Laboratory strategies for the identification of Burkholderia species: From classical phenotyping to advanced genomic and proteomic approaches
Giorgio Silva-Santana, Francisca Letícia Sousa de Sales, Marcelo Luiz Lima Brandão

TL;DR
This paper reviews methods for identifying Burkholderia species, comparing classical techniques with modern genomic and proteomic approaches.
Contribution
The paper provides a comprehensive review of current strategies for identifying Burkholderia species, emphasizing the need for standardized methods.
Findings
Classical methods like selective media and biochemical tests are useful for initial screening but lack resolution for closely related species.
Molecular methods such as MLST and WGS offer high-resolution identification and are recommended for detailed epidemiological monitoring.
A tiered strategy combining initial screening with advanced genomic techniques is suggested for improved accuracy and standardization.
Abstract
The genus Burkholderia, particularly the Burkholderia cepacia complex (Bcc), encompasses Gram-negative bacteria of recognized ecological, biotechnological, and clinical relevance. Accurate identification of species within this complex is challenging due to high phenotypic and genetic similarity, impacting clinical diagnostics and epidemiological surveillance. This review addresses phenotypic, proteomic, and molecular methods for species- and clone-level identification, highlighting their advantages and limitations. Classical methods, including selective media, Burkholderia cepacia selective agar (BCSA), Oxidation-Fermentation Polymyxin Bacitracin Lactose agar (OFPBLA), and Pseudomonas cepacia agar (PCA), and biochemical tests are useful for initial screening but have limited resolution for differentiating closely related Bcc species. Automated systems, such as VITEK® 2, provide rapid…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
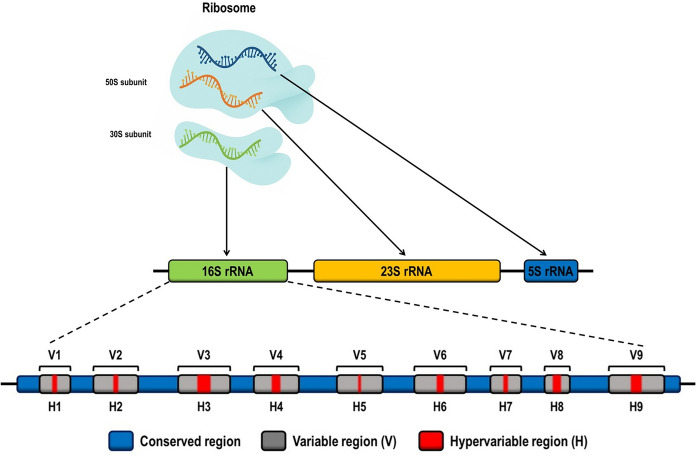 Figure 1
Figure 1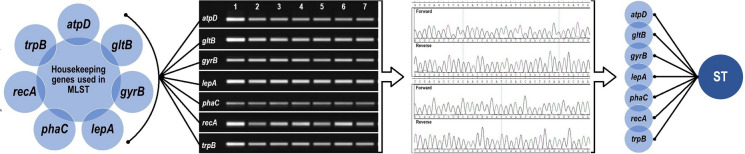 Figure 2
Figure 2 Figure 3
Figure 3 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Identification and Susceptibility Testing · Burkholderia infections and melioidosis · Cystic Fibrosis Research Advances
Introduction
The genus Burkholderia, within the class Betaproteobacteria, represents a widely distributed bacterial group with well-established ecological, biotechnological, and clinical relevance (Espinosa-Victoria et al. 2020; Parte et al. 2020; LPSN, 2025). Originally described as Pseudomonas cepacia by Burkholder in 1950, it was later reclassified based on phenotypic and genomic criteria, currently encompassing 36 validated species (Yabuuchi et al. 1992; Parte et al. 2020; LPSN, 2025). These microorganisms inhabit soils, aquatic ecosystems, and hospital surfaces, engaging in complex ecological interactions and acting as opportunistic pathogens in humans and other animals (Compant et al. 2008; Eberl and Vandamme 2016).
Among the clinically most relevant groups is the Burkholderia cepacia complex (Bcc), which is responsible for infections in immunocompromised patients, particularly those with cystic fibrosis (CF) (Tavares et al. 2020; Gutiérrez Santana and Coria Jiménez 2024). These microorganisms are frequently associated with nosocomial infections and exhibit multidrug resistance (MDR), complicating epidemiological control and clinical management (Shaban et al. 2020). The genus’s genomic complexity, including multiple chromosomes, pronounced genomic plasticity, and mobile genetic elements, limits the effectiveness of traditional phenotypic methods (Depoorter et al. 2020; Scoffone et al. 2021).
Classical isolation and screening methods, such as Burkholderia cepacia selective agar (BCSA), Oxidation-Fermentation Polymyxin Bacitracin Lactose agar (OFPBLA), and Pseudomonas cepacia agar (PCA), as well as biochemical tests, have low cost and are useful for initial screening but lack sufficient resolution to differentiate closely related Bcc species (Henry et al. 1999; Coenye et al. 2001; Vanlaere et al. 2009). Molecular and proteomic techniques, including sequencing of housekeeping genes (gyrB and recA), Matrix-Assisted Laser Desorption/Ionization–Time of Flight Mass Spectrometry (MALDI-TOF MS), Fourier-Transform Infrared Spectroscopy (FTIR), and Multilocus Sequence Typing (MLST), enable precise species- and clone-level identification, facilitating epidemiological surveillance and detailed clonal typing (Wenning and Scherer 2013; Jin et al. 2020; Uribe et al. 2023; Calderaro and Chezzi, 2024).
Whole-genome sequencing (WGS) represents the current gold standard, simultaneously providing data on identification, antimicrobial resistance, virulence factors, and clonal typing, although it requires high investment, specialized infrastructure, and bioinformatic processing (De Maio et al. 2019; Calderaro and Chezzi 2024). Therefore, methodological choices should balance cost, analytical capacity, and clinical or research objectives, with a stepwise strategy recommended: accessible methods for initial screening and advanced technologies, such as MLST and WGS, for research centers and genomic surveillance (Deng et al. 2014; Guo et al. 2014; Calderaro and Chezzi 2024).
In this context, the present article aims to critically review the main strategies for species identification within the genus Burkholderia, encompassing classical phenotypic methods as well as innovative techniques such as Raman spectroscopy, FTIR, MALDI-TOF MS, MLST, and comparative genomics analysis. The objective is to elucidate the advantages and limitations of each approach and to highlight the importance of methodological standardization for improving clinical diagnosis, epidemiological surveillance, and the management of infections caused by these emerging pathogens.
The genus Burkholderia
The genus Burkholderia belongs to the phylum Proteobacteria, class Betaproteobacteria, order Burkholderiales, and family Burkholderiaceae (Parte et al. 2020; LPSN, 2025). These bacteria are Gram-negative bacilli, obligate aerobes, motile, and incapable of fermenting carbohydrates (Coenye and Vandamme 2003; Mahenthiralingam et al. 2005). Currently, the genus comprises 36 validated species (Parte et al. 2020), recognized for their ecological versatility. They inhabit diverse environments, including soil, freshwater bodies, industrial and hospital surfaces, and medical devices (Compant et al. 2008). Their interactions with hosts are highly varied, ranging from pathogenic to symbiotic relationships that affect plants, insects, and humans (Kaltenpoth and Flórez 2020).
When originally described as P. cepacia, this species was characterized as a Gram-negative, flagellated, strictly aerobic bacillus capable of utilizing a wide range of organic compounds as carbon and nitrogen sources for energy and growth (Burkholder 1950). It was identified as a phytopathogen responsible for onion rot, distinguished from fungal infections and Pseudomonas allicola by producing a characteristic yellow pigmentation and a distinct odor (Burkholder 1950).
By the end of the twentieth century, the discovery of new species related to P. cepacia prompted Yabuuchi et al. (1992) to propose the creation of a new genus. This reclassification was based on analyses of 16S rRNA gene sequences, DNA-DNA hybridization, membrane lipid and fatty acid composition, as well as phenotypic characteristics. The new genus was named Burkholderia in honor of W. H. Burkholder (Yabuuchi et al. 1992; Bazani et al. 2024). Subsequent DNA-rRNA hybridization studies led to the taxonomic transfer of P. cepacia and six other species from the former rRNA group II of Pseudomonas to the new genus, with B. cepacia designated as the type species (Yabuuchi et al. 1992; Bach et al. 2023; Bazani et al. 2024).
With advances in molecular biology and comparative genomics, the taxonomy of the genus Burkholderia underwent further revision. Based on genomic analyses, Cloutier et al. (2018) proposed dividing the genus into two main groups: one comprising species pathogenic to humans, such as B. pseudomallei and members of the Bcc, as well as phytopathogenic species like B. plantarii and others associated with endosymbiotic relationships with fungi; and another group consisting of non-pathogenic species, commonly isolated from natural environments or in symbiosis with plants (Cloutier et al. 2018).
This division was later supported by studies employing genomic phylogeny, conserved molecular markers such as Conserved Signature Indels (CSIs), metrics like Average Nucleotide Identity (ANI), and digital DNA-DNA hybridization (dDDH) (Bach et al. 2023). These genomic approaches, recognized by international bacterial taxonomy organizations, revealed the need for an even deeper subdivision of *Burkholderia *sensu lato (s.l.). This culminated in the creation of new genera such as Paraburkholderia, Caballeronia, Mycetohabitans, Trinickia, Robbsia, and Pararobbsia (Estrada-de Los Santos et al. 2018; Lin et al. 2020; Bach et al. 2023).
Species of ecological and agricultural importance, previously classified within Burkholderia but non-pathogenic and known for their potential to promote plant growth, synthesize antimicrobial compounds, and perform bioremediation of pollutants, were transferred to the genus Paraburkholderia. This reclassification reflects increasing concern for taxonomic accuracy, especially due to phenotypic overlap between pathogenic and environmental groups (Sawana et al. 2014; Scoffone et al. 2021; Bach et al. 2023). In a pharmaceutical industry facility, Aguiar et al. (2025) reported the isolation of two Burkholdeiria spp. strains that are possible new species after full gene 16S rRNA sequencing analysis (Aguiar et al. 2025).
Burkholderia cepacia complex: from genovars to pathogenic species
In 2001, Coenye et al. (2001) observed that isolates previously identified as B. cepacia exhibited distinct phenotypes, low DNA-DNA hybridization levels (30–50%), and high similarity in 16S rRNA gene sequences (98–100%). These findings led to the reclassification of these isolates as genomovars, which were later grouped within the Bcc (Coenye et al. 2001; Bevivino et al. 2002; Bazani et al. 2024).
With advances in polyphasic taxonomy, the Bcc came to include several species with distinct genomes, initially categorized as genomovars I–X. Over time, these genomovars were reclassified as separate species, such as B. cepacia, B. multivorans, B. cenocepacia, B. stabilis, B. vietnamiensis, B. dolosa, B. ambifaria, B. anthina, B. pyrrocinia, and B. ubonensis (Coenye and Vandamme 2003; Ramette et al. 2005; Lauman and Dennis 2021). Additionally, the identification of related microorganisms in natural environments has contributed to expanding the number of recognized species within the complex (Lauman and Dennis 2021).
Currently, the Bcc comprises approximately 24 species and 17 genomovars, including the aforementioned species as well as newly described ones such as B. aenigmatica, B. arboris, B. catarinensis, B. contaminans, B. diffusa, B. lata, B. latens, B. metallica, B. paludis, B. pseudomultivorans, B. puraquae, B. seminalis, B. stagnalis, and B. territorii (Bellich et al. 2020; Depoorter et al. 2020; Schoch et al. 2020).
The Bcc is widely recognized for its clinical significance, comprising species with high pathogenic potential associated with a range of serious infections (Baylan et al. 2012; Ghafil et al. 2022). In particular, infections in patients with CF and chronic granulomatous disease (CGD) are prominent, as well as respiratory, urinary, and systemic infections in immunocompromised individuals such as frail elderly patients, people living with the human immunodeficiency virus (HIV), and patients undergoing chemotherapy (Baylan et al. 2012; Tavares et al. 2020; Lauman and Dennis 2021).
This bacterial complex is also a significant agent of hospital-acquired infections, especially in intensive care units (ICUs). The Bcc ranks among the most frequently isolated non-glucose-fermenting pathogens and is considered one of the most clinically important groups, second only to the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.), all notorious for their high antimicrobial resistance (Padma et al. 2023; Miller and Arias 2024).
In some countries, such as India, nosocomial outbreaks caused by Bcc species account for approximately 8.8% of hospital-acquired infections. These MDR pathogens represent a significant challenge to infection control programs (Shaban et al. 2020). Moreover, characteristics such as resistant biofilm formation and the production of insoluble exopolysaccharides, as demonstrated for B. cenocepacia, reinforce the persistence and resistance potential of the Bcc in hospital environments, contributing to the difficulty of eradicating outbreaks (Bellich et al. 2020; Silva-Santana et al. 2025).
