Protein kinase-related tumors in the pediatric population: Updated review on an emerging group with emphasis on the more rarely involved kinases
Uta Flucke, Yvonne M. H. Versleijen-Jonkers, Thomas Mentzel, Annette M. Mueller, Laura S. Hiemcke-Jiwa, Rita Alaggio

TL;DR
This paper reviews protein kinase-related tumors in children, emphasizing molecular classification over traditional methods to improve diagnosis and treatment.
Contribution
The paper highlights rare protein kinases involved in pediatric tumors and advocates for molecular stratification in diagnosis.
Findings
Molecular techniques reveal overlapping mesenchymal lesions linked to protein kinases.
Molecular classification is more clinically relevant than morphological classification for these tumors.
Rare kinases like RAF proteins are involved in activating similar oncogenic pathways.
Abstract
Advanced and widespread molecular techniques have deepened our understanding of mesenchymal lesions, revealing considerable overlap among morphologically defined entities now known to be related to protein kinases (PKs). This paradigm shift is important for understanding oncogenesis and also in terms of treatment options and prognosis. Therefore, it is preferable to stratify these tumors molecularly instead of morphologically, as the different categories have clinical implications. Molecular analyses are an essential and integrated part of the diagnostic workup of tissue specimens, especially those of young patients. Involved PKs range from receptor tyrosine kinases (neurotrophic tyrosine receptor kinase [NTRK]1, 2, 3; anaplastic lymphoma kinase [ALK]; proto-oncogene 1 [ROS1]; proto-oncogene [RET]; and proto-oncogene/hepatocyte growth factor receptor [MET]; etc.) to intracytoplasmic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
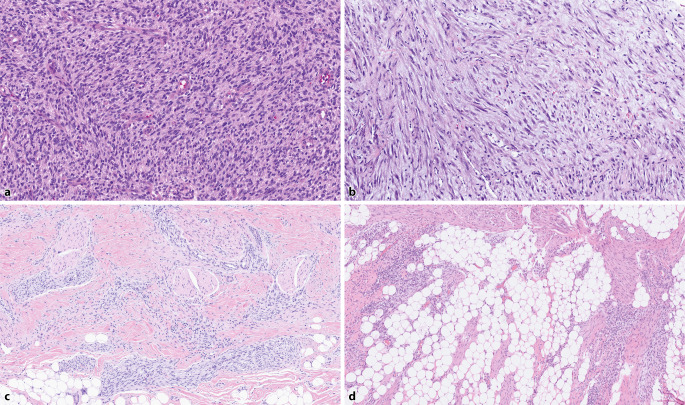 Figure 1
Figure 1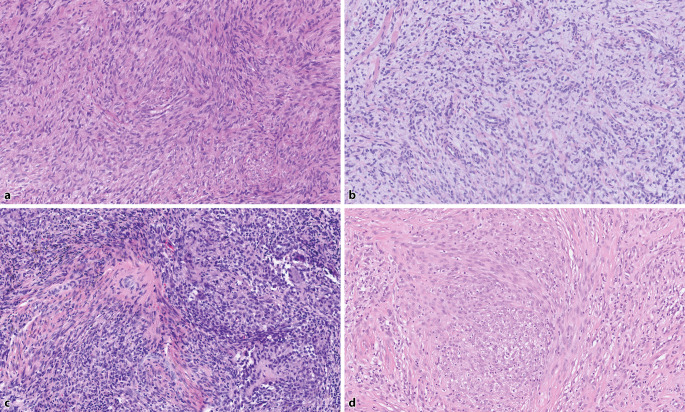 Figure 2
Figure 2 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSarcoma Diagnosis and Treatment · Soft tissue tumor case studies · Tuberous Sclerosis Complex Research
During recent decades, advanced and widespread molecular techniques have greatly deepened our understanding of mesenchymal lesions, revealing considerable overlap among morphologically defined entities now known to be related to protein kinases (PKs).
This paradigm shift is not only important for understanding the oncogenesis of these tumors but also has a huge impact on treatment and prognosis. Therefore, attempts are made to (sub)classify tumor entities based on molecular findings, thereby opening up the possibility of therapeutically inhibiting the identified activated PK. As such, molecular analyses are an essential and integrated part of the diagnostic workup of tissue specimens, especially of those from tumors of young patients.
