune checkpoint inhibitors may be a viable treatment for HL-related VBDS.
Abstract
Vanishing bile duct syndrome (VBDS) is a rare, and often fatal complication of Hodgkin lymphoma (HL), characterized by progressive intrahepatic bile duct loss and severe cholestasis. Management remains ill-defined, particularly in patients with refractory HL and significant hepatic dysfunction. We present a case of a young woman with biopsy-proven VBDS who experienced disease progression and worsening cholestasis despite second-line chemotherapy, corticosteroids, and brentuximab vedotin. Salvage therapy with pembrolizumab was initiated, resulting in a complete metabolic remission of HL and normalization of liver function. Notably, the patient did not experience immune-related hepatic adverse events. To our knowledge, this is the first report of HL-related VBDS successfully treated with programmed death-1 blockade. This case suggests that immune checkpoint inhibitors may be a viable…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
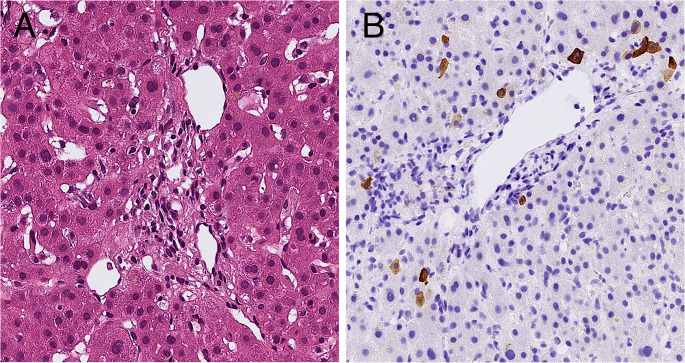 Figure 1
Figure 1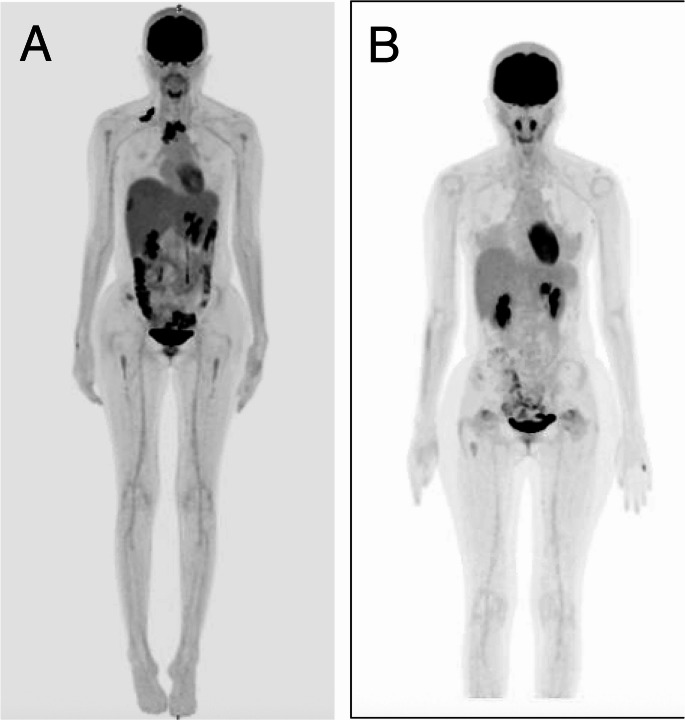 Figure 2
Figure 2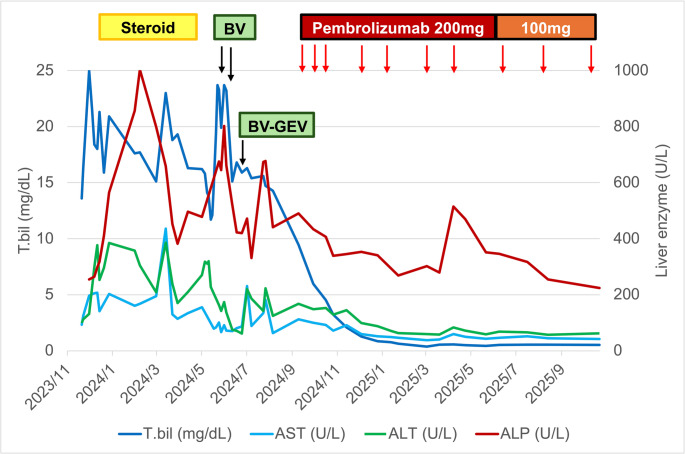 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLiver Diseases and Immunity · Cancer Immunotherapy and Biomarkers · Liver Disease and Transplantation
Introduction
Jaundice occurs in approximately 3–13% of patients with Hodgkin lymphoma (HL) and may arise from diverse etiologies, including hepatic infiltration, biliary obstruction, viral reactivation, drug-induced liver injury, hemolysis, or paraneoplastic syndromes [1, 2]. Vanishing bile duct syndrome (VBDS), a rare paraneoplastic manifestation characterized by progressive intrahepatic ductopenia and severe cholestasis, carries a particularly poor prognosis [3]. Currently, there is no established standard of care for HL-related VBDS [4].