Genetics of Burkholderia: Evolutionary Challenges
Species of the genus Burkholderia possess highly complex and large genomes, typically organized into two or three chromosomes, with total sizes ranging from 6 to 9 Mb (Bochkareva et al. 2018). For example, B. pseudomallei has two replicons measuring 4.07 Mb and 3.17 Mb, totaling approximately 7.24 Mb and encoding between 3460 and 2395 proteins per replicon (Holden et al. 2004). In contrast, B. mallei contain two chromosomes of 3.51 Mb and 2.33 Mb, summing to around 5.84 Mb, with approximately 5535 genes in total (Nierman et al. 2004).
On average, genomes of Burkholderia species contain about 5000 genes and exhibit multiple gene duplications, reflecting their high genomic plasticity (Holden et al. 2004; Tavares et al. 2020; Scoffone et al. 2021). A distinctive feature is the high guanine-cytosine (G + C) content, ranging from 66 to 69%, which confers increased structural stability to the DNA (Bochkareva et al. 2018).
These genomes also display elevated levels of recombination and structural variability, including an abundant presence of insertion sequence (IS) elements, genomic islands, constituting 5–6% of the genome, and chromosomal rearrangements such as inversions, transpositions, duplications, and deletions (Loutet and Valvano 2010; Bochkareva et al. 2018). Notably, Bochkareva et al. (2018) demonstrated that these rearrangements are nonrandom, suggesting strong selective pressure to preserve genomic organization, especially to prevent intra-replichore inversions that transfer large gene blocks between leading and lagging strands during replication (Bochkareva et al. 2018).
Another key characteristic of these microorganisms is their remarkable adaptive capacity, enabling rapid accumulation of mutations in response to environmental stresses, both in vitro and during infectious processes (Drevinek et al. 2010; Loutet and Valvano 2010; Seng et al. 2023). This genotypic and phenotypic plasticity allows Burkholderia species to exploit diverse metabolic pathways, facilitating colonization of various ecological niches and development of resistance to multiple environmental challenges, including antimicrobial agents (Tavares et al. 2020; Lagage et al. 2023).
Accurate identification of species within the genus remains challenging due to their high genetic and phenotypic diversity, which directly impacts the reliability of data concerning their ecological distribution and adaptive behaviors. To overcome these difficulties, the Burkholderia Genome Database (http://www.burkholderia.com) serves as a valuable resource, offering comprehensive information on all sequenced genomes of the genus (Winsor et al. 2008). This database provides detailed genomic annotations, gene content data, chromosomal structures, and tools for comparative analyses, making it indispensable for research on genetic diversity, evolution, and gene function in Burkholderia species (Loutet and Valvano 2010; Bochkareva et al. 2018; Scoffone et al. 2021).
Phenotypic identification methods
Selective culture media
Precise species identification within the genus Burkholderia is essential, as several species behave as opportunistic pathogens causing severe and potentially fatal infections in both immunocompromised and immunocompetent hosts (Inglis et al. 2005). The rising emergence of antimicrobial resistance among these bacteria underscores the need for rapid and reliable diagnostic methods (Trespidi et al. 2020). However, the high degree of genetic and phenotypic similarity among Burkholderia species presents significant challenges for definitive species-level discrimination, particularly in clinical microbiology laboratories (Jin et al. 2020; Wang et al. 2020).
Various selective culture media have been developed and employed to isolate and identify Burkholderia species, each tailored for specific applications. Among them, Burkholderia cepacia selective agar (BCSA) stands out as one of the most widely used media for isolating members of the Bcc from respiratory samples of CF patients (Henry et al. 2001). BCSA demonstrates sensitivity up to 100% after 72 h of incubation, outperforming the 96% and 84% sensitivities reported for Oxidation-Fermentation Polymyxin Bacitracin Lactose agar (OFPBLA) and Pseudomonas cepacia agar (PCA), respectively (Henry et al. 1999; Marrs et al. 2021). In addition to its superior sensitivity, BCSA effectively suppresses the growth of non-Bcc microorganisms, a critical factor to prevent cross-contamination in samples collected from non-sterile clinical specimens or cases of polymicrobial infections (Henry et al. 1999).
The OFPBLA medium is recognized for its utility in differentiating Burkholderia from other respiratory pathogens and is particularly valuable for screening CF patients (Gilligan et al. 2003). However, its sensitivity is lower (84% at 48 h) and exhibits limited inhibition of non-target organisms, such as fungi and other Gram-negative bacteria, which limits its specificity (Henry et al. 1999).
Similarly, PCAT/PCA, modified with antimicrobials to distinguish Burkholderia from Pseudomonas in clinical and environmental samples, shows an approximate sensitivity of 74% at 48 h. However, its specificity is reduced due to the growth of unwanted microorganisms such as Pandoraea spp., Stenotrophomonas maltophilia, Cupriavidus spp., and rapidly growing mycobacteria (Mycobacterium chelonae, M. peregrinum, M. septicum, M. smegmatis) (Sandlin 1974; Henry et al. 1999).
In regions endemic for melioidosis, Ashdown agar is widely used for detecting B. pseudomallei, although it exhibits low sensitivity for B. mallei (Ashdown 1979; Peacock et al. 2005). Additionally, non-selective media such as Triple Sugar Iron (TSI) and MacConkey agar can assist in preliminary isolation of Burkholderia and in differentiating other Gram-negative bacilli in both clinical and environmental contexts (Rastogi et al. 2019; De et al. 2022) (Table 1).Table 1. Efficiency and limitations of selective media in the characterization of BurkholderiaMediumCompositionCharacteristicsLimitationsReferencesBCSACasein and soybean peptones: nutrientsGLC: energy sourcePMB, GEN, and TIC: inhibit competing Gram-positive and Gram-negative bacteriaCV: inhibits Gram-positive bacteriaOpaque and gray/yellow colonies.Morphological differentiation among Bcc speciesNot 100% specificMay allow growth of other resistant bacteria(Henry et al. 2001)PCATGLY: carbon sourcePMB and TIC: selective antibioticsTCA: enhances selectivityCV: inhibits Gram-positive bacteriaAdapted for B. cepaciaMay allow growth of other Bcc speciesLower selectivity compared to BCSA(Sandlin 1974)OFPBLALAC: fermentable substratePMB and BAC: inhibit competing bacteriaPR: indicator of LAC fermentationBurkholderia do not ferment LAC, resulting in yellow colorationOther non-LAC-fermenting bacteria may complicate identification(Gilligan et al. 2003)Ashdown agarGLY: carbon sourceCV: inhibits Gram-positive bacteriaGEN: inhibits competing Gram-negative bacteriaNR: indicator for acid-producing coloniesB. pseudomallei forms rough or smooth colonies, often orange to brown in colorLess suitable for isolation of other Bcc species(Ashdown 1979)TSIGLC, LAC, and SUC: fermentable sugarsSTS: substrate for hydrogen sulfide productionPR: pH indicatorDifferentiation of enterobacteria by sugar fermentation and H_2_S productionBurkholderia spp. produce bright yellow coloniesNon-selectiveAllows growth of various bacteria(Forbes et al. 2007; Rastogi et al. 2019; De et al. 2022)MacConkey agarLAC: fermentable substrateBS and CV: inhibit Gram-positive bacteriaNR: pH indicatorBurkholderia spp. do not ferment LAC and produce purple pigmentsDifficulty differentiating among Bcc species due to lack of LAC fermentation(MacFaddin 1985; Rastogi et al. 2019; De et al. 2022)BCSA, Burkholderia cepacia selective agar; PCAT, Pseudomonas cepacia agar modified with antimicrobials; OFPBLA, Oxidation-Fermentation Polymyxin Bacitracin Lactose agar; TSI, Triple Sugar Iron; BAC, bacitracin; BS, bile salts; CV, crystal violet; GEN, gentamicin; GLC, glucose; GLY, glycerol; LAC, lactose; NR, neutral red; PMB, polymyxin B; PR, phenol red; STS, sodium thiosulfate; SUC, sucrose; TCA, trichloroacetic acid; TIC, ticarcillin
Although selective media do not guarantee unequivocal species-level identification of Burkholderia, they remain essential tools in clinical microbiology laboratories. Beyond facilitating isolation from complex samples, these media support epidemiological surveillance and investigations into virulence factors and antimicrobial resistance within Burkholderia populations (Henry et al. 2001).
Biochemical tests: conventional and automated systems
Conventional biochemical methods, when combined with antimicrobial susceptibility testing (AST), enable identification of Burkholderia at the genus level and, in some cases, at the species level. However, assays based on catalase activity, gluconate, malate, phenylacetate, and leucine arylamidase exhibit low discriminatory power in differentiating species within the Bcc from those outside the complex (Henry et al. 2001; Krejcí and Kroppenstedt 2006; Vanlaere et al. 2009).
The high phenotypic similarity between Burkholderia species and other genera of non-fermenting Gram-negative bacilli, such as Cupriavidus, Ralstonia, Achromobacter, Brevundimonas, Comamonas, Pandoraea, and Delftia, often leads to misidentification (Coenye et al. 2001). As an initial screening approach, intrinsic resistance to polymyxin B has been employed to differentiate Burkholderia isolates from other environmental bacteria (Laffineur et al. 2002; Loutet and Valvano 2011). However, this characteristic alone is insufficient and must be complemented by additional biochemical tests and, preferably, molecular tools such as target gene sequencing to improve diagnostic accuracy (Laffineur et al. 2002; Mahenthiralingam et al. 2005).
The difficulty in distinguishing species within the Bcc partly stems from the genomic plasticity and metabolic diversity of the genus. This plasticity is directly linked to their large genomes, often organized into multiple replicons and enriched with genes associated with environmental adaptation (Loutet and Valvano 2010; Scoffone et al. 2021; Rodríguez-Cisneros et al. 2023). Some species, such as B. contaminans, exhibit relatively distinctive phenotypes, including yellow pigmentation and production of antifungal compounds (e.g., occidiofungin and pyrrolnitrin) when cultured on Yeast Extract Peptone Dextrose (YPD) medium. However, these traits are not exclusive and may vary depending on environmental conditions (O’Rourke et al. 2020; Yang et al. 2021; Rodríguez-Cisneros et al. 2023).
Automated systems such as the VITEK® 2 (bioMérieux, France) are widely used for microorganism identification, including Burkholderia species (Santana et al. 2024). This system utilizes disposable cards, such as the GN card for Gram-negative bacteria, that contain between 41 and 47 biochemical tests monitored by Advanced Colorimetry™. These tests assess enzymatic and fermentative reactions, comparing the resulting profiles against an internally maintained and periodically updated database (bioMérieux VITEK® 2 Systems, 2023; BIOMÉRIEUX VITEK® 2 Systems, 2023). The system can identify up to 500 microorganisms and perform antimicrobial susceptibility testing (AST) for approximately 180 agents, following standards established by the Clinical and Laboratory Standards Institute (CLSI), the European Committee on Antimicrobial Susceptibility Testing (EUCAST), and the Food and Drug Administration (FDA). In Brazil, the Brazilian Committee on Antimicrobial Susceptibility Testing (BRCAST) can also be used as a standard (Vasala et al. 2020; bioMérieux VITEK® 2 Systems, 2023; BIOMÉRIEUX VITEK® 2 Systems, 2023).
The VITEK® 2 GN card evaluates various metabolic activities, including hydrolysis of β-N-acetyl-glucosaminidase (BNAG) and β-N-acetyl-galactosaminidase (NAGA), as well as metabolism of sugars such as D-cellobiose, D-glucose, and D-sorbitol. It also assesses arylamidase activities like L-proline arylamidase (ProA) and tyrosine arylamidase (PyrA). These reactions are essential for intra-Bcc differentiation. The average identification time for Gram-negative bacteria ranges from 2 to 4 h, while preliminary AST results are typically available within 6 to 8 h (O'Hara and Miller 2003; bioMérieux VITEK® 2 Systems, 2023; BIOMÉRIEUX VITEK® 2 Systems, 2023).
Despite its versatility, the VITEK® 2 system’s performance in identifying Bcc species varies considerably: it achieves overall accuracy of approximately 53% for Bcc members, with accuracy reaching 89% for B. multivorans, but only 38% for virulent genovar III strains such as B. cenocepacia J2315, C5424, and HI2424 (Alby et al. 2013; Wong et al. 2020). Accuracy is higher for clinical isolates (55%) compared to environmental isolates (39%). For B. pseudomallei, accuracy rates range between 63 and 81% (Gassiep et al. 2019).
Integration with the VITEK® MS PRIME system (MALDI-TOF MS) significantly improves identification accuracy, reaching up to 99.8%, making it a promising tool for rapid and reliable Burkholderia identification (Wong et al. 2020; bioMérieux VITEK® 2 Systems, 2023; BIOMÉRIEUX VITEK® 2 Systems, 2023). Comparative studies, such as the performed by Alby et al. (2013), have shown that the two MALDI-TOF systems MALDI Biotyper® (Bruker) or VITEK® MS (bioMérieux) outperform purely phenotypic methods, achieving accuracy rates above 90%, even for challenging environmental isolates (Alby et al. 2013; Santana et al. 2024).