However, morphological suspicion/recognition seems to be the first step toward arriving at a diagnosis, as the spectrum of involved kinases is broad, and the aberrations comprise fusion genes, tandem repeat duplications, point mutations, and amplifications—all activating the kinase domain [1–7]. The most typical morphological growth patterns are presented in Table 1. Often, a hemangiopericytoma (staghorn)-like vasculature is present with perivascular hyalinization, which can be a diagnostic clue [5, 8].Table 1. Common morphological, immunohistochemical, and genetic findings of protein kinase-related mesenchymal tumors (modified from Xu et al. [8] and the 2023 World Health Organization criteria for pediatric tumors)Tumor typeMorphology, immunohistochemistry, geneticsInfantile fibrosarcoma (IFS)More or less primitive myoid spindle cells with monomorphic oval to tapered nuclei and inconspicuous cytoplasm; arrangement in sheets and fascicles; hemangiopericytoma-like vasculature; variable expression of SMA, desmin, CD34, S100ETV::NTRK3IFS-likeSee IFS; alternative kinase fusion, e.g., NTRK1, 2, 3; ALK; LTK; RET; EGFR-KDD; BRAF; MET; FGFR1Inflammatory myofibroblastic tumor (IMT)More or less vague bundles of myofibroblastic cells, background loose–collagenous, SMA ±, desmin +/−, ALK +/−ALK (~50–70%), ROS1, RET, NTRK, PDGFRB, IGF1RIMT-likeSee IMT; CD34 +/−, S100 +/−Lipofibromatosis (LPF)/LPF-like neural tumor (LPF-NT)Irregular bundles of slender myofibroblastic cells mainly located in subcutaneous fat with a collagenous background, univacuolated small fat cells at the interface, SMA +/−, S100 +/−, CD34 +/−NTRK1, 2, 3; EGFR; ALK; ROS; RET; LTK; FGFR1; PDGFRBMalignant peripheral nerve sheath tumor (MPNST)-likeOften cellular, long fascicles of monomorphic spindle cells, S100 +/−, CD34 +/−NTRK1, 2, 3; LTK; MET; RET; ALK; ROS; (B)RAFAdult fibrosarcoma (FS)-likeCellular, fascicular, herringbone, S100 −, CD34 +/−For genetic changes, see MPNST-likeMyofibroma/myopericytomaMyoid spindle cells with oval or tapered nuclei, hemangiopericytoma-like (staghorn) vasculature with hyalinization, SMA +, desmin −/+PDGFRBMyofibroma/myopericytoma/hemangiopericytoma-likeSee myopericytoma; CD34 +/−, S100 +/−NTRK1, 2, 3; RETDermatofibrosarcoma protuberans (DFSP)-likeInfiltrative storiform growth pattern, monomorphic spindle cells, CD34 +, S100 +/−EGFR exon 20, **ALK, FGFR1Myxoid neoplasmsVariable (mostly less) cellular, vague bundles/storiform arrangement monomorphic spindle and/or epithelioid cells set in a myxoid stromaNTRK1, 2, 3; EGFR ex 20; ALK; RET; FGFR1Epithelioid neoplasmsSheets/nests epithelioid/ganglion-like cells/histiocytoid, SMA +/−, desmin +/−, CD34 +/−, S100 +/−NTRK1, 2, 3; ALK; RETFibrous hamartoma of infancy (FHI)Bundles of spindle cells (between fat), nests/sheets primitive spindle to round cells, fibrous areas with giant cells (giant cell fibroblastoma-like) CD34 +/−, SMA +/−EGFR exon 20 mutationsFHI-likeSee FHI; S100 +/−, CD34 +/−MET, FGFR1**SMA smooth muscle actin
*NTRK1-/2-/3-*associated tumors are the prototype and are extensively described in the literature. The first to be identified was the ETV6::NTRK3 gene fusion in infantile fibrosarcoma and congenital mesoblastic nephroma, which represented a huge step toward the new (sub)classification of such neoplasms (Fig. 1; [5, 9, 10]).Fig. 1a Infantile fibrosarcoma with ETV6::NTRK3, ×10 magnification. This classic example shows vague bundles and sheets of primitive myofibroblastic cells with monomorphic nuclei. b Inflammatory myofibroblastic tumor with FN1::ALK, ×10 magnification. This is a myxoid example consisting of slightly polymorphic myofibroblasts arranged loosely and in bundles, with an obviously abundant amphophilic cytoplasm. c Fibrous hamartoma of infancy-like pattern in an otherwise typical infantile fibrosarcoma with EGFR-KDD, ×5 magnification, comprising nests of primitive cells and bundles of mature slender myofibroblasts. d Lipofibromatosis pattern in an otherwise typical infantile fibrosarcoma with EGFR-KDD, ×5 magnification
Pathways that become dysregulated by ligand-independent kinase activity with autophosphorylation of tyrosine or serine/threonine are PI3K-AKT, JAK-STAT, RAS-MAPK, CRKL-C2G, and MEKK2/3-MEK5-ERK [11–13].
Involved cells seem to be precursor cells, and, probably due to their developmental stage when the genetic aberration originates, they may be more or less well differentiated, which is morphologically reflected by primitive-looking or more developed neoplasms with a (myo)fibroblastic, pericytic [3, 14], or even vascular phenotype [15]. Primitive cartilage can also be present [4, 16–18].