Pembrolizumab, a programmed death-1 (PD-1) immune checkpoint inhibitor (ICI), has demonstrated robust efficacy in relapsed or refractory classical HL, particularly following brentuximab vedotin (BV) failure [5, 6]. To our knowledge, we report the first case of HL-related VBDS successfully treated with anti-PD-1 therapy.
Case
A 23-year-old woman with no significant past medical history was diagnosed with Lugano stage IV nodular sclerosis classical Hodgkin lymphoma. She received induction chemotherapy with ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine), but subsequent positron emission tomography-computed tomography (PET-CT) demonstrated progressive disease. Salvage chemotherapy with DHAX (dexamethasone, high-dose cytarabine, and oxaliplatin) was initiated as a bridge to autologous stem cell transplantation.
Two weeks after initiating DHAX, the patient developed jaundice, tea-colored urine, and generalized pruritus. She denied fever, abdominal pain, arthralgias, myalgias, or acholia. Laboratory evaluation revealed a cholestatic pattern of liver injury: total bilirubin of 5.6 mg/dL, direct bilirubin of 3.5 mg/dL, aspartate aminotransferase (AST) of 119 U/L, alanine transaminase (ALT) of 278 U/L, and alkaline phosphatase (ALP) of 461 U/L. Workup for viral hepatitis, autoimmune liver disease, and metabolic etiologies was unremarkable. She reported no recent travel, animal exposure, alcohol consumption, or use of herbal supplements. Concomitant medications included prophylactic dose of cotrimoxazole (sulfamethoxazole 400 mg/trimethoprim 80 mg) and acyclovir 200 mg daily.
Abdominal computed tomography (CT) scan showed mild periportal edema and minimal ascites, with no evidence of hepatosplenomegaly or lymphadenopathy. Endoscopic retrograde cholangiopancreatography showed normal intrahepatic and extrahepatic biliary anatomy, without evidence of obstruction, stricture, or choledocholithiasis. A liver biopsy demonstrated cholestatic injury with extensive loss of small intralobular bile ducts in the majority of portal tracts, consistent with VBDS (Fig. 1a, b).
Fig. 1. Liver biopsy findings. (a) Portal tract without bile duct (H&E, 29.48x). (b) Immunohistochemistry for cytokeratin 7 (CK7) shows absence of small bile ducts (CK7, 20.54×)
Therapy with prednisolone (30 mg daily) and ursodeoxycholic acid (600 mg daily) was initiated. This was followed by a prolonged tapering of dexamethasone for five months. Cotrimoxazole was discontinued approximately five months after the initial presentation. Despite these regimen, clinical and biochemical cholestasis persisted. A repeat liver biopsy performed after the corticosteroid course confirmed persistent cholestasis and ductopenia. Crucially, no Reed-Sternberg cells or atypical lymphoid infiltrates were identified. Although mild histologic improvement in ductopenia was noted compared to the initial biopsy, liver function remained severely impaired.
Follow-up PET-CT demonstrated disease progression (Fig. 2a). The patient was transitioned to BV as salvage therapy, initiated at a reduced dose of 1.2 mg/kg due to hepatic dysfunction. Following the first cycle, her serum bilirubin level acutely increased to 23.7 mg/dL, then gradually decreased to 15.1 mg/dL. Subsequent cycles were administered at the dose of 1.5 mg/kg, but hyperbilirubinemia persisted. In view of stable disease on interim imaging, gemcitabine and vinorelbine (GEV) were added to augment cytoreduction. The BV-GEV regimen comprised BV (1.8 mg/kg, day 1), gemcitabine (800 mg/m², day 1, 4), vinorelbine (5 mg/m², day 1), and oral prednisolone (100 mg daily, day 1–4). During the second cycle of this combination, the patient developed new-onset abdominal pain with worsening cholestatic markers (ALP and GGT). BV was discontinued due to suboptimal therapeutic response and exacerbation of hepatic cholestasis.