Nevertheless, some limitations remain. For instance, a study in Malaysia reported that up to 35% of B. pseudomallei isolates were misidentified as B. cepacia by the VITEK® 2 system. Intraspecies biochemical variability, such as absence of BNAG and NAGA activities, was directly linked to misidentification (13% of strains lacking BNAG activity were incorrectly classified) (Podin et al. 2013). Another factor influencing accuracy is the culture medium: strains grown on Columbia Horse Blood agar (CHBA) exhibited significantly higher correct identification rates compared to those cultured on Trypticase Soy agar (TSA) (Lowe et al. 2006).
Although the VITEK® 2 database is continuously updated, accurate identification depends on the representativeness of target species within the database and the stability of biochemical profiles expressed by tested strains (bioMérieux, 2025). Therefore, especially in critical clinical contexts, combining biochemical, molecular, and proteomic methods, such as MALDI-TOF MS alongside target gene sequencing, is recommended to ensure robust and precise identification (Guo et al. 2014; Wong et al. 2020).
Molecular identification methods
Molecular methods have proven essential for the accurate identification of species within the genus Burkholderia, effectively overcoming the inherent limitations of traditional phenotypic and biochemical techniques (Esmaeel et al. 2016). Among these approaches, pulsed-field gel electrophoresis (PFGE) stands out as a widely used tool in epidemiological studies and outbreak investigations, as it enables discrimination of bacterial strains based on their genomic DNA fragmentation patterns (Zulkefli et al. 2016; Ribot et al. 2019).
Another increasingly adopted technique is MALDI-TOF MS, which allows for rapid and accurate analysis of ribosomal protein mass profiles. This methodology has gained widespread application in clinical microbiology and research settings, facilitating the reliable identification of Burkholderia species (Vandamme et al. 2017; Wong et al. 2020).
Sequencing of target genes, such as 16S rRNA, 23S rRNA, recA, hisA, groEL, rpsU, fur, among others, also stands out for its high sensitivity and specificity (Chakravorty et al. 2007; Yoon et al. 2017). The 16S rRNA gene, in particular, contains nine hypervariable regions (V1–V9), which exhibit considerable diversity across different bacterial species and are extensively explored for diagnostic and phylogenetic purposes (Yoon et al. 2017; Fergusson et al. 2020; Chalita et al. 2024). However, no single hypervariable region can discriminate all bacterial species; therefore, combined analysis of multiple regions, such as V2, V3, and V6, is recommended to maximize discriminatory power (Chakravorty et al. 2007).
In addition, genes such as recA and hisA have been employed to improve resolution among closely related Bcc species, owing to their higher interspecies variability compared to the 16S rRNA gene (Fergusson et al. 2020). The implementation of strategies such as MLST, which targets multiple housekeeping loci, has further enhanced strain typing capabilities and the discrimination of specific lineages (Yoon et al. 2017).
The integration of these molecular approaches provides a robust and comprehensive strategy, overcoming the limitations of conventional methods and ensuring faster, more accurate species identification (Ho et al. 2019; Safir et al. 2023). This combined approach is particularly valuable in clinical, environmental, and epidemiological contexts, where precise pathogen detection and monitoring are critical for public health and infection control (Iida and Takemoto 2018; Wong et al. 2020).
Pulsed-field gel electrophoresis: restricted epidemiology
Pulsed-field gel electrophoresis (PFGE) is widely recognized as a robust tool in molecular epidemiology due to its high discriminatory power in differentiating bacterial strains within the same species (Goering 2010). The method is based on the digestion of bacterial genomes with restriction enzymes, generating DNA fragments of varying sizes that are subsequently separated on agarose gels subjected to an alternating electric field. This process results in characteristic banding patterns, referred to as pulsotypes, that allow for detailed analysis of genomic differences among strains (Zulkefli et al. 2016; Lopez-Canovas et al. 2019).
Despite its high resolution, PFGE is sensitive to genomic rearrangements, which can complicate data interpretation in long-term studies or in highly recombinant bacterial populations (Lopez-Canovas et al. 2019). For this reason, the technique is particularly well suited for outbreak investigations in geographically or clinically restricted settings, such as localized hospital infections (Kaufmann 1998; Zulkefli et al. 2016).
A notable example of PFGE application occurred in Rio de Janeiro, Brazil, during an outbreak of primary bloodstream infections caused by Bcc species across three hospitals. PFGE analysis identified six distinct pulsotypes, one of which accounted for 65% of the cases (11 out of 17 patients). The outbreak was traced to the intravenous administration of contaminated bromopride, and the subsequent withdrawal of this product from the market was pivotal in containing the event. This case underscored the importance of epidemiological surveillance and the rigorous implementation of infection control measures (Martins et al. 2010).
Another relevant case was reported between 2011 and 2015 at the National Bone Marrow Transplant Center in Tunis, Tunisia, where intermittent outbreaks of infections associated with the infusion of hematopoietic stem cells (HSCs) contaminated with B. cepacia occurred. PFGE analysis of 23 strains isolated between 2007 and 2015 revealed five distinct genetic clusters, with one predominant cluster comprising 18 strains recovered from HSC bags, patient blood cultures, and environmental samples, including water containers and water baths. The investigation identified the water used in the water bath during thawing of the bags as the source of contamination. Implementation of a dry bath thawing method in May 2015 effectively controlled the outbreak, with no new cases reported at least until 2020 (Raddaoui et al. 2022).
These episodes clearly demonstrate the effectiveness of PFGE in elucidating transmission patterns, clonal relationships, and in facilitating the containment of hospital outbreaks caused by Burkholderia species. They highlight the essential role of this technique in molecular epidemiology and in the development of prevention and control strategies for nosocomial infections (Zulkefli et al. 2016; Raddaoui et al. 2022).
16S and 23S rRNA genes: limitations in bacterial identification
The 16S rRNA gene is a fundamental molecular marker in microbiology, widely used for the identification and characterization of bacteria (Woese and Fox 1977; Weisburg et al. 1991). It encodes the small (16S) ribosomal subunit, a key component of the protein synthesis machinery. Although highly conserved across bacterial taxa, this gene contains hypervariable regions (V1–V9) that enable differentiation between genera and, in many cases, between species (Chakravorty et al. 2007; Pruesse et al. 2007) (Fig. 1). Owing to these features, 16S rRNA sequencing has been extensively employed for bacterial identification in clinical and environmental samples, particularly in contexts where rapid and accurate pathogen detection is critical (Park et al. 2021; Botan et al. 2024; Santana et al. 2024).Fig. 1. Schematic representation of ribosomal genes in the bacterial genome. The 70S ribosome is composed of the small 30S subunit (containing the 16S rRNA) and the large 50S subunit (containing the 23S and 5S rRNAs). The 16S rRNA gene comprises nine hypervariable regions (V1–V9) interspersed with conserved regions, widely used for bacterial identification and phylogeny. In many bacteria, the 16S, 23S, and 5S genes are organized within the same ribosomal operon (rrn) (Fukuda et al. 2016)
However, in the identification of species within the genus Burkholderia, particularly those belonging to the Bcc, the 16S rRNA gene exhibits significant limitations. The high sequence conservation among Bcc species reduces its discriminatory power, often resulting in genus-level identifications or taxonomic ambiguities (Furlan et al. 2019; Jin et al. 2020; Santana et al. 2024).
As an alternative, the 23S rRNA gene, which encodes the large (23S) ribosomal subunit, has been explored due to its higher nucleotide variability, which provides improved taxonomic resolution among closely related species (Pei et al. 2009; Walker et al. 2020). This gene plays a central role in peptide bond formation during translation and is also a key target in antimicrobial resistance studies (Ng et al. 2002; Gaynor and Mankin 2003).
Combined analysis of the 16S and 23S rRNA genes, or the use of the 16S–23S intergenic spacer (ITS), has demonstrated a significant increase in identification accuracy, particularly in complex clinical and environmental samples. The ITS region, due to its greater variability compared to the adjacent ribosomal genes, serves as a highly discriminatory marker for differentiating phylogenetically close species (Nagpal et al. 1998; Chakravorty et al. 2007). In species such as B. pseudomallei, for instance, ITS analysis has enabled discrimination of geographically distinct lineages, revealing phylogenetic patterns not detectable by 16S rRNA analysis alone (Liguori et al. 2011).
Nevertheless, despite these advantages, the 23S rRNA gene also presents limitations in resolving species within the Bcc due to conserved segments within certain regions of the gene (Depoorter et al. 2020). However, it remains useful for broader phylogenetic inference and for characterizing intra-genus diversity. Additionally, specific point mutations in the 23S rRNA, such as the A2058G substitution, have been associated with resistance to macrolides, lincosamides, and streptogramin B antibiotics, by interfering with antibiotic binding to the ribosome (Ng et al. 2002; Gaynor and Mankin 2003; Jin et al. 2020).
To overcome the limitations of ribosomal genes, alternative target genes such as recA, rpoB, and groEL have been widely used, as they encode essential proteins with greater interspecies variability, providing refined taxonomic resolution for Burkholderia, particularly in the context of the Bcc (Vermis et al. 2002; Woo et al. 2002). In more challenging cases, whole-genome sequencing (WGS) emerges as the most precise and comprehensive approach, enabling not only species- and strain-level identification but also the analysis of genes related to virulence, antimicrobial resistance, and genomic evolution (Botan et al. 2024).
Multi-locus sequence typing: evolution and epidemiology
The use of molecular methods based on housekeeping genes is widely recommended in geographic, epidemiological, and longitudinal studies due to their high conservation and genomic stability (Zulkefli et al. 2016). Among these methodologies, MLST stands out, as it analyzes internal fragments of seven essential housekeeping genes involved in core cellular metabolism, which are characterized by low mutation rates. These features confer high discriminatory power to the technique for differentiating bacterial strains and assessing genetic diversity in epidemiological contexts (Maiden et al. 1998, 2013; Urwin and Maiden 2003; Jolley et al. 2018).
In the genus Burkholderia, the seven housekeeping genes commonly used in the MLST scheme are: atpD, gltB, gyrB, lepA, phaC, recA, and trpB (Maiden 2006) (Fig. 2). This approach has proven particularly effective in characterizing bacterial strains, supporting the phylogenetic delineation of new species within the Bcc, as well as in constructing detailed phylogenetic trees (Vanlaere et al. 2009). Among the main advantages of MLST are its high reproducibility and international standardization, factors that facilitate cross-study comparisons and enable interlaboratory collaborations, which are crucial for molecular surveillance programs and outbreak control (Spilker et al. 2009; Jolley et al. 2018) (Table 2).Fig. 2. Representative scheme of the steps in the Multilocus Sequence Typing (MLST) technique. MLST is a molecular typing method used to characterize microorganisms based on the analysis of constitutive genes (housekeeping). The main steps include: 1. Extraction of total genomic DNA from the microorganism’s cells. 2. PCR amplification of the selected constitutive genes (atpD, gltB, gyrB, lepA, phaC, recA, and trpB). 3. Sequencing of the PCR products in both forward and reverse directions to obtain high-fidelity allelic sequences. 4. Comparison of the obtained sequences with MLST databases for allele assignment. 5. Definition of the allelic profile and determination of the sequence type (ST)Table 2. Adaptive and pathogenic functions of the genes used in the Multilocus Sequence Typing schemeGeneProteinsFunctionRelevanceAdaptive ImportancePathogenic ImportanceReferencesatpDATPβCatalyzes ATP synthesis via OXPHOSCytoplasmic membrane-localized, essential for energy generationCritical for survival in low O_2_ and/or nutrient conditionsFacilitates survival in CF lungs, where oxygenation is limited(Spilker et al. 2009; Zulkefli et al. 2016)gltBGltBCatalyzes the conversion of Gln and α-KG to GluNitrogen metabolism and amino acid biosynthesisFacilitates efficient nitrogen source utilization, promoting colonization in diverse ecological nichesContributes to host environment adaptation and secondary metabolite production(Spilker et al. 2009; Han et al. 2023)gyrBGyrBSupercoils DNA, facilitating replication, transcription, and repairDNA integrity maintenanceFacilitates adaptation to genomic stress and DNA repair processesMutations confer FQ resistance, enabling survival in AM environments(Yamamoto and Harayama 1995; Champoux 2001; Cheng and Currie 2005)lepALepAFacilitates ribosome movement during protein synthesisCrucial for protein synthesis under environmental stressFacilitates efficient protein production, essential for survival under nutrient scarcityAssociated with virulence processes and AMT resistance, aiding host stress adaptation(Manno et al. 2004)phaCPhaCCatalyzes the polymerization of 3-hydroxyalkanoates into PHACrucial for carbon storage under nutrient stressEnables adaptation to environments with variable carbon sourcesFacilitates colonization of diverse ecological niches and is explored in biotechnology for bioplastics(Oliveira-Filho et al. 2022)recARecADNA repair and HRCrucial for genomic stress response, enabling DNA strand ExchangeCrucial for stress adaptation and DNA damage repairContributes to AMR and survival in antimicrobial environments(Roca and Cox, 1990; Alam et al. 2016)trpBTrpBCatalyzes conversion of IGP to TrpCritical for tryptophan biosynthesis, an essential amino acidEnables survival in environments with limited tryptophan sourcesContributes to host immune response modulation and persistence in environments like CF patients' lungs(Rhodes and Schweizer 2016)AM, antimicrobial; AMR, antimicrobial resistance; AMT, antimicrobial therapy; ATPβ, ATP synthase β subunit; CF, cystic fibrosis; FQ, fluoroquinolones; Gln, glutamine; GltB, NADPH-dependent glutamate synthase large subunit; Glu, glutamate; GyrB, DNA gyrase B subunit; HR, homologous recombination; IGP, indole-3-glycerol phosphate; LepA, GTPase involved in ribosome translocation; OXPHOS, oxidative phosphorylation; PHA, polyhydroxyalkanoates; PhaC, polyhydroxyalkanoate synthase (PHA synthase); RecA, RecA protein (recombinase); Trp, tryptophan; TrpB, tryptophan synthase β subunit; α-KG, alpha-ketoglutarate
Data generated by MLST are extensively used in epidemiological surveillance, analysis of the global dissemination of bacterial strains, and investigation of antimicrobial resistance mechanisms. Allelic characterization enables tracking the occurrence and persistence of specific lineages across different geographic regions and over time, contributing to the understanding of pathogen evolution and dissemination dynamics (Maiden 2006; Maiden et al. 2013).