The aim of this review is to report on the variety of protein kinases involved in the oncogenesis of mesenchymal tumors, mainly of pediatric patients, with a focus on the more rarely documented PKs: EGFR, ALK, LTK, PDGFRB, MET, (B)RAF, RET, FGFR, ABL, and IGF1R. The morphological and immunohistochemical hallmarks of these tumors are presented, including their overlaps, and the usefulness of detecting them is discussed. The presented tabular algorithms aim to provide a helpful overview of this complex and emerging topic (Tables 1 and 2).Table 2. Altered genes encoding for protein kinases and the corresponding morphologyInvolved geneHistology/tumor typeNTRK1, 2, 3IFS(-like), IMT, LPF-like, LPF-NT, adult fibrosarcoma, MPNST-like, myopericytoma-like, epithelioid fibrous histiocytoma/superficial ALK-rearranged myxoid spindle cell neoplasm, round cell, epithelioid cell, myxoid neoplasmsEGFR exon 20FHI, LPF, DFSP-like, myxoid spindle cell neoplasm, congenital peribronchial myofibroblastic tumorEGFR-KDDIFS, LPF-likeEGFR rearrangementLPF/LPF-NTEGFLPF/LPF-NTALKIMT, IFS, LPF-like/LPFNT, DFSP-like, 34+ plaque like dermal fibroma, spindle cell/epithelioid/myxoid neoplasms, epithelioid fibrous histiocytoma/superficial ALK-rearranged myxoid spindle cell neoplasm, hemangiomaROSIMT, LPF-like/LPFNT, MPNST-likeLTKIFS-like, MPNST-like, LPF-like/LPF-NTMETFHI-like, IFS-like, MPNST-like, LPF-like/LPF-NTBRAF/RAF1 (cRAF)IFS-like, fibromatosis-like, IMT-like, MPNST-like, round cellsFGFR1IFS-like, LPF-like/LPFNT, DFSP-like, biphasic (primitive/spindle cells), epithelioid (files), myxoid, collagenousABLSoft tissue angiofibroma-like, solitary fibrous tumor-like, perineurioma-likeRETIMT, IFS-like, MPNST-like, LPF-like/LPF-NT, myofibroma-like, MPNST-like, epithelioid fibrous histiocytoma/superficial ALK-rearranged myxoid spindle cell neoplasm, myxoid, epithelioid/round cell neoplasmsPDGFRBMyofibroma, myopericytoma, dermatomyofibroma, IMT, LPFIGF1RIMTIFS infantile fibrosarcoma, LPF lipofibromatosis, PPF-NT lipofibromatosis-like neural tumor, MPNST malignant peripheral nerve sheath tumor; FHI fibrous hamartoma of infancy, DFSP dermatofibrosarcoma protuberans, IMT inflammatory myofibroblastic tumor
EGFR-related lesions
The epidermal growth factor receptor (EGFR), a member of the ErbB family of tyrosine kinases, is altered in different mesenchymal tumors. Fusion genes, exon 20 mutations, and kinase domain duplication (KDD) activate the kinase domain and its downstream cascade to induce oncogenesis [3]. Table 3 shows the involved entities with the corresponding genetic aberrations.Table 3. Entities/tumor types and their corresponding EGFR alterationsTumor typeEGFR exon 20 mutationsEGFR-KDD*EGFR *rearrangementFibrous hamartoma of infancyX––LipofibromatosisXXXInfantile fibrosarcoma–X–CD34/S100+ neoplasm––XDFSP-like neoplasmX––Myxoid spindle cell neoplasmX––Mesoblastic nephroma–X–Congenital peribronchial myofibroblastic tumor–X–DFSP dermatofibrosarcoma protuberans, EGFR epidermal growth factor receptor, *KDD *kinase domain duplication
Fibrous hamartoma of infancy
Fibrous hamartoma of infancy (FHI) was first described in a series by Reye in 1954 and later on confirmed as being a separate entity with benign behavior by Enzinger in 1965, who coined the used term [19].
The lesion mainly arises in male patients with a mean age of 15 months (range birth–14 years) [19–21]. Fibrous hamartoma of infancy is localized subcutaneously, commonly in the shoulder girdle. Other reported sites are the trunk, head and neck region, genital region, pelvic limb girdle, and the (lower) extremities [20, 21]. Changes of the overlying skin (discoloration, hypertrichosis, edema, tethering) are rare [20, 21]. The neoplasms usually grow slowly. Rapid growth is mainly observed in early lesions. Recurrences are identified in a few cases, commonly after incomplete excision but also after a couple of years. Long-term clinical follow-up indicates a benign clinical course [19–21].
Grossly, the lesions are described as being poorly delineated, variably fibrous, and myxoid, including adipose tissue [21].
Histology shows an organoid triphasic pattern characterized by fibrocollagenous trabeculae resembling fetal tendons, desmoid fibromatosis or lipofibromatosis, and nests of primitive mesenchymal cells arranged loosely or in a whorl-like manner in a highly vascularized myxoid matrix. There is haphazardly entrapped fat, with a variable content that can exceed 50% of the tumor mass. In some cases, a variable fibrous stroma that may show slit-like pseudovascular artefacts lined by prominent stromal cells similar to giant cell fibroblastoma is observed [19–21].
A few cases have been reported to show malignant transformation with cellular areas of primitive spindle and round cells with monomorphic nuclei and frequent mitotic figures [21]. Given the EGFR kinase activation in FHI and also in infantile fibrosarcoma (see below), a coincidence of different growth patterns within one tumor is not surprising.
Immunohistochemically, the neoplasms are variably positive for smooth muscle actin (SMA) and CD34, with S100 expression in the fat cells only [20, 21]. While EGFR is reported to be positive in most cases, this marker is not highly consistent and should be used with caution [3, 22].
From a genetic point of view, EGFR exon 20 insertion/duplication mutations are identified [3, 22].
Differential diagnoses are lipoblastoma, especially when fibrous or fibromyxoid areas are prominent. However, beside CD34, these cases also express desmin and the pleomorphic adenoma gene (PLAG1) due to PLAG1 rearrangement [22–24]. Triphasic MET-related lesions are another differential diagnosis (see below).