Fig. 2. Positron emission tomography–computed tomography (PET-CT) scans. (a) PET-CT before brentuximab vedotin treatment shows active disease. (b) PET-CT after pembrolizumab therapy shows complete metabolic response
Immunotherapy with pembrolizumab (200 mg every three weeks) was subsequently initiated. In the following cycles, serum bilirubin levels dramatically declined and normalized after four cycles of pembrolizumab (Fig. 3). Clinically, the patient’s symptoms resolved. Restaging PET-CT after seven doses of pembrolizumab demonstrated a complete metabolic response (Fig. 2b). The intervals of pembrolizumab were lengthened gradually, and she remains in the complete metabolic remission with close clinical surveillance.
Fig. 3. Liver function test results during treatment course. Treatments include corticosteroid monotherapy (Steroid), brentuximab vedotin (BV), BV combined with gemcitabine and vinorelbine (BV-GEV), and Pembrolizumab. AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALP, alkaline phosphatase; T.bil, total bilirubin
Discussion
This case demonstrates that in a patient with primary refractory classical HL complicated by VBDS, pembrolizumab was effective not only in controlling the underlying lymphoma but also in reversing the paraneoplastic hepatic dysfunction.
VBDS is a rare acquired disorder characterized by progressive destruction of intrahepatic bile ducts, leading to ductopenia. The association with HL was first described by Hubscher et al. in 1993 [3]. Since then, numerous cases have been reported, as summarized in Table 1. Although the precise pathogenesis remains unclear, proposed mechanisms include immune-mediated injury, paraneoplastic cytokine release, and T-cell cross-reactivity against biliary epithelial antigens [7–10].
Table 1. Reported literature involving the association between vanishing bile duct syndrome and hodgkin’s lymphomaAuthor/YearPathologyHL treatmentOutcome/cause of deathBouroncle/1962 [2]ICCTDeath/Hepatic failureICCTDeath/Hepatic failureJuniper/1963 [25]ICSteroid aloneDeath/Hepatic failureGroth/1972 [26]ICCTDeath/Hepatic failurePerera/1974 [27]ICRT + steroidRemissionICRT + steroidRemissionICRT + steroidDeath/Hepatic failurePiken/1979 [28]ICCTDeath/UnknownTrewby/1979 [29]ICCTRemissionLymphoma infiltrationNo treatmentDeath/Hepatic failureLymphoma infiltrationCTDeath/Unknownmild portal hepatitisNo treatmentDeath/Hepatic failuremixed inflammatory and atypical histocytesCTRemissionICCTDeath/SepsisLieberman/1986 [30]ICNo treatmentDeath/Respiratory arrestBirrer/1993 [31]ICCTDeath/SepsisHubscher/1993 [3]VBDSCTDeath/Hepatic failureVBDSCT + RTDeath/Hepatic failureVBDSCTDeath/Hepatic failureJansen/1994 [32]ICRTDeath/Hepatic failureWarner/1994 [33]ICYesRemissionGottrand/1997 [34]VBDSNo treatmentDeath/Hepatic failureCrosbie/1997 [14]VBDSCTRemissionDe Medeiros/1998 [35]VBDSCTRemissionVBDSCTDeath/Hepatic failureYalcin/1999 [36]ICNo treatmentSepsisICCT + RTRemissionDourakis/1999 [37]Hepatocellular necrosisCTDeath/Hepatic failureYusuf/2000 [38]VBDSNo treatmentDeath/Hepatic failureRossini/2000 [39]VBDSCTDeath/HepaticAllory/2000 [40]VBDSYesRemissionOzkan/2001[41]VBDSNo treatmentDeath/Hepatic failureRipoll/2002 [42]VBDSCT + RTDeath/Hepatic failureVBDSCT + RTRemissionKomurcu/2002 [43]VBDSCT, PBSCTDeath/Hepatic failureLiangpunsakul/2002 [44]Cholestatic hepatitisCTRemissionGuliter/2004 [45]VBDSCTDeath/SepsisCórdoba/2005 [46]VBDSCTRemissionHan/2005 [47]VBDSNo treatmentSpontaneous resolution of VBDS, Tumor recurrenceSchmitt/2006 [48]VBDSNo treatmentDeath/SepsisBarta/2006 [49]ICRTRemissionDeBenedet/2008 [50]VBDSCTDeath/unknownLeeuwenburgh/2008 [51]VBDSCTRemissionBallonoff/2008 [52]VBDSCT + RTRemissionPass/2008 [53]VBDSCT + RTDeath/unknownVBDSCT + RTprogressive VBDS; HL remissionUmit/2009 [54]VBDSSteroid aloneUnknownWong/2013 [4]VBDSCT, PBSCTRemissionAleem/2013 [55]VBDSCTDeath/Hepatic failureNader/2013[56]VBDSCTDeath/Hepatic failure and SepsisRota/2014 [57]VBDSCTRemissionBakhit/2017[58]VBDSCT + RTRemissionGupta/2016 [16]VBDSBV + CTRemissionVBDSBV + CTRemissionFong/2019 [17]VBDSBV + CTRemissionPapakonstantinou/2021[18]ICBV + CTRemissionIshitsuka/2022 [19]VBDSBVRemissionMorgan/2023 [20]VBDSBV + CTRemissionMa/2025 [21]VBDSBV + CTRemissionOur caseVBDSPembrolizumabRemission*, no reported regimen; BV, brentuximab vedotin; CT, chemotherapy; HL, Hodgkin lymphoma; IC, intrahepatic cholestasis; PBSCT, peripheral blood stem cell transplantation; RT, radiotherapy; VBDS, vanishing bile duct syndrome