In this context, the importance of the PubMLST repository (https://pubmlst.org/) is particularly noteworthy, as it stores allelic profiles, gene sequences, and associated metadata. The platform also offers bioinformatics tools for phylogenetic analysis and visualization of evolutionary and epidemiological relationships among strains. The use of PubMLST has been fundamental in identifying global dissemination patterns, conducting biodiversity studies, and strengthening collaborative public health initiatives (Maiden 2006; Jolley et al. 2012, 2018).
atpD gene
The atpD gene encodes the β subunit of ATP synthase, an essential enzyme located in the cytoplasmic membrane of bacteria from the genus Burkholderia, including species within the Bcc (Table 2). This enzyme plays a fundamental role in the synthesis of adenosine triphosphate (ATP) from adenosine diphosphate (ADP) and inorganic phosphate, utilizing the proton gradient generated by the electron transport chain during oxidative phosphorylation, a process vital for cellular bioenergetics (Zulkefli et al. 2016; Nakano et al. 2023; UniProt Consortium 2025).
In pathogenic lineages of the Bcc, ATP synthase is critical for bacterial survival under hostile conditions, such as environments with low oxygen availability. Studies have demonstrated that B. cenocepacia is capable of growing under microaerophilic conditions, with oxygen concentrations ranging from 0.1 to 5%, levels comparable to those found in the thick mucus of CF patients. Under these conditions, increased antibiotic resistance is observed, and sustained activity of ATP synthase is essential to maintain cellular viability (Pessi et al. 2013).
In this context, maintaining energy homeostasis through ATP synthase is crucial for metabolic adaptation and bacterial persistence in chronic infections, contributing to the high intrinsic antimicrobial resistance characteristic of Bcc species (Spilker et al. 2009). Additional evidence indicates that ATP synthase is also involved in oxidative stress responses and in the regulation of overall cellular homeostasis (Hamad et al. 2011).
The atpD gene is widely employed as a molecular marker in MLST schemes due to its high degree of conservation, low mutation rate, and functional importance. These characteristics confer high reliability for its use in phylogenetic and epidemiological studies, enabling species differentiation within the Bcc and facilitating the tracking of pathogenic lineage evolution (Baldwin et al. 2005; Zulkefli et al. 2016).
Furthermore, alterations in atpD expression or mutations within the gene can compromise the structure and functionality of ATP synthase, negatively affecting bacterial bioenergetic efficiency. Such changes are associated with resistance mechanisms targeting the respiratory chain (oxidative phosphorylation). The link between energy metabolism and antimicrobial resistance is particularly relevant in chronic infections, where metabolic adaptation supports bacterial persistence within the host (Spilker et al. 2009; Li et al. 2024).
Recent studies indicate that exposure of B. cepacia to agents such as bismuth salts can induce alterations in the expression of genes involved in the oxidative phosphorylation complex, including atpD, resulting in reduced ATP production and impaired metabolic competence. These findings suggest that modulation of ATP synthase may serve both as a mechanism of antimicrobial action and as part of bacterial tolerance strategies (Li et al. 2025a). Moreover, specific ATP synthase inhibitors, such as oligomycin, can trigger adaptive responses that may lead to cross-resistance or increased bacterial tolerance, particularly in biofilm-associated contexts and chronic infections, a phenomenon already described in various pathogens, including Burkholderia species (Mackieh et al. 2023; Li et al. 2025a).
gltB gene
The gltB gene encodes the large subunit of NADPH-dependent glutamate synthase (NADPH-GS), a key enzyme in bacterial nitrogen metabolism (Table 2). This enzyme catalyzes the conversion of glutamine and α-ketoglutarate into two molecules of glutamate, a critical step in amino acid biosynthesis, nitrogen assimilation, and the regulation of central metabolic pathways (Merrick and Edwards 1995; Han et al. 2023). By integrating nitrogen metabolism with the tricarboxylic acid (TCA) cycle, NADPH-GS activity enables the incorporation of inorganic nitrogen into organic compounds, thereby promoting the synthesis of proteins and other essential metabolites (Fortunato et al. 2023; Han et al. 2023).
In species such as B. cepacia and B. vietnamiensis, the gltB gene is associated with the ability to efficiently utilize various nitrogen sources, including ammonia, nitrate, and organic nitrogen compounds, in heterogeneous environments. This metabolic flexibility supports adaptation and colonization across diverse ecological niches, from soils rich in organic matter to hospital environments characterized by fluctuating nutrient availability (Sawana et al. 2014; Han et al. 2023; Liu et al. 2025) (Table 3).Table 3. Amplification and sequencing scheme of the seven housekeeping and auxiliary genes used in Burkholderia identification1. Multi Locus Sequence TypingNucleotide Sequence (5’-3’)Nucleotide Sequence (5’-3’)AmplificationThermal cycling conditionsSequencingGenesPrimersInitial DEN(Nº of cycles)DEN(Nº of cycles)ANN(Nº of cycles)EXT(Nº of cycles)Final EXT(Nº of cycles)PrimersAS (pb)References1301atpDF – GATCGTACAGTGCATCGG94 °C/2 min94 °C/1 min58 °C/1 min72 °C/2 min72 °C/5 minF – GTTCATCTGGCCGTACAC1.395[Baldwin et al. 2005]R – ATCGTGCCGACCATGTAGR – AACTGACGCTCGAAGTCCF – ATGAGTACTRCTGCTTTGGTAGAAGG95 °C/2 min94 °C/30 s56 °C/30 s72 °C/60 s72 °C/5 minSPA0.756[Spilker et al. 2009]R – CGTGAAACGGTAGATGTTGTCGgltBF – CGCTCGAAGATCAAGCAG94 °C/2 min94 °C/1 min58 °C/1 min72 °C/2 min72 °C/5 minF – CTTCTTCTTCGTCGCCGA4,704[Baldwin et al. 2005]R – GGGAACACCTTCACGAACR – TTGCCGACGTAGTCGTTGF – CTGCATCATGATGCGCAAGTG95 °C/2 min94 °C/30 s58 °C/30 s72 °C/60 s72 °C/5 minSPA0.652[Spilker et al. 2009]R – CTTGCCGCGGAARTCGTTGGgyrBF – CGACAACTCGATCGACGA94 °C/2 min94 °C/1 min58 °C/1 min72 °C/2 min72 °C/5 minF – ATCGTGATGACCGAGCTG2.475[Baldwin et al. 2005]R – GACAGCAGCTTGTCGTAGR – CGTTGTAGCTGTCGTTCCF – ACCGGTCTGCAYCACCTCGT95 °C/2 min94 °C/30 s60 °C/30 s72 °C/60 s72 °C/5 minSPA0.738[Spilker et al. 2009]R – YTCGTTGWARCTGTCGTTCCACTGClepAF – CGACGGCAAGGTCTACAA94 °C/2 min94 °C/1 min58 °C/1 min72 °C/2 min72 °C/5 minF – GGCATCAAGGAACTGACG1.794[Baldwin et al. 2005]R – AGCATGTCGACCTTCACGR – CTGCGGCATGTACAGGTTF – CTSATCATCGAYTCSTGGTTCG95 °C/2 min94 °C/30 s55 °C/30 s72 °C/60 s72 °C/5 minSPA0.975[Spilker et al. 2009]R – CGRTATTCCTTGAACTCGTARTCCphaCF – CTCAGCGAATTGCGTACG94 °C/2 min94 °C/1 min58 °C/1 min72 °C/2 min72 °C/5 minF – AGACGGCTTCAAGGTGGT0.741[Baldwin et al. 2005]R – CCGTTCAGCGAGAAGTCGR – ACACGGTGTTGACCGTCAF – GCACSAGYATYTGCCAGCG95 °C/2 min94 °C/30 s58 °C/30 s72 °C/60 s72 °C/5 minSPA0.525[Spilker et al. 2009]R – CCATSTCSGTRCCRATGTAGCCrecAF – GATAGCAAGAAGGGCTCC94 °C/2 min94 °C/1 min58 °C/1 min72 °C/2 min72 °C/5 minF – TGACCGCCGAGAAGAGCAA1.071[Baldwin et al. 2005]R – CTCTTCTTCGTCCATCGCCTC^C^R – GACCGAGTCGATGACGATF – AGGACGATTCATGGAAGAWAGC95 °C/2 min94 °C/30 s58 °C/30 s72 °C/60 s72 °C/5 minSPA0.704[Spilker et al. 2009]R – GACGCACYGAYGMRTAGAACTTtrpBF – GATCTACCTGAAGCGCGA94 °C/2 min94 °C/1 min58 °C/1 min72 °C/2 min72 °C/5 minF – CTGGGTCACGAACATGGA1.194[Baldwin et al. 2005]R – GTGTGCATGTCCTTGTCGR – CCGAATGCGTCTCGATGAF – CGCGYTTCGGVATGGARTG95 °C/2 min94 °C/30 s58 °C/30 s72 °C/60 s72 °C/5 minSPA0.787[Spilker et al. 2009]R – ACSGTRTGCATGTCCTTGTCG2. Auxiliary genes in identification1301hisAF – AGGACCCGGCGGCGAT95 °C/2 min95 °C/30 s67 °C/45 s72 °C/60 s72 °C/10 minSPA: A-442_for and A-442_rev0.442[Papaleo et al. 2010]R – TGCAGCATCCCGTCGCG1401groELNS94 °C/60 s55 °C/60 s72 °C/120 s72 °C/10 minSPA: LPW374_for and LPW377_rev1.641[Woo et al. 2002]F – AGAAGACATCATGGCCGTLPW375 – TTTTCACGGTCGTAGTCCR – ATTACATGTCCATGCCCALPW376 – TTCCAAGACCAGCGACAALPW400 – ATGGAAGAGCCGCTGCGC1351rpsUF – GTGGAGCTTCTTCGGCAGCAT94 °C/10 min94 °C/60 s59 °C/60 s72 °C/60 s72 °C/10 minSPA: fup-1_for and fup-2_rev0.210[Frickmann et al. 2012]R – ATGACGACGATTCTTTTGAA1301fur1.F – ATGACCAATCCGACCGATCTCAA96 °C/2 min96 °C/60 s55 °C/60 s72 °C/60 s72 °C/2 minSPA: JD490_for and JD491_rev0.429[Lynch and Dennis 2008]1.R – TCAGTGCTTGCGITNIGGGCAGTT2.F – GGCNGAAGACGTCTACCGG96 °C/2 min96 °C/60 s63 °C/60 s72 °C/60 s72 °C/2 minConventional PCR, rapid diagnostic analysis, not sequenced0.1172.R – TCGAAGTTGCTGCGCGAC3.F – CTAAAGGCCACCCTACCGCGG0.3963.R – TCAGTGCTTGCGGTGGGG4.F – AGCAGAGCCCCGTGCGG96 °C/2 min96 °C/60 s65 °C/60 s72 °C/60 s72 °C/2 min0.3424.R – GGTGGGGGCAGTTTTCGGTG5.F – TGACCAATCCGACCGATCTCA96 °C/2 min96 °C/60 s60 °C/60 s72 °C/60 s72 °C/2 min0.3375.R – ATCGCCTGCTGGCGGCTC6.F – CNACCGTCTATCGCGTGCTC0.2756.R – TCAGTGCTTGCGGTGGGG7.F – TGACCAATCCGACCGATCTCA0.2617.R – CGTGGTGGGAGCCTTCGTTG8.F – CCCNGTGCGTCACCTGACT0.1798.R – CGTGGTGCGAACCTTCATTCAA9.F – TGACCAATCCGACCGATCTCA0.0989.R – CAGGTGACGCACGGGGCTC10.F – GGCNGAAGACGTCTACCGG0.23710.R – ATCGCCTGCTGGCGGCC**(i)** For PCR reactions, the annealing temperature can be adjusted within a range of 2 to 5 °C above or below the primers’ melting temperature (Tm), providing greater flexibility for optimizing amplification conditions. (ii) The primers used in the MLST scheme described by Spilker et al. (2009) contained degenerate base codes (e.g., R, Y, S, W), which were strategically designed to ensure efficient amplification of various species within the Bcc. (iii) In sequencing the groEL gene, besides the primers used for amplification, three additional internal primers were employed during sequencing to ensure full coverage of the target region and to enhance the quality and continuity of the sequence reads obtained. (iv) For amplification of the fur gene, in addition to the universal primer pair (JD490_forward and JD491_reverse), capable of amplifying the gene in all Burkholderia species, Lynch and Dennis (2008) also used species-specific primers aimed at rapid and differential identification of complex members without the need for sequencing. These included: 2. Specific for B. cepacia; 3. Specific for B. dolosa; 4. Specific for B. multivorans; 5. Specific for B. cenocepacia (subgroups IIIA, IIIB, IIID); 6. Specific for B. stabilis, B. multivorans, B. cenocepacia, and B. dolosa; 7. Specific for B. vietnamiensis; 8. Specific for B. ambifaria; 9. Specific for B. anthina, B. multivorans, B. cenocepacia, B. stabilis, and B. pyrrocinia (one strain); 10. Specific for B. pyrrocinia. (v) Sequencing of the amplified products was conducted using the fluorescent dye terminator method (BigDye Terminator, PE Biosystems, Foster City, California, USA), as described by Baldwin et al. (2005). Although the exact thermal cycling conditions for the sequencing reactions were not specified by the authors, given the widespread use of the Sanger method with BigDye in studies by Spilker et al. (2009), Papaleo et al. (2010), Woo et al. (2002), Frickmann et al. (2012), and Lynch and Dennis (2008), it is presumed that the manufacturer’s recommended parameters were followed. These typically include: denaturation at 96 °C for 10 s; annealing between 50 °C and 55 °C for 5 s; and extension at 60 °C for 4 min, repeated over approximately 25 cycles.AS, amplicon size; F, forward; PCR, polymerase chain reaction; R, reverse; DEN, denaturation; ANN, annealing; EXT, extension; SPA, same primers as amplification
In clinical contexts, particularly in chronic lung infections associated with CF, gltB plays a central role in bacterial metabolic adaptation to the host environment. Its expression promotes the production of glutamate, which functions not only as a biosynthetic intermediate but also as a precursor for glutathione, a key antioxidant involved in cellular defense against oxidative stress (D'Orazio et al. 2012; Li et al. 2025a). This metabolic pathway contributes to bacterial resistance to adverse conditions such as hypoxia, acidosis, and nutrient limitation, hallmarks of the thick pulmonary mucus observed in CF patients (Spilker et al. 2009; Didelot et al. 2016).