Other lesions with EGFR aberrations
Other lesions with *EGFR *exon 20 mutations are lipofibromatosis (characterized by bundles of myofibroblastic cells with tapered nuclei traversing adipose tissue), dermatofibrosarcoma protuberance (DFSP)-like (with storiformly arranged myofibroblasts), and myxoid spindle cell neoplasm (showing a multinodular growth pattern of monomorphic spindle cells set in a myxoid matrix; Fig. 2; [3, 25]).Fig. 2a Spindle cell neoplasm with TPM3::NTRK1, ×10 magnification, showing vague short bundles and whorls of primitive-looking myofibroblastic spindle cells. b Spindle cell neoplasm with TMF1::RAF1, ×10 magnification In this myxoid lesion, a loose arrangement of primitive myofibroblastic spindle cells is seen. Note the entrapped collagen. c Spindle cell neoplasm with ETV6::BRAF, ×10 magnification. The primitive-looking cells are arranged in sheets possessing oval to elongated nuclei. There is a variable fibrous background, an inflammatory reaction, and deposition of hemosiderin. d Epithelioid fibrous histiocytoma with *ALK *rearrangement comprising epithelioid and plump spindle cells with round to oval nuclei and an amphophilic cytoplasm. Note the collagenous background
*EGFR *kinase domain duplication (KDD) has been found in mesoblastic nephroma, infantile fibrosarcoma, lipofibromatosis-like lesions, and congenital peribronchial myofibroblastic tumor (CPMT). All share morphological features (monomorphic primitive spindle cells arranged in more or less well-defined fascicles). However, the additional presence of primitive cartilage is typical for CPMT and may occur in mesoblastic nephroma and infantile fibrosarcoma [4, 7, 16–18, 26].
EGFR fusion genes have been described in lipofibromatosis and also in the case report of a young adult with a CD34-/S100-positive tumor in his lumbar region consisting of confluent nests of slightly pleomorphic polygonal primitive cells. The nests were surrounded by a collagenous stroma [27].
In the molecularly heterogeneous group of lipofibromatosis, EGF fusions are also identified, showing that this growth pattern can be associated with the occurrence of different kinases and their ligands [28].
ALK-related lesions
The anaplastic lymphoma kinase (ALK) oncogene, located on chromosome 2p23, encodes for a cell surface receptor tyrosine kinase belonging to the insulin receptor kinase superfamily and, together with LTK, to a unique subfamily. It was first discovered as a fusion partner in anaplastic large cell lymphoma [29]. Its recurrent involvement in inflammatory myofibroblastic tumor (IMT) has been known since 1999 [30]. Whereas fusion genes are the predominant genetic aberration, it has been demonstrated that gene amplification, which also results in elevated protein expression, is an alternative mechanism in rare cases [31].
Nowadays it is known that mesenchymal lesions harboring an ALK rearrangement show a broad clinicopathologic spectrum, with affection of pediatric and adult patients and occurrence in skin, superficial and deep soft tissue, bone, viscera, and the central nervous system (CNS) [2, 31–33]. However, young patients are more often involved. Beside the well-known entities, such as IMT or epithelioid fibrous histiocytoma, descriptive terms were coined to attribute to the morphology, as all neoplasms have a similar genetic background [34–36]. The tumors reported to date and their features are shown in Table 4. Their morphology varies within and between tumor types, ranging from epithelioid/histiocytoid to myofibroblastic spindle cells organized in sheets, nests, bundles, and whorls in a loose, myxoid, and/or collagenous matrix with possible hyalinization. The nuclei are relatively monomorphic, either ovoid, tapered, or round with prominent nucleoli. The latter is seen in the epithelioid cells possessing also an obvious amphophilic cytoplasm. Lesions with high-grade features have enlarged nuclei, mitotic activity, and necrosis. A hemangiopericytoma-like vasculature can be prominent and therefore diagnostic [32–34, 37]. Immunohistochemically, ALK expression is a perfect surrogate marker. Depending on the partner genes, subcellular localization of the ALK chimeric protein can be membranous, intracytoplasmic, or perinuclear. Other variably expressed markers are SMA, desmin, CD34, and S100 (Table 4; [13, 33]). Exceptionally, cases without ALK alteration show ALK expression [38].
Alternatively affected receptor tyrosine kinases are ROS, RET, and NTRK1/2/3, with NTRK3 being the prototypical gene involved in oncogenesis of infantile fibrosarcoma. This provides evidence that there is morphological overlap between protein kinase-related lesions [13, 32, 33, 37, 39].
A comprehensive review of ALK-rearranged mesenchymal tumors was recently published by Agaimy [33].
Inflammatory myofibroblastic tumor
Inflammatory myofibroblastic tumor (IMT) is the prototype of an ALK-rearranged mesenchymal tumor with myofibroblastic properties and is a member of the intermediate prognostic group (locally aggressive, rarely metastasizing) [33]. It was originally described in the lung under various terms such as inflammatory pseudotumor, plasma cell granuloma, pseudosarcomatous myofibroblastic proliferation, or inflammatory myofibrohistiocytic proliferation. It became apparent that these tumors can also occur at extrapulmonary sites, such as the mesentery and other intraabdominal/retroperitoneal locations, the small pelvis including the genitourinary tract, the head and neck region including the upper respiratory tract and the meninges, the mediastinum including the heart, the breast, and the extremities [40, 41]. The sex distribution of the patients is roughly equal, and the age range is broad, from 3 months to 46 years (mean 9 years) [40, 41]. Patients present with a mass with or without pain. Systemic symptoms are reported in 1/3 of patients, presenting with fever; weight loss; malaise; and/or peripheral blood abnormalities like anemia, thrombocytosis, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia caused by cytokine mediators [41].