Accurate liver assessment is critical in Hodgkin lymphoma. Picardi et al. [11, 12]established Color Doppler ultrasound-guided core-needle biopsy (CNB) as a safe, high-yield technique for distinguishing lymphoma infiltration from infection or toxicity, even in patients with severe coagulopathy. Beyond diagnosis, ultrasound is effective for surveillance; a randomized trial [13] confirmed its utility in detecting relapse during first remission. Consequently, ultrasonography may be integrated into management algorithms to ensure both safe histological characterization and effective, radiation-free monitoring.
While HL-related VBDS was potentially reversible in some case reports, the overall prognosis remains poor, primarily due to hepatic failure or sepsis (Table 1). The management of HL- related VBDS remains challenging. Conventional modalities, including chemotherapy, radiotherapy, corticosteroids, and supportive care, have demonstrated limited efficacy. While some experts advocate dose-attenuated regimens in patients with hepatic dysfunction, others support full-dose chemotherapy when clinically feasible [4, 11].
BV, an anti-CD30 antibody drug conjugate, has demonstrated efficacy in relapsed HL [15]. However, its application in patients with severe hepatic impairment remains controversial. Several case reports suggest that BV monotherapy or BV combined with chemotherapy may help transition HL patients with severe liver dysfunction to partial hepatic recovery for potential subsequent intensive treatment. Nevertheless, BV alone often does not achieve complete response, particularly in refractory cases [16–21].
In contrast, ICIs such as pembrolizumab and nivolumab have established robust efficacy in relapsed or refractory HL. In our patient, pembrolizumab was initiated after the failure of both conventional chemotherapy and BV. This resulted in the complete resolution of both cholestasis and the lymphoma, without precipitating hepatic immune-related adverse events (irAEs). Although hepatotoxicity has been reported in up to 6% of patients receiving PD-1 inhibitors, it typically manifests as transaminase elevation rather than cholestasis. Paradoxically, VBDS itself has been described as a rare immune-mediated complication of pembrolizumab [22–24]. However, in this case, VBDS was attributable to HL as a paraneoplastic syndrome, and pembrolizumab achieved a complete metabolic response and resolution of cholestasis by controlling the underlying malignancy.
This apparent paradox underscores the heterogeneity of VBDS pathogenesis. The syndrome may arise from diverse mechanisms, and optimal management must therefore be individualized according to the underlying etiology. This mechanistic variability may explain why pembrolizumab can precipitate VBDS as an irAE in some cases, yet provide therapeutic resolution in HL-associated VBDS. Clinicians should carefully weigh the clinical context when selecting therapy and remain vigilant for immune-mediated sequelae. Nonetheless, our case suggests that PD-1 inhibition represents a promising and well-tolerated salvage option for selected patients with HL-related VBDS, particularly who are not eligible for conventional therapy.
Conclusion
HL-related VBDS is a rare, life-threatening complication that presents significant therapeutic challenge. Our case highlights the potential role in pembrolizumab to bring refractory HL into remission even in the context of VBDS, without inducing hepatic irAEs. Our report captures this importance. Further studies are warranted to validate the safety and efficacy of immune checkpoint inhibitors in this type of clinical conundrum.