Studies have demonstrated that NADPH-GS contributes to maintaining cellular redox balance in Burkholderia species such as B. thailandensis. The expression of gltB is regulated by redox-sensitive mechanisms, including the transcriptional regulator OhrR, which responds to oxidative stressors such as cumene hydroperoxide (CHP) by inducing the expression of antioxidant genes, including ohr. This redox regulation plays a crucial role in intrinsic antimicrobial resistance, particularly under conditions of high selective pressure, such as during prolonged antibiotic therapy (Pande et al. 2018).
gyrB gene
The gyrB gene encodes the B subunit of DNA gyrase, a type II topoisomerase essential for introducing negative supercoils into DNA, a critical topological modification required to maintain the structural and functional integrity of the bacterial genome (Champoux 2001). This enzymatic activity facilitates key cellular processes such as DNA replication, transcription, and repair. DNA gyrase is indispensable for bacterial survival and proliferation, particularly under adverse environmental and physiological conditions (Reece and Maxwell 1991; Collin et al. 2011). The enzyme operates by introducing transient double-strand breaks in the DNA, thereby relieving torsional stress and reorganizing genome topology. This process is ATP-dependent, with hydrolysis catalyzed by the Bergerat domain located in the GyrB subunit (Champoux 2001; Collin et al. 2011).
Mutations in gyrB are frequently associated with bacterial resistance to topoisomerase-targeting antibiotics, particularly fluoroquinolones. In B. pseudomallei, evidence indicates that alterations within the fluoroquinolone-binding site of GyrB contribute to intrinsic resistance by reducing drug affinity and compromising therapeutic efficacy. Such mutations interfere with the stabilization of the DNA–gyrase–fluoroquinolone complex, diminishing enzymatic inhibition and enhancing bacterial survival. This resistance mechanism is especially relevant in chronic infections, where selective pressure promotes the persistence of resistant strains (Cheng and Currie 2005; Malik et al. 2012).
Beyond its role in antimicrobial resistance, gyrB is widely employed as a molecular marker for the identification and characterization of Burkholderia species. Due to its higher nucleotide variability compared to the 16S rRNA gene, gyrB sequencing enables the discrimination of closely related species, such as B. cepacia and B. vietnamiensis (Tabacchioni et al. 2008). Phylogenetic analyses show that gyrB sequences cluster isolates into groups corresponding to distinct Bcc species, providing superior taxonomic resolution compared to 16S rRNA sequencing-based methods (Yamamoto and Harayama 1995; Spilker et al. 2009). This approach is particularly valuable in clinical settings, where accurate pathogen identification is essential for guiding therapeutic decisions (Cheng and Currie 2005).
Evidence also suggests that gyrB contributes to the adaptive capacity of Burkholderia in hostile microenvironments, such as the respiratory tract of CF patients, characterized by chronic hypoxia, inflammation, and continuous antimicrobial exposure. Modulation of DNA supercoiling allows the bacterium to rapidly adjust gene expression in response to these environmental stresses (Spilker et al. 2009; Mira et al. 2011). This genetic plasticity, the ability to reorganize transcriptional profiles in response to external stimuli, favors bacterial persistence and the progression of chronic infections, reinforcing the functional importance of gyrB in Burkholderia pathogenicity (Cheng and Currie 2005; Mira et al. 2011).
lepA gene
The lepA gene encodes the LepA protein, a highly conserved GTPase that plays a fundamental role in regulating translation in bacterial cells. LepA associates with the ribosome and acts as a proofreading factor by reversing incorrect translocations during polypeptide chain elongation. This process, known as back-translocation, ensures the fidelity of messenger RNA (mRNA) reading by properly repositioning the ribosome, utilizing energy derived from guanosine triphosphate (GTP) hydrolysis (Manno et al. 2004; Qin et al. 2006; Shoji et al. 2010).
Studies indicate that LepA, also referred to as elongation factor 4 (EF4), contributes to translational quality control and can rescue mispositioned ribosomes, thereby promoting efficient protein synthesis even under cellular stress conditions (Youngman and Green 2007; Shoji et al. 2010). Deletion of lepA increases bacterial susceptibility to stressors such as tetraphenylphosphonium and certain antibiotics, suggesting its involvement in adaptation to adverse conditions (Shoji et al. 2010).
In Burkholderia species, the LepA protein contributes significantly to metabolic adaptation by facilitating efficient protein translation under restrictive environmental conditions. This function is particularly relevant in oligotrophic environments, such as the host milieu, where maintaining high-fidelity protein synthesis is essential for bacterial survival and niche colonization. Beyond ribosomal regulation, LepA is involved in cellular responses to abiotic stresses, including antibiotic exposure and nutrient limitation, preserving translational efficiency during physiological challenges (Manno et al. 2004; Estrada-de los Santos et al. 2013).
Additional evidence suggests that this GTPase participates in rapid adaptive responses mediated by riboswitches and short-latency regulatory circuits, commonly observed in bacteria adapted to resource-limited environments (Estrada-de los Santos et al. 2018). The presence of such mechanisms in environmental and symbiotic lineages of the Burkholderia s.l. complex further reinforces the role of LepA in evolutionary adaptation to diverse microbial lifestyles (Manno et al. 2004; Estrada-de los Santos et al. 2013).
In pathogenic Bcc species, lepA expression has been linked to mechanisms of virulence and antimicrobial resistance. LepA promotes the production of factors involved in cell adhesion, biofilm formation, and immune evasion, critical processes in chronic pulmonary infections, such as those observed in CF patients. Furthermore, evidence suggests that LepA contributes to tolerance against translation-targeting antibiotics, such as aminoglycosides, by maintaining translational fidelity even under chemical and physical stress (Manno et al. 2004; Fasnacht and Polacek 2021).
phaC gene
In dozens of Burkholderia s.l. genomes, including B. cepacia and B. thailandensis, the phaC gene is present in one or more copies, typically arranged in operons such as phaCABR, which encode PHA synthase (PhaC), β-ketothiolase (PhaA), acetoacetyl-CoA reductase (PhaB), and phasin (PhaP). These operonic configurations reflect an adaptive and versatile metabolic organization, consistent with the broad capacity of these bacteria to synthesize polyhydroxyalkanoates (PHAs) from sugars or fatty acids via multiple pathways, including Entner-Doudoroff, pentose phosphate, and β-oxidation (Alvarez-Santullano et al. 2021).
The PHA synthase enzyme, encoded by phaC, catalyzes the polymerization of 3-hydroxyalkanoate (3-HA) monomers into PHAs, which can accumulate as homopolymers, such as poly(3-hydroxybutyrate) (PHB), or as copolymers such as P(3HB-co-3HV) or P(3HB-co-4HB), depending on substrate availability and the specific class of synthase involved. In Burkholderia, PHA accumulation occurs predominantly under conditions of carbon excess combined with nitrogen, phosphorus, or oxygen limitation, an adaptive mechanism that enables cells to store energy and carbon reserves for survival under stress (Rodrigues et al. 2000; Alvarez-Santullano et al. 2021). PHA production by Burkholderia s.l. strains, such as B. cepacia ATCC 17759, B. thailandensis E264^T^, and Paraburkholderia sacchari LMG 19450^ T^ (preveolsy B. sacchari), has been extensively documented, with reported PHB yields of up to 7.5 g/L from waste oils, as well as various copolymers produced from co-substrates such as odd-chain fatty acids, levulinic acid, or γ-butyrolactone (Alvarez-Santullano et al. 2021).
Beyond its ecological significance, the phaC gene holds considerable biotechnological relevance, as its modulation enables the tuning of polymer physicochemical properties, including flexibility, biodegradability, and thermal resistance. This versatility is further enhanced by the functional diversity of PHA synthases in Burkholderia s.l., which are primarily classified into classes I, III, and IV. These enzymes act on short-chain-length (scl-PHAs) and medium-chain-length (mcl-PHAs) monomers, supporting the production of biopolymers with diverse industrial applications (Alvarez-Santullano et al. 2021; Oh et al. 2025).
The modulation of phaC has been a major focus of metabolic engineering strategies aimed at maximizing PHA production. For instance, the Burkholderia Oh_219 strain was genetically modified with a phaC variant combined with the phaJ gene from P. aeruginosa, enabling efficient production of the copolymer P(3HB-co-3HHx) from crude glycerol, with yields reaching 15.3 g/L under fed-batch fermentation conditions. This underscores the potential of Burkholderia-based chassis for sustainable bioplastic production (Oh et al. 2025).
Furthermore, advances in the use of low-cost substrates, such as agro-industrial residues and crude glycerol, are improving the economic feasibility of industrial-scale PHA production, increasing its competitiveness relative to conventional petroleum-based polymers (Garcia et al. 2013; Mai et al. 2024).
recA gene
The recA gene encodes the RecA protein, a highly conserved recombinase essential for DNA damage repair, homologous recombination, and the preservation of genome integrity (Table 2). In response to genotoxic lesions, such as those induced by ultraviolet (UV) radiation or alkylating agents, RecA mediates homologous strand pairing and strand exchange, thereby activating the SOS (save our souls) response (Roca and Cox, 1990; Maslowska et al. 2019). This coordinated pathway induces expression of DNA repair genes, transiently inhibits cell division, and increases tolerance to genetic stress (Maslowska et al. 2019; Podlesek and Žgur Bertok 2020).
During the SOS response, RecA assembles into nucleoprotein filaments on single-stranded DNA (ssDNA), functioning as a coprotease that stimulates LexA repressor autoproteolysis. Cleavage of LexA derepresses ~ 50 genes involved in survival under DNA damage conditions, including translesion DNA polymerases (Maslowska et al. 2019; del Val et al. 2021). Additionally, RecA acts as a molecular motor to restore stalled replication forks, a critical function under stresses such as UV exposure, reactive oxygen species, or intercalating antibiotics (del Val et al. 2021).
In bacteria of the genus Burkholderia, particularly B. cepacia, the RecA protein plays a pivotal role in survival under hostile environmental conditions. Studies involving recA mutants of the G4 strain demonstrated a significant loss of viability following exposure to UV radiation, hydrogen peroxide, and chlorinated compounds, thereby confirming the reliance on RecA-mediated homologous recombination for the effective repair of damaged DNA (Yeager et al. 2001). Additionally, evidence indicates that RecA also regulates genes involved in bacterial virulence and persistence, further underscoring its multifunctional role in the pathogenicity of this genus (Roca and Cox, 1990).