The tumors can be huge. One large study reports a mean diameter of 6.0 cm (range 1–17 cm). The masses are circumscribed and firm, white or tan, showing a whorled, fleshy, or myxoid cut surface with focal hemorrhage, necrosis, and calcification in a minority of cases [40].
Histology in most IMTs is inhomogeneous and does not correlate with clinical behavior. It can be granulation tissue-like and/or tissue culture-like, resembling nodular fasciitis. Other patterns are compact spindle cell proliferation or scar tissue-like. The constituent myofibroblasts are variable in size and shape and are mainly arranged in vague fascicles. They are spindly or polygonal (ganglion-like), with round, oval, or elongated nuclei and sometimes prominent nucleoli (ganglion-like). The cytoplasm is inconspicuous or abundant and amphophilic (ganglion-like). The stroma may be loose/myxoid and/or collagenous. The inflammatory infiltrate can be more or less prominent, consisting of lymphocytes, plasma cells, eosinophils, and neutrophils [33, 40, 41].
Immunohistochemically, SMA and desmin are variably expressed, and ALK expression corresponds with the ALK genetic abnormality. It seems from the literature that up to 70% of IMTs are *ALK-*related [33, 41]. Other involved genes are ROS, RET, and NTRK, among other rarely involved genes (Table 1; [41]).
Differential diagnosis includes ALK-positive histiocytosis, which shows strong and diffuse CD163-positive staining. PU1.1, a nuclear marker of histiocytes, may be useful to differentiate background histiocytes from fibroblastic/myofibroblastic neoplastic cells in the case of IMT with abundant histiocytes [33].
Cutaneous hemangioma with epithelioid features and ALK rearrangement
These vascular lesions were reported recently. They are exceptional within the group of ALK-related lesions, mostly showing a myofibroblastic or, more rarely, a histiocytic phenotype. The age range of patients is 2–38 years.. Reported sites are the abdominal wall, extremities, shoulder, and head and neck region.
Histologically, lesions are circumscribed, nodular, and surrounded by an epidermal collarette. The vascular channels are lined by epithelioid endothelial cells with round, relatively monomorphous nuclei with fine chromatin and small nucleoli. The mitotic count is 4–6/10 high-power fields (HPFs). Cells have abundant eosinophilic cytoplasm showing focally intracytoplasmic vacuoles. There is maturation toward the periphery, with more distinct vessels lined by flattened endothelial cells. Pericytes are present. There is a chronic inflammatory reaction.
Immunohistochemically, lesions are positive for ERG, CD31, and ALK. Surrounding pericytes are highlighted by SMA. Other markers are negative, e.g., human herpesvirus 8 (HHV8), glucose transporter type 1 (GLUT1), and pankeratin AE. None of the control cases, including epithelioid hemangiomas and epithelioid angiomatous nodules, stained for ALK. All cases harbored an ALK fusion gene. Follow-up was uneventful.
Differential diagnoses include epithelioid angiomatous nodule and epithelioid hemangioma harboring FOS or FOSB rearrangement, with immunohistochemical expression of the corresponding proteins being negative in ALK-rearranged hemangiomas. Epithelioid hemangioendothelioma is less likely [13, 15].Table 4. Tumor entities/types with ALK rearrangementTumor typeClinical findingsHistologyPositive immunohistochemical resultsInflammatory myofibroblastic tumor/inflammatory myofibroblastic sarcoma^#^Children, adults^#^Intraabdominal*^#^, intrathoracic*^#^, H&N*, extremities*(Epithelioid^#^) myofibroblasts, ganglion-like +/−Bundles +/−*Sheets^#^*Collagen +/−Myxoid −/+ inflammationSMA, desmin, ALK (50–70% of all cases, corresponding to rearrangement)Infantile fibrosarcoma-likeChildrenExtremities, H&N, visceralHaphazardly primitive spindle cells, bundles of myofibroblastic spindle cells, herringbone pattern +/−Collagen +/−Myxoid +/−HPC-like vasculatureSMA +/−, S100 +/−, CD34 +/−Dermatofibrosarcoma protuberans-like/CD34 + plaque-like dermal fibromaChildren, adultsMainly skin, extremities, limb girdles, trunk, H&NInfiltrative, storiform, vague bundles, (myo)fibroblastic spindle cellsCD34 +S100 −ALK +Lipofibromatosis-likeChildren, adultsExtremities, limb girdleSheets, nests, bundles spindle cells infiltrating fat and deeper soft tissueCollagen +/−Myxoid +/−HPC-like vasculatureS100 +/−CD34 +/−ALK +Spindle cell/epithelioid/myxoid neoplasmsChildren, adultsTrunk, groin, paratesticular, extremities, H&N, CNSSheets epithelioid cells, bundles, whorls, haphazard spindle cells, enlarged or pleomorphic nuclei −/+Collagen +/−Myxoid +/−HPC-like vasculatureALK +SMA −/+Caldesmon −/+ H3K27me3 +/−Epithelioid fibrous histiocytoma/superficial ALK-rearranged myxoid spindle cell neoplasmChildren, adultsExtremities, trunk, H&NSheets