Activation of the recA gene is likewise critical in the bacterial response to genotoxic antibiotics such as fluoroquinolones. In B. thailandensis, recA deletion resulted in a fourfold increase in susceptibility to ciprofloxacin (Ulrich et al. 2013; Mercolino et al. 2022). Similarly, inactivating mutations in recA in other bacterial species markedly enhance the activity of fluoroquinolones, positioning the RecA protein as a potential target for adjuvant therapies. For instance, studies with Escherichia coli demonstrated that recA deletion, when combined with inhibition of redox detoxification genes, can increase ciprofloxacin susceptibility by up to 30-fold (Diaz-Diaz et al. 2021).
A study published by Alam et al. (2016) reinforces the protective role of RecA, demonstrating that specific inhibitors of this recombinase, such as phthalocyanine tetrasulfonate (PcTs) derivatives, can block the induction of the SOS response triggered by bactericidal antibiotics, thereby enhancing their efficacy and preventing both the acquisition of resistance mutations and the horizontal transfer of mobile genetic elements (Alam et al. 2016).
RecA also plays a fundamental role in horizontal gene transfer (HGT). By mediating homologous recombination, it facilitates the stable integration of exogenous DNA into the bacterial genome, promoting genetic variability and enhancing adaptive potential. The SOS response mediated by RecA stimulates not only mutagenesis but also the mobilization of mobile genetic elements, including integrons and conjugative plasmids (Alam et al. 2016; Sun 2018; Yakimov et al. 2021). In clinical settings characterized by intense antimicrobial pressure, such as hospitals, this activity fosters the dissemination of resistance genes, ultimately compromising the effectiveness of conventional therapies (von Wintersdorff et al. 2016).
trpB gene
The trpB gene encodes the β subunit of the enzyme tryptophan synthase, which catalyzes the final step in the biosynthesis of tryptophan, an essential amino acid in higher organisms (Table 2). This enzyme facilitates the conversion of indole-3-glycerol phosphate (IGP) into L-tryptophan, utilizing precursors derived from the tricarboxylic acid (TCA) cycle (Buller et al. 2016). Beyond its critical role in protein synthesis, tryptophan serves as a precursor for several bioactive molecules, including serotonin, melatonin, and metabolites of the kynurenine pathway, all of which are involved in immunological, neurophysiological, and redox processes (Gupta et al. 2023).
In species of the genus Burkholderia, the capacity to synthesize tryptophan provides a significant adaptive advantage, particularly in oligotrophic environments such as nutrient-poor soils. Species like B. cepacia and B. vietnamiensis rely on the trpB-mediated pathway to enhance their ecological competitiveness (Liu et al. 2019). Moreover, tryptophan biosynthesis is frequently linked to the production of indole-derived secondary metabolites, such as indole-3-acetic acid (IAA), which function as signaling molecules or defense agents in microbial interactions, particularly in response to antimicrobials (Li 2023).
In clinical contexts, trpB is associated with the pathophysiology of Bcc species, especially in chronic pulmonary infections affecting patients with CF. The production of tryptophan and its derivatives contribute to modulation of the host immune response, thereby facilitating bacterial persistence (Rhodes and Schweizer 2016). Metabolites of the kynurenine pathway, such as quinolinic acid and 3-hydroxykynurenine, can suppress T-lymphocyte responses and modulate macrophage activity, promoting an immunosuppressive microenvironment within the lungs (Pamart et al. 2024).
Activation of the indoleamine 2,3-dioxygenase (IDO) pathway in pulmonary inflammatory cells further contributes to the accumulation of these immunomodulatory metabolites, a mechanism exploited by Burkholderia species to sustain chronic infections and evade adaptive immune responses (Viberg et al. 2017). Simultaneously, the tryptophan pathway also plays a role in the oxidative stress response, commonly encountered during chronic infections. In B. thailandensis, exposure to β-lactam antibiotics such as piperacillin induces oxidative stress and triggers the expression of biosynthetic genes, including those involved in the production of tryptophan derivatives as part of an adaptive response (Li et al. 2021).
Owing to its multifunctional roles, ranging from basic amino acid biosynthesis to the regulation of virulence and immune evasion, the trpB gene emerges as a promising target for novel therapeutic strategies. Inhibitors of tryptophan synthase have the potential to disrupt the synthesis of tryptophan and its downstream metabolites, thereby attenuating the virulence and antimicrobial resistance of opportunistic pathogens (Buller et al. 2016; Michalska et al. 2019).
Auxiliary genes employed in bacterial identification
Precise identification of species within the Bcc presents a significant challenge due to high intraspecies genetic diversity and pronounced interspecies phenotypic similarity, factors that compromise the effectiveness of conventional biochemical methods, even when automated (Medina-Pascual et al. 2012; Devanga Ragupathi and Veeraraghavan 2019).
As an alternative, molecular analyses based on sequencing of the 16S and 23S rRNA genes have been widely employed for phylogenetic characterization. However, this approach offers limited resolution for discriminating closely related species within the Bcc, owing to the low nucleotide variability in these highly conserved regions (Vandamme and Dawyndt 2011; Scoffone et al. 2021).
To overcome these limitations, multigene approaches have demonstrated greater effectiveness. Combining ribosomal genes with additional molecular markers, such as gyrB, recA, hisA, and rpsU, provides enhanced taxonomic resolution among species within the complex. For instance, sequencing of the recA gene allows for accurate differentiation of up to 19 Burkholderia species, including distinct genotypes within the Bcc (Devanga Ragupathi and Veeraraghavan 2019; Mahenthiralingam et al. 2000).
Studies have shown that hisA is particularly useful for distinguishing Bcc species based on specific sequence variations, while rpsU exhibits strong accuracy in differentiating Burkholderia from related genera, albeit with lower discriminatory power among species within the Bcc. The combined use of these molecular targets constitutes a robust strategy for species identification (Devanga Ragupathi and Veeraraghavan 2019).
recA gene (housekeeping)
The recA gene is widely used as a molecular marker in phylogenetic studies of the genus Burkholderia, including species within the Bcc, due to its high taxonomic resolution capacity (Mahenthiralingam et al. 2005; Payne et al. 2005; Estrada de los Santos et al. 2013; Depoorter et al. 2020; Aguiar et al. 2025). Although it retains its essential biological function, recA exhibits sufficient interspecies variability to accurately distinguish closely related species, making it particularly valuable for taxonomic, epidemiological, and clinical applications (Eisen et al. 1995; Karlin et al. 1995; Mahenthiralingam et al. 2000).
Studies have shown that recA sequence analysis enables species-level discrimination of up to 65% of clinical Bcc isolates, substantially contributing to epidemiological investigations (Cesarini et al. 2009). Comparisons with 16S rRNA sequencing indicate that recA provides superior phylogenetic resolution, allowing the differentiation of all Bcc members through sequence-based approaches (Mahenthiralingam et al. 2000; Payne et al. 2005).
A pertinent example of this applicability was provided by Wong et al. (2020), who used recA sequencing to accurately identify B. contaminans ST102 in clinical isolates from diverse sources, including pulmonary, renal, ocular, and soft tissue infections, as well as from medical devices. Accurate species identification in this case underscored the efficacy of recA as a discriminative molecular marker (Wong et al. 2020).
Furthermore, recA sequencing has enabled the tracking of the wide geographic distribution of the B. contaminans Sequence Type (ST) 102 lineages, which has been documented in both CF and non-CF patients across the European Union, the United States of America (USA), Russia, and China, as recorded in the PubMLST database (Savi et al. 2019; Wong et al. 2020). This molecular marker has proven essential for monitoring clonal dissemination and elucidating the evolutionary dynamics of Bcc species (Araujo et al. 2016; Nunvar et al. 2016; Wong et al. 2020).
The importance of recA was also highlighted during the multistate outbreak of bloodstream infections caused by Bcc in the USA between September 2016 and January 2017. The outbreak affected 162 patients across 59 long-term care facilities and was traced to contamination of prefilled syringes containing sterile saline produced by a single manufacturer. Given the high genetic similarity among Bcc species, analyses incorporating recA sequencing and WGS were necessary to identify B. cepacia and B. arboris as the etiologic agents. The investigation also revealed multiple clones associated with different geographic locations, underscoring the critical importance of high-resolution molecular tools for outbreak tracking (Brooks et al. 2019).
Complementary methods such as SNaPBceBcon (SNaP, Single Nucleotide Polymorphism; Bce, Burkholderia cepacia complex; Bcon, Burkholderia-conserved), developed by Araujo et al. (2016), have also demonstrated high accuracy in distinguishing B. cepacia from B. contaminans. This assay, based on single nucleotide polymorphisms (SNPs) selected from MLST data, achieved a discrimination index of 0.94 and was validated using clinical and environmental isolates collected over two decades. The combined characterization of isolates using recA profiles and SNaPBceBcon enabled the identification of clonally recurring lineages and highlighted the microevolutionary adaptation of variants during chronic infections (Araujo et al. 2016).
Therefore, the use of recA has been firmly established as a robust and versatile tool for molecular studies within the Bcc, with direct implications for epidemiological surveillance, outbreak control, and clinical diagnostics. Its adoption is particularly relevant in scenarios where traditional methods fail to provide adequate discrimination among highly related species (Araujo et al. 2016; Wong et al. 2020).
hisA gene
The hisA gene encodes the enzyme 1-(5′-phosphoribosyl)-5-[(5′-phosphoribosylamino)methyleneamino] imidazole-4-carboxamide isomerase, which plays an essential role in the histidine biosynthetic pathway. This enzyme catalyzes a critical intermediate step, converting phosphoribosyl pyrophosphate (PRPP) into imidazole glycerol phosphate (IGP), thereby contributing to the formation of the imidazole ring in amino acids. Although functionally conserved across bacterial species, the nucleotide sequence of hisA exhibits sufficient interspecies variability to serve as a valuable molecular marker in phylogenetic studies, particularly within the genus Burkholderia (Mahenthiralingam et al. 2005; Papaleo et al. 2010; Scoffone et al. 2021).
Within the Bcc, hisA has become established as a complementary marker to genes widely used in MLST, such as gyrB and recA, offering enhanced taxonomic resolution and improved refinement in epidemiological surveillance (Baldwin et al. 2005). Phylogenomic studies have demonstrated that hisA possesses sufficient variability to distinguish closely related taxa within the Bcc, making it highly useful for identifying clinically relevant species (Spilker et al. 2009; Papaleo et al. 2010; Jin et al. 2020).
Papaleo et al. (2010) evaluated the utility of hisA for identifying Bcc species by sequencing a 442 bp fragment in 78 strains representing the 17 known species at the time. Phylogenetic analyses using Neighbor-Joining and Maximum Likelihood methods demonstrated that this short gene fragment was sufficient to separate all complex species, including the four B. cenocepacia lineages (IIIA–IIID), with high phylogenetic consistency. Notably, the study also developed an 11-nucleotide SNP code that enables rapid and accurate species discrimination without the need for constructing phylogenetic trees (Papaleo et al. 2010).
Jin et al. (2020) expanded on this approach by analyzing both recA and hisA genes in 116 genomes representing 17 Bcc species. The hisA gene demonstrated high discriminatory power, correctly identifying 115 of 116 isolates (99.1% success rate), with the single exception attributed to genome quality or incomplete annotation. Discrepancies observed between the recA and hisA phylogenetic tree topologies suggest that recombination and polymorphisms play important roles in the evolutionary dynamics of these markers, further underscoring the value of multilocus approaches for robust phylogenetic analyses (Jin et al. 2020).
In addition to its taxonomic relevance, hisA also holds important ecological implications. Although the histidine biosynthesis pathway is energetically costly, requiring approximately 41 ATP molecules per histidine synthesized, it provides adaptive advantages to Burkholderia strains inhabiting oligotrophic environments such as soil and the rhizosphere, where free amino acids are scarce. These adaptations may contribute to the genus’s capacity to colonize environmental niches while also acting as opportunistic human pathogens (Coenye et al. 2001; del Duca et al. 2021).
More recently, Li et al. (2025b) reinforced the applicability of hisA as a diagnostic target by demonstrating its effectiveness in next-generation sequencing (NGS)-based platforms. The genetic variability of this gene enables tracking of epidemic strains and enhances molecular identification methods, with direct implications for the surveillance of hospital- and community-acquired infections caused by Bcc species (Li et al. 2025b).
Thus, the study of hisA extends beyond phylogenetic classification, providing valuable insights into microbial ecology, metabolic evolution, and the adaptive dynamics of Burkholderia in challenging niches. Its combination with other genetic markers enhances taxonomic resolution and represents a promising strategy for both scientific research and clinical and environmental applications (Coenye et al. 2001).
groEL gene
The groEL gene encodes the GroEL chaperone protein, which plays a critical role in proper protein folding and the maintenance of cellular homeostasis. In addition to its essential biological function, groEL has been widely explored as a molecular marker due to its genetic variability among bacterial species, particularly within the Bcc (Woo et al. 2002; Suppiah et al. 2010).
In contrast to the highly conserved 16S rRNA gene, whose low variability limits its discriminatory power among closely related species, the groEL gene exhibits numerous SNPs and insertion/deletion events, providing higher phylogenetic resolution (Mahenthiralingam et al. 2000; Woo et al. 2002). These characteristics support its application in species differentiation within the Bcc and in the construction of robust phylogenetic trees, with direct implications for taxonomy, epidemiological surveillance, and the understanding of ecological adaptation in pathogenic lineages (Scales et al. 2022).
The superiority of groEL over 16S rRNA has been demonstrated in studies involving both environmental and clinical strains, in which accurate species-level identification within the Bcc is essential for infection diagnosis and control (Mahenthiralingam et al. 2000). A comparative study involving B. thailandensis and B. pseudomallei using the Basic Local Alignment Search Tool (BLAST) for nucleotide sequence (BLASTn) and amino acid sequence (BLASTp) for analyses, revealed protein identity levels above 99% between GroEL proteins from these species and other members of the Burkholderia genus, including B. pseudomallei, B. mallei, B. cepacia, B. vietnamiensis, and B. fungorum. Notably, the GroEL protein sequence of B. thailandensis shared more than 99.5% identity with its homolog in B. pseudomallei, highlighting a high degree of protein structural conservation between these species. However, analysis of the nucleotide sequences of groEL revealed greater discriminatory power: B. thailandensis strains exhibited intraspecies identity above 99.8%, while B. pseudomallei strains showed over 99.5% identity; in contrast, identity between the two species dropped to less than 97.6%. Thus, despite the functional conservation of the GroEL protein, DNA-level differences are sufficient to differentiate the species. These findings reinforce the effectiveness of groEL as a molecular marker for distinguishing closely related species, particularly in cases where more conserved genes, such as 16S rRNA, lack sufficient resolution (Woo et al. 2002).
Recent studies have also demonstrated the use of groEL in clinical settings. Periaiah et al. (2024) evaluated 60 clinical isolates resistant to polymyxin B, cultured on MacConkey agar, using three identification methods: polymerase chain reaction (PCR) targeting groEL, semi-automated systems (VITEK® 2 and VITEK® MS), and conventional biochemical tests. All methods showed complete concordance at the genus level (Burkholderia); however, significant discrepancies were observed at the species level. Conventional phenotypic tests correctly identified only 28.2% of the isolates at the species level, whereas both groEL-PCR and the automated systems demonstrated substantially higher accuracy (Periaiah et al. 2024).
The integration of groEL sequencing with proteomic methods such as MALDI-TOF MS has proven effective in enhancing and accelerating the identification of Bcc species. This combined approach helps to overcome the individual limitations of each technique, thereby improving diagnostic reliability and turnaround time, particularly in high-risk hospital settings (Mahenthiralingam et al. 2000; Vianna et al. 2021).
rpsU gene
The rpsU gene, which encodes the ribosomal protein S21 of the 30S ribosomal subunit, is highly conserved among Gram-negative bacteria. Nevertheless, it exhibits sufficient variability to serve as a molecular marker for identifying the Burkholderia genus in both clinical and environmental samples, particularly in early-stage molecular screening strategies. PCR assays targeting rpsU have demonstrated high specificity, making them especially valuable in contexts requiring rapid pathogen detection, such as hospital outbreaks and respiratory infections in patients with CF (Frickmann et al. 2014; Ostermann et al. 2016).
The application of the rpsU gene has proven useful in both epidemiological outbreak investigations and hospital-based microbiological diagnostics. By sequencing fragments of this gene, it is possible to distinguish the Burkholderia genus from other phylogenetically related taxa, such as Ralstonia, Cupriavidus, and Pandoraea, which are frequently misidentified by conventional phenotypic methods. Additionally, rpsU enables the identification of clinically relevant phylogenetic clusters, including species of particular clinical concern, such as B. pseudomallei and B. mallei, both members of the B. pseudomallei complex (Bpc). Accurate differentiation of these pathogens is critical for both clinical management and biosafety (Frickmann et al. 2012, 2014).
In a comprehensive study, Frickmann et al. (2014) analyzed 84 reference strains from various Burkholderia species, originating from several international collections (ATCC, DSMZ, JCM, LMG, and NCTC) and clinical samples. Sequencing of the rpsU gene revealed four main phylogenetic clusters: rpsU-I, grouping species such as B. plantarii, B. glumae, B. cocovenenans, and B. gladioli; rpsU-II, corresponding to the Bpc, which included B. mallei, B. pseudomallei, and B. thailandensis; rpsU-III, the largest and most heterogeneous cluster, encompassing most clinically relevant Bcc species, such as B. cepacia, B. multivorans, B. cenocepacia, B. vietnamiensis, B. ambifaria, B. anthina, B. pyrrocinia, B. dolosa, among others; and rpsU-IV, composed of environmental species such as B. sacchari, B. xenovorans, B. fungorum, and B. phymatum. Intra-cluster sequence homology exceeded 92%, demonstrating the strong phylogenetic clustering potential of rpsU at both genus and subgenus levels (Frickmann et al. 2014).
However, despite its phylogenetic potential and its ability to differentiate between complexes and distinct genera, the rpsU gene exhibits significant limitations in resolving closely related species within the Bcc. Specifically, B. cenocepacia and B. multivorans displayed 100% sequence homology in their rpsU genes, indicating a lack of discriminatory polymorphisms and rendering species differentiation impossible when relying on this marker alone. Similarly, B. cepacia and B. cenocepacia clustered within rpsU-III, further compromising the accuracy of species-level identification based solely on rpsU (Frickmann et al. 2014).
Given these limitations, the use of multilocus approaches has been widely recommended. Combining rpsU with genes such as recA and hisA, which exhibit greater interspecific variability, has proven effective for enhancing taxonomic resolution within the Bcc. While rpsU is particularly suited for broad screening and genus-level differentiation, recA and hisA are crucial for the precise discrimination of clinically important species such as B. cepacia, B. contaminans, B. ambifaria, and B. vietnamiensis (Frickmann et al. 2012, 2014; WHO, 2018).
fur gene
The fur gene (ferric uptake regulator) has been extensively investigated as a molecular marker within the genus Burkholderia due to its essential role in iron homeostasis, an element critical to bacterial metabolism, virulence, and resistance to oxidative stress (Andrews et al. 2003; Troxell and Hassan 2013; Schmidt et al. 2018). Although functionally conserved across various Gram-negative bacteria, fur exhibits sufficient sequence variability to enable the distinction of species or phylogenetic complexes within Burkholderia, offering, in this regard, greater discriminatory power than traditionally used genes such as 16S rRNA (Roca and Cox, 1990; Lynch and Dennis 2008; Butt and Thomas 2017).
The high conservation of ribosomal genes such as 16S rRNA among species within the Bcc limits their applicability for taxonomic and diagnostic purposes. In this context, fur gene sequencing has demonstrated superior performance, particularly for clinically significant species such as B. cenocepacia and B. multivorans, which are notorious for causing severe infections in patients with CF and immunocompromised individuals (Lynch and Dennis 2008; Wong et al. 2020).
In the study conducted by Lynch and Dennis (2008), clinical and environmental isolates representing various Burkholderia species were analyzed using PCR with fur-specific primers, followed by multiple sequence alignment with ClustalW and phylogenetic tree construction using the Molecular Evolutionary Genetics Analysis (MEGA) software. The results showed that fur gene-based phylogenetic trees produced topologies more consistent with the recognized taxonomy of the Burkholderia genus than those based on 16S rRNA gene sequencing. Species within both the Bcc and Bpc were correctly grouped into well-supported clades, enabling the accurate differentiation of closely related species. This proved particularly relevant for the identification of B. pseudomallei, the etiologic agent of melioidosis, and B. cenocepacia, which is frequently associated with poor clinical outcomes in CF patients (Lynch and Dennis 2008).
However, the use of fur as a standalone marker is not without limitations. Studies have shown that sequence similarity with homologous genes from other genera, particularly within the order Burkholderiales, can compromise specificity in complex microbiota environments (Lynch and Dennis 2008). To mitigate this limitation, the adoption of multigene approaches is recommended, combining fur with markers such as recA, hisA, and groEL. Alternatively, the combination with MALDI-TOF MS may be employed to enhance identification accuracy and reduce false-positive rates (Gautam et al. 2017; Fergusson et al. 2020).
Mass Spectrometry in bacterial identification
Recently, automated MALDI-TOF MS, implemented in platforms such as MALDI Biotyper® and VITEK® MS Prime, has become a rapid, sensitive, and cost-effective tool for bacterial identification in clinical laboratories. This technique is based on the analysis of ribosomal proteins and chaperones (heat shock proteins), enabling reliable genus-level identification and, species-level discrimination, particularly for bacteria with distinctive proteomic profiles. However, in groups with high genomic similarity, such as the Bcc, overlapping spectral profiles between closely related species may occur, reducing resolution (Depoorter et al. 2020; Fergusson et al. 2020; Wong et al. 2020).
The platform’s software generates a confidence score based on the spectral similarity between the analyzed isolate’s profile and the spectra present in the library (Seng et al. 2013; MALDI-TOF Bruker Daltonics, 2023 ). In the Bruker Biotyper® standard, the score scale is interpreted as follows: values equal to or greater than 2.0 indicate reliable identification up to the species level; values between 1.7 and 1.99 indicate reliable identification only up to the genus level; and values below 1.7 are considered insufficient for reliable identification (Fehlberg et al. 2013; Seng et al. 2013; MALDI-TOF Bruker Daltonics, 2023). This classification is essential for the critical evaluation of identification results obtained in the studies discussed here, as it directly impacts accuracy at different taxonomic levels (Patel 2015).
Recent studies report accuracy rates ranging from 97 to 100% for Burkholderia genus identification by MALDI-TOF MS. However, species-level accuracy varies considerably (23–83%), depending on the platform (MALDI Biotyper® or VITEK® MS) and the breadth and curation quality of the spectral libraries employed (Gautam et al. 2017; Wong et al. 2020).
The VITEK® MS PRIME V3.3 system (bioMérieux), with a database updated in 2023, contains 4,320 microbial spectra, including 20 Burkholderia species: B. ambifaria, B. anthina, B. arboris, B. cenocepacia, B. cepacia, B. contaminans, B. diffusa, B. dolosa, B. gladioli, B. lata, B. latens, B. mallei, B. metallica, B. multivorans, B. pseudomallei, B. pyrrocinia, B. stabilis, B. thailandensis, B. ubonensis, and B. vietnamiensis. Notable environmental and pathogenic species include B. pseudomallei and B. mallei, both classified as biosafety level 3 (BSL-3) agents. According to the manufacturer’s recommendations, identification of these species mandates immediate referral of isolates to reference laboratories in accordance with national biosafety protocols (bioMérieux, 2023; MALDI-TOF Bruker Daltonics, 2023).
Despite this broad taxonomic coverage, the system shows significant limitations in intra-species discrimination within the Bcc. Cross-identifications have been documented, such as B. arboris misidentified as B. lata, B. cepacia as B. lata, B. contaminans as B. lata, and B. cenocepacia as B. diffusa, reflecting substantial proteomic overlap among closely related species. These findings underscore the difficulty of differentiating species that share ANI values within the genomic “gray zone” (95–96%) (Bach et al. 2023).
Specific studies illustrate these limitations. In an analysis by Fehlberg et al. (2013), 57 clinical Bcc isolates were evaluated using the MALDI-TOF MS (MALDI Biotyper®). Genus-level identification was successful in 100% of cases; however, only 76.9% of isolates were correctly identified at the species level, resulting in a 23.1% error rate. Among the most significant errors were misidentifications of all B. contaminans strains and 33.3% of B. cepacia strains (Fehlberg et al. 2013).
In a more comprehensive study, Wong et al. (2020) analyzed 122 isolates representing 22 Bcc species using both the MALDI Biotyper® and VITEK® MS platforms. MLST, including recA gene sequencing, served as the gold standard. The MALDI Biotyper® achieved 100% accuracy at the genus level but only 26% at the species level (score > 2.0). When considering only the first suggested result (“first hit”), accuracy increased to approximately 57.5%. Accuracy varied among species: B. multivorans showed an 80% success rate, B. cenocepacia 50%, while eight of the 23 evaluated species were not correctly identified by this criterion. In contrast, the VITEK® MS platform reached 97% genus-level and 67% species-level accuracy.
One of the major challenges in MALDI-TOF MS identification is the underrepresentation of rare or phylogenetically close species in commercial spectral libraries. This limitation may result in identification failures (no-ID) or incorrect assignments to clinically distinct species, especially for environmentally derived microorganisms, emerging strains, or isolates from poorly studied geographic regions (Seng et al. 2013).
To address these issues, various approaches have been employed to improve taxonomic resolution. Supplementing commercial libraries with spectra from local or rare strains, creating so-called “custom” or “in-house” databases, has proven highly effective. Studies show that such databases markedly increase the rate of correct identification by reducing recurrent errors present in original commercial libraries (Depoorter et al. 2020; Fergusson et al. 2020; Costa et al. 2022; de Miranda et al. 2023).
In this context, Fergusson et al. (2020) developed a spectral library specifically for the MALDI Biotyper® platform, targeting Burkholderia species and related genera such as Caballeronia and Paraburkholderia. This library, openly available on Dryad (10.5061/dryad.pk0p2ngjx), includes 95 spectra (MSP, Main Spectra Profiles) and was validated with 49 environmental isolates, correctly identifying all isolates at the genus level, even when specific species spectra were absent. This approach represents an effective and practical solution for laboratories with limited resources for curating in-house libraries (Fergusson et al. 2020; Linington 2020).