epithelioid cells +/− Round nuclei, binucleation +/−Amphophilic cytoplasm, whorls nests, sheets, bundles histiocytoid spindle cells −/+Collagen +/−Myxoid +/−(HPC-like vasculature/lipofibromatosis-like)CD34 +/− S100 −/+ SMA −/+ EMA +/−ALK +Myxoid fibroblastic tumor of the vocal cordAdolescents, adults, male>>femaleVariable cellularity and variable myxoid background, cytological range: bland-looking spindle cells to epithelioid cells; ganglion-like with enlarged, pleomorphic nuclei, nuclear pseudoinclusions, prominent (capillary) vasculatureSMA +/−S100 −CD34 −Desmin −ALK +Cutaneous hemangioma with epithelioid featuresChildren, adultsH&N, extremities, trunkCircumscribed/nodular epithelioid endothelial cells, peripheral maturationERG +CD31 +SMA + (pericytes)ALK +H&N head and neck region, CNS central nervous system, HPC Hemangiopericytoma, SMA smooth muscle actin, EMA epithelial membrane antigen, ALK anaplastic lymphoma kinase, ERG avian v-ets erythroblastosis virus E26 oncogene homolog
ROS1
The *ROS1 *gene encodes for an RTK that belongs to the sevenless subfamily of tyrosine kinase insulin receptor genes. The resulting type I integral membrane protein is, when altered, mainly involved in the oncogenesis of inflammatory myofibroblastic tumor (IMT; see above). A lipofibromatosis-like/lipofibromatosis-like neural tumor pattern has been reported in exceptional cases [39].
LTK-related lesions
Leucocyte receptor tyrosine kinase (LTK) is a member of the ALK/LTK family of genes. It has a homology with ALK and has been reported to be activated by a fusion transcript upregulating the kinase domain and, consecutively, the PI3K/AKT and MAPK pathways [42].
Three neoplasms were recently described with the identification of a fusion gene. Patients’ age ranged from 3 months to 17 years. Lesions originated in the muscle of the entire leg, the toe, or finger. (Long-term) follow-up was uneventful in two cases. The most extended neoplasm recurred locally.
Grossly, the tumors were described as infiltrative whitish lesions [42]. Histomorphologically, the infiltrative lesions were either composed of sheets of primitive monomorphic spindle cells or more differentiated spindle cells arranged in fascicles within a fibrous background. In two cases, a prominent hemangiopericytoma-like vasculature with perivascular fibrosis was present.
Immunohistochemically, like other PK-related lesions, CD34 and S100 were variably present, with positivity for smooth muscle markers in the more maturated myofibroblastic lesion.
At the RNA expression and methylation levels, cases were similar to DFSP and a tumor with RAF1 rearrangement, confirming that PK-related lesions are a family of tumors. However, other PK-related tumors like infantile fibrosarcoma and inflammatory myofibroblastic tumors clustered apart. Of note, such a clustering analysis depends on the included entities/cases for comparison and is therefore a matter of perspective [42, 43].
MET-related lesions
The hepatocyte growth factor receptor (HGFR) encoded by *MET *is a tyrosine kinase receptor. It seems to be rarely involved in the oncogenesis of PK-related mesenchymal tumors. Among the four reported tumors, patients’ age ranged from birth to 4 months. Tumors were located superficially and deep: subcutis of the lumbar area, pelvic soft tissue, thigh, trunk, and masseter muscle. The size ranged from 5.2 to 20 cm. Histologically, these tumors have a triphasic pattern like fibrous hamartoma of infancy (see above) and/or an infantile fibrosarcoma‑/MPNST-like appearance. Immunohistochemically, variable expression of S100 and CD34 has been reported. Patients have a good prognosis [39, 44, 45].
BRAF/RAF1(cRAF)-related lesions
The RAF proteins are cytosolic serine/threonine kinases activating the MAPK cascade downstream of RAS. The corresponding genes can be altered by mutations or fusion genes.
RAF1 (cRAF)
RAF1-rearranged tumors have mostly been reported in infants. Tumors were located in the arm, thigh, and kidney, with a size of up to 12 cm. The described cases were primitive looking, with variable cellularity like IFS/CMN (cellular mesoblastic nephroma), or had a more mature myofibroblastic morphology, with a fibromatosis-like appearance showing long fascicles of slender spindle cells. Nuclei were oval or tapered, monomorphous, and bland looking; S100 and CD34 were variably positive [12].
BRAF
Patients with *BRAF-*altered tumors ranged in age from birth to 32 years. Tumors originated in the soft tissues of the extremities, the small pelvis, and the retroperitoneum including the rectum wall and the kidney as well as in the mediastinum and the spinal, paraspinal, and head and neck regions [4, 17, 46]. When involved, skin can be ulcerated [46]. A neoplasm occurring in bone (mastoid) of a 14-year-old boy has also been described [6].