Additionally, national microbiology reference networks have coordinated the compilation of expanded databases and the standardization of interpretive protocols. These collective efforts have been instrumental in enhancing diagnostic capabilities for uncommon microorganisms and improving epidemiological surveillance and outbreak response involving Bcc species (Rocca et al. 2025).
These advances parallel profound taxonomic revisions within the genus Burkholderia, driven by genetic sequencing data (16S rRNA and housekeeping genes), phylogenomic analyses, and metrics such as ANI and dDDH. A comparative analysis of 612 core genome genes, along with concatenated sequences of atpD, gltB, gyrB, lepA and recA, confirmed the division of the genus into Burkholderia sensu stricto (s.s.), encompassing human and animal pathogens, and six new genera: Paraburkholderia (Sawana et al. 2014), Caballeronia (Dobritsa and Samadpour 2016), Robbsia (Lopes-Santos et al. 2017), Mycetohabitans and Trinickia (Estrada-de Los Santos et al. 2018), and Pararobbsia (Lin et al. 2020).
Additional studies applying ANI and average amino acid identity (AAI) have reinforced the validity of these taxonomic divisions, particularly for endophytic and soil-associated microorganisms (Estrada-de Los Santos et al. 2018; Mannaa et al. 2018). This reorganization has been further consolidated by the formal transfer of species, such as B. andropogonis to R. andropogonis (Lopes-Santos et al. 2017; Bach et al. 2023) and B. alpina to P. alpina (Lin et al. 2020; Bach et al. 2023).
These updates not only improve taxonomic accuracy, by better delineating microorganisms with distinct pathogenic, symbiotic, or ecological profiles, but also carry important clinical, ecological, and biosafety implications. The clear distinction between Burkholderia s.s. and environmental genera supports clinical diagnostics, laboratory risk management, and the understanding of microbial ecological interactions (Lopes-Santos et al. 2017; Estrada-de Los Santos et al. 2018; Mannaa et al. 2018; Lin et al. 2020).
Raman Spectroscopy in bacterial identification
Raman spectroscopy (RS) is an advanced analytical technique based on the interaction between incident light and the molecular vibrations of a sample, resulting in vibrational spectra that serve as molecular fingerprints. These spectral signatures are highly specific and have proven valuable for the precise identification of microorganisms, even within complex biological matrices (Choo-Smith et al. 2001; Pahlow et al. 2015; Ho et al. 2019).
The applications of RS span diverse fields, including clinical diagnostics, environmental microbiology, food safety, and biotechnology (Stöckel et al. 2015; Nicolson et al. 2021). In the clinical context, the technique offers several notable advantages: it is non-destructive, reagent- and dye-free, does not require bacterial enrichment, and can be performed directly on the sample. However, the primary limitation of conventional RS remains its low sensitivity, due to the inherently small Raman scattering cross-section (Uysal Ciloglu et al. 2020).
To address this limitation, Surface-Enhanced Raman Scattering (SERS) was developed. This technique employs nanostructured metallic substrates, typically silver or gold nanoparticles, capable of amplifying the Raman signal by up to 10^14^–10^15^ times, thereby enabling the detection of individual bacterial cells (Pahlow et al. 2015; Hassan et al. 2024). In addition to traditional metals, hybrid substrates incorporating graphene, semiconductors, and biopolymers have also been explored to further enhance selectivity, sensitivity, and reproducibility in complex biological systems (Ho et al. 2019; Hassan et al. 2024).
Classic studies by Choo-Smith et al. (2001) demonstrated that bacterial microcolonies after approximately six hours of growth exhibit low intra-structural biochemical heterogeneity, making them ideal for generating robust and representative spectral databases. In contrast, older colonies (12–24 h) display intrinsic biochemical gradients, such as higher glycogen content in outer layers, elevated RNA levels in deeper layers, and variations in carotenoid pigments (as observed in S. aureus), which, if not controlled, can compromise the accuracy of spectral classification (Choo-Smith et al. 2001).
RS has shown great promise for the detection of high-risk pathogens, such as species within the Burkholderia genus. In the study by Moawad et al. (2019), a machine learning–based methodology was developed to differentiate B. mallei, B. pseudomallei, and related species. The authors employed heat inactivation, rather than the formaldehyde-based inactivation used in earlier protocols by Stöckel et al. (2015), thus enabling safe sample handling under Biosafety Level 1 (BSL-1) conditions. The panel included 12 isolates of B. mallei, 13 of B. pseudomallei, and 11 isolates of other Burkholderia species (Stöckel et al. 2015; Moawad et al. 2019). Spectral analysis, combined with Support Vector Machine (SVM) classification algorithms and dimensionality reduction via Principal Component Analysis (PCA), yielded sensitivities greater than 95% for B. mallei and B. pseudomallei, and between 90 and 100% for other species within the genus (Moawad et al. 2019).
In addition to SVM, other algorithms have also been incorporated into spectral analyses. For example, Partial Least Squares Discriminant Analysis (PLS-DA) has been used to enhance discrimination between phylogenetically related strains. As discussed by Ho et al. (2019), combining RS with supervised learning methods enables differentiation even at the intraspecies variant level, based on subtle differences in vibrational spectra. This capability is particularly relevant for the early detection of MDR or clinically high-risk clones (Ho et al. 2019; Moawad et al. 2019).
These findings reinforce the potential of RS, particularly when combined with artificial intelligence (AI), to deliver rapid, accurate, and culture-independent diagnostics. The model proposed by Moawad et al. (2019) enables single-cell–level analyses within a reduced timeframe, with direct applications in clinical practice and epidemiological surveillance. Nevertheless, despite these promising results, the application of RS for bacterial identification remains largely at the research stage. To date, no commercially available and clinically validated Raman spectral libraries exist for routine use in clinical microbiology, and the databases currently employed are predominantly developed in-house. Furthermore, most of the results reported thus far derive from single-center studies, underscoring the need for multicenter validation, methodological standardization, and expansion of spectral databases prior to the broad implementation of this approach in routine laboratory settings (Choo-Smith et al. 2001; Stöckel et al. 2015; Ho et al. 2019; Nicolson et al. 2021).
Fourier transform infrared spectroscopy: principles and applications
Fourier-transform infrared spectroscopy (FTIR) is recognized as one of the most robust, versatile, and informative spectroscopic techniques for molecular characterization, combining rapid result acquisition with high discriminatory power (Neves et al. 2024). Its principle is based on the interaction of infrared radiation with matter, specifically on the selective absorption of this radiation by chemical bonds characteristic of the molecules present in the sample (Kamnev and Tugarova 2023). Each bond vibrates at specific frequencies, recorded as peaks in the absorption spectrum, forming a unique molecular "fingerprint" (spectral fingerprint) (Kassem et al. 2023). This specificity allows not only the differentiation of distinct compounds but also the detection of subtle biochemical variations in strains of the same species, a crucial aspect for applications in microbiology and industrial quality control (Lin 2021; Candela et al. 2025; Muchaamba and Stephan 2024).
Although the principles of infrared spectroscopy date back to the 1950s, its practical application was consolidated in 1969 with the launch of the first spectrometer equipped with Fourier transform by Digilab Inc. (Digilab Spectrum Software, Marlborough, MA, USA) (Griffiths 2017). This advancement was made possible by the development of fast and reliable computational systems, such as the PDP-8 (1965), capable of converting interferometric signals into interpretable spectra. FTIR overcame the limitations of traditional dispersive techniques by offering greater sensitivity, improved spectral resolution, and enhanced calibration accuracy (Johnston 1990; Griffiths and de Haseth, 2007; Griffiths 2017).
In the microbiological field, FTIR has stood out in the infra-specific phenotypic typing of bacteria. The spectral region between 1,200 and 900 cm⁻^1^, known as the fingerprint region, predominantly contains signals originating from polysaccharides in bacterial surface structures. This region directly reflects the chemical composition of the cell wall and capsule, which varies significantly among species and strains (Paul et al. 2023). In Gram-negative bacteria, these signals are related to the O, K, and H antigens, whereas in Gram-positive bacteria, they mainly correspond to polysaccharide capsules (Rahn et al. 2003). Recent studies demonstrate that analyzing this region allows highly accurate differentiation of serotypes, serogroups, and clinically important clones such as E. coli ST131, carbapenemase-producing K. pneumoniae (KPC), and carbapenem-resistant A. baumannii, with accuracy comparable to genotypic methods like PFGE and MLST (Silva et al. 2020; Neves et al. 2024; Novais et al. 2024).
In industry, FTIR is strategic for sectors such as pharmaceutical, food, and cosmetics, where rapid detection of contamination, adulteration, and process deviations is critical to ensuring final product quality. Its applications range from real-time monitoring of production lines to verification of authenticity and integrity of raw materials (Saji et al. 2024). When combined with proprietary spectral databases, FTIR enables tracking of seasonal, geographical, and temporal variations in microbial populations, optimizing risk management and product quality control (Cebi et al. 2023; Souza et al. 2024).
Among the operational advantages, analytical speed stands out: the entire process, from sample collection to result interpretation, can be completed in under three hours. This agility results from simplified sample preparation, often requiring only a single cultivated bacterial colony, and the elimination of expensive reagents, reducing costs compared to molecular methods or mass spectrometry (Davis and Mauer 2010). Despite the high initial investment, the durability of the systems, low cost per analysis, and scalability justify its adoption in high-throughput laboratories (Novais et al. 2019).
The recent development of specialized software, such as IR Biotyper® (Bruker), which incorporates automated spectral analysis, multivariate algorithms, and machine learning, has increased the reliability and standardization of FTIR (Fredes-García et al. 2025). Integration with genomic data and other omics approaches expands its capabilities, enabling not only microbial identification but also the prediction of virulence phenotypes, antimicrobial resistance, and environmental adaptation (Picard et al. 2021; de Souza et al. 2025).
Attenuated total reflection mode (ATR-FTIR) has proven to be particularly robust for microbial identification and typing (Kassem et al. 2023). In a pioneering study, Lam et al. (2019) created a comprehensive clinical spectral database composed of 263 reference strains that generated 789 spectra, representing 65 yeast species, including emerging pathogens such as Candida auris and Trichosporon asahii. In the prospective evaluation, 318 clinical isolates from 38 laboratories were tested, achieving 100% accuracy at the genus level and 99.7% accuracy at the species level. Analysis was completed in under 2 min per sample, using only a single colony directly from the agar plate without prior preparation, delivering performance equal to or exceeding that of MALDI-TOF MS, but without acid extraction steps or the use of consumables (Lam et al. 2019).
Beyond the high performance achieved in the study, the technique proved particularly effective in discriminating morphologically and genetically close species, such as C. albicans and C. dubliniensis. This discriminatory ability was attributed to the inclusion of an adequate number of reference strains per species in the construction of the database, as well as the careful and optimized selection of spectral regions with greater discriminatory power. The authors emphasized that the continuous maintenance and expansion of the spectral library, incorporating newly isolated strains and emerging species, are essential to preserve the accuracy and long-term relevance of the technique (Lam et al. 2019).
The versatility of FTIR in bacterial typing was thoroughly explored by Novais et al. (2019), who demonstrated its potential to differentiate Gram-negative bacteria (E. coli and K. pneumoniae) and Gram-positive bacteria (S. aureus and L. monocytogenes) based on unique spectral signatures. The region from 1200 to 900 cm⁻^1^, predominantly associated with surface polysaccharides (including capsules and O and K antigens), proved particularly useful for differentiating serotypes and capsular types, with direct implications for epidemiology, surveillance, and understanding pathogen adaptation to different hosts. Compared to genotypic methods such as PFGE or MLST, the technique showed much shorter turnaround times (hours versus days) and drastically reduced costs (less than US50–100 for full sequencing), establishing itself as an economically viable and operationally attractive alternative for routine epidemiological surveillance programs (Novais et al. 2019).
In the context of environmental and opportunistic pathogenic bacteria, Fang et al. (2013) applied FTIR to analyze B. seminalis isolates from different sources such as apricot, water, and rice rhizosphere. Clear variations were observed in the protein-to-lipid ratios: 2.25 for apricot isolates, 1.95 for water isolates, and 1.65 for rhizosphere isolates, reflecting biochemical adaptations to specific environmental conditions. Additionally, a strong ester C=O band was consistently identified at approximately 1736 cm⁻^1^, a spectral signal characteristic of the presence of polyhydroxyalkanoates (PHAs), intracellular polymers widely produced by bacteria under conditions of carbon excess and nutrient limitation (Sindhu et al. 2011; Fang et al. 2013; Kourmentza et al. 2017).
This band, located in the range of 1720–1750 cm⁻^1^, is recognized as a robust marker for PHA detection by FTIR. Studies with Bacillus megaterium and B. cereus identified intense peaks near 1736 cm⁻^1^ directly related to the ester-carbonyl bonds of these polymers (Kourmentza et al. 2017), while other works confirmed the presence of bands between 1720 and 1724 cm⁻^1^ associated with the same chemical functionality in different bacterial species (Sindhu et al. 2011).
Thus, the results of Fang et al. (2013