Lesions showed IFS-like and also IMT-like morphology consisting of monomorphic spindle cells, either primitive or with a more myofibroblastic phenotype possessing oval or elongated/tapered nuclei. A more or less fascicular architecture with infiltrative growth is common. Areas of round cells may be present. In the background, collagen with variable myxoid changes can be variably seen, but it is often scarce combined with high cellularity. The vascular pattern is often hemangiopericytoma-like, which can be a diagnostic clue in receptor protein kinase-related tumors mainly with an IFS-like pattern. Heterologous cartilage may occur [4, 17].
Immunohistochemically, SMA expression is seen to a variable degree, with desmin being positive in exceptional cases; CD34 and S100 positivity is observed in some cases [4, 17].
Most cases harbor a BRAF fusion gene. Alternatively, a point mutation can occur, including p.V600D, p.V600E, and p.L485F. Tandem duplications and compound deletions have also been reported. In exceptional cases, a combined *BRAF *fusion or a BRAF fusion gene in addition to a point mutation has been identified [4, 7, 17, 46]. Tumors may bear additional mutations, e.g., in P53 or APC [46].
In terms of follow-up, lesions may recur, also after years, but there is no apparent progression after long-term follow-up in most cases. However, aggressive behavior has been documented in isolated cases [17, 46].
Whether there is a link to metanephric stromal tumors [46] needs to be explored more thoroughly.
FGFR1-related lesions
The fibroblast growth factor receptor (FGFR) family comprises the RTKs. Normally, they become activated by binding of fibroblast growth factors. In soft tissue tumors, fusion genes are found, leading to hyperfunctioning of the kinase [1–4].
The reported patients were infants. Tumors were located in deep gluteal or pelvic/perirectal soft tissue, with involvement of the rectal wall. The size of the tumors ranged from 4.2 to 11 cm. Neoplasms were mainly highly cellular. They consisted of primitive spindles arranged in vague bundles, whorls, and/or sheets. Infiltration of the fat was lace-like/DFSP-like. Epithelioid cells arranged in sheets or cords were also present. Less cellular areas were collagenous or myxoid. Cellular atypia was observed after chemotherapy. Mitotic figures were scarce. Immunohistochemical analysis revealed CD34 expression without S100 staining. Patchy desmin expression was observed in rare instances. Locally aggressive behavior has been documented [47].
ABL-related lesions
The ABL family of kinase genes comprises proto-oncogenes 1 and 2 encoding for non-receptor tyrosine kinases localized in the nucleus and cytoplasm. They fuse with a variety of genes in hematological malignancies. Fusion genes in mesenchymal tumors are rare.
Lesions occurred mainly in pediatric patients (age range 7–76 years) and were localized in the trunk, limb girdles, and proximal and distal extremities, superficially or deep. Reported sizes were up to 11 cm. These tumors were misinterpreted as soft tissue angiofibroma, solitary fibrous tumors, and perineurioma.
Histologically, neoplasms are circumscribed or infiltrative and consist of bland-looking monomorphic spindle cells arranged in sheets, bundles, and whorls, with a fibrous/fibromyxoid background. When the cells have long cytoplasmic processes, they resemble perineurial cells. Mitotic activity is low.
Immunohistochemistry reveals positivity for CD34, S100, EMA, GLUT1, and claudin, with the latter three markers being characteristic of perineurioma. However, lesions mainly show morphological, immunohistochemical, and molecular features of PK-related lesions, as mentioned in Table 1.
Locally aggressive behavior seems to be an exception [48, 49].
RET-related lesions
The rearranged during transfection (RET) gene encodes a receptor tyrosine kinase for members of the glial cell line-derived neurotropic factor family of extracellular signaling molecules. The RET fusion genes are responsible for autonomic activation of the kinase, leading to oncogenesis of mesenchymal neoplasms with the typical characteristics reported for PK-related lesions.
Tumors occur in children, including newborns and infants. Adults may also be affected. Neoplasms range in size from 2.9 to 13 cm. They are located in superficial and deep soft tissue and may affect bone (chest wall). Reported sites are the proximal and distal extremities, gluteal region, trunk, neck, and visceral organs (lung, kidney).
Morphologically, neoplasms are described as IFS-like, MPNST-like, and LPF-like/LPF-NT, with sheets/bundles and whorls of monomorphic primitive spindle cells. The background is variable and can be collagenous or myxoid. A hemangiopericytoma vasculature can be prominent. In immunohistochemistry, expression of CD34, S100, and SMA was variably identifiable. Of note, ALK and ERG expression is possible.
Rarely occurring high-grade morphology with a high nuclear–cytoplasmic (NC) ratio and necrosis correlate with malignant behavior. Metastases can develop in the lung, lymph nodes, soft tissue, or CNS [38, 50].
PDGFRB-related lesions
Platelet-derived growth factor receptor beta (PDGFRB) and its paralog alpha belong to the class III family, along with c‑KIT, colony-stimulating factor 1 receptor, and FMS-like tyrosine kinase 3 receptor. They play a role in various disorders, including soft tissue tumors [51].
Pericytic tumors
Pericytic tumors comprise myofibroma, myopericytoma, angioleiomyoma, and glomus tumors. Myofibroma, myopericytoma, and angioleiomyoma are on a morphological continuum. The age range is wide, with myofibromas being very common in (early) childhood. Myofibromas occur solitarily or, less commonly, multiply (myofibromatosis) in skin/subcutis, deep soft tissue, bone, and viscera. The latter can be life threatening. Rarely, hereditary tumors are reported [52].
Morphology is typically biphasic in myofibroma, with primitive-looking areas and maturated myofibroblasts in a bluish matrix. Myopericytomas show sheets and concentric growth of primitive or maturated myofibroblasts around blood vessels and a variable collagenous background. In both tumors, an HPC-like vasculature may be obvious. The latter is also characteristic of angioleiomyomas and glomus tumors, surrounded by either leiomyocytes or glomus cells. Such lesions rarely originate in the pediatric population. Immunohistochemically, SMA and desmin are variably expressed [52, 53].
PDGFRB point mutations have been reported in all mentioned lesions. They occur in the juxtamembrane (exon 12) or kinase domain (exon 14), leading to activation of the kinase [53].
Myofibromatosis has also been shown to bear a PDGFRB fusion gene [54]. Other reported fusion genes are *MTCH2-FNBP4, FN1-TIMP1, COL4A1-VEGFD *(with and without PDGFRB mutation) [55].
Dermatomyofibroma
Dermatomyofibroma, first reported by Hügel in 1992, is a benign skin lesion occurring rarely in children, with neoplasms originating in the neck, shoulder girdle, trunk, and extremities. They are plaque-like and consist of fascicles of uniform slender myofibroblasts with elongated nuclei. The fascicles are parallel to the epidermis. Elastic fibers are preserved, and skin adnexal structures are spared. When infiltrating the subcutis, there are similarities with lipofibromatosis [51, 56].
Immunohistochemically, SMA, calponin, and CD34 may be positive [51, 56]. PDGFRB point mutations similar to those seen in pericytic tumors have been identified [51].
IMT, lipofibromatosis
*PDGFRB *fusions are more rarely documented in IMTs (see above) and also in lipofibromatosis [28, 57].
IGF1R-related lesions
Insulin-like growth factor 1 receptor (IGF1R) is a receptor tyrosine kinase that binds insulin-like growth factor. The receptor is involved in physiological and pathological processes. It has an anti-apoptotic effect.
It can be an alternative fusion gene to ALK and has been reported in an inflammatory myofibroblastic tumor occurring in the duodenum of a middle-aged woman [58].
Conclusion
This review provides an update of the variety of protein kinases involved in the oncogenesis of mesenchymal tumors, mainly in pediatric patients, with a focus on the more rarely documented PKs—EGFR, ALK, LTK, PDGFRB, MET, (B)RAF, RET, FGFR, ABL, and IGF1R. The morphological and immunohistochemical hallmarks of such tumors, including their overlap, are highlighted, in order to facilitate detection of such tumors because of the therapeutic consequences.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1van Spronsen R, Kester LA, Knops RRG et al. Infantile fibrosarcoma with an EGFR kinase domain duplication: Underlining a close relationship with congenital mesoblastic nephroma and highlighting a similar morphological spectrum. Ann Diagn Pathol. 2022;57:151885.10.1016/j.anndiagpath.2021.15188535032896 · doi ↗ · pubmed ↗
- 2Vidrine DW, Berry JF, Garbuzov A et al. DCTN 1-ALK gene fusion in inflammatory myofibroblastic tumor (IMT) of the CNS. Childs Nerv Syst. 2021;37(7):2147–51.10.1007/s 00381-021-05219-334014367 · doi ↗ · pubmed ↗
- 3Vallese S, Barresi S, Hiemcke-Jiwa L et al. Spindle Cell Lesions with Oncogenic EGFR Kinase Domain Aberrations: Expanding the Spectrum of Protein Kinase-Related Mesenchymal Tumors. Mod Pathol. 2024;37(9):100539.10.1016/j.modpat.2024.10053938880352 · doi ↗ · pubmed ↗
- 4Kao YC, Fletcher CDM, Alaggio R et al. Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol. 2018;42(1):28–38.10.1097/PAS.0000000000000938 PMC 573046028877062 · doi ↗ · pubmed ↗
- 5Al-Ibraheemi A, Zhou Y, Rullo E, Alaggio R. What is new in fibroblastic/myofibroblastic tumors in children. Virchows Arch. 2025;486(1):127–41.10.1007/s 00428-024-03964-939499317 · doi ↗ · pubmed ↗
- 6Suurmeijer AJH, Xu B, Torrence D, Dickson BC, Antonescu CR. Kinase fusion positive intra-osseous spindle cell tumors: A series of eight cases with review of the literature. Genes Chromosomes Cancer. 2024;63(1):e 23205.10.1002/gcc.23205 PMC 1125099237782551 · doi ↗ · pubmed ↗
- 7Wegert J, Vokuhl C, Collord G et al. Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nat Commun. 2018;9(1):2378.10.1038/s 41467-018-04650-6PMC 600630929915264 · doi ↗ · pubmed ↗
- 8Xu B, Suurmeijer AJH, Agaram NP, Antonescu CR. Head and Neck Mesenchymal Tumors with Kinase Fusions: A Report of 15 Cases With Emphasis on Wide Anatomic Distribution and Diverse Histologic Appearance. Am J Surg Pathol. 2023;47(2):248–58.10.1097/PAS.0000000000001982 PMC 984657836638315 · doi ↗ · pubmed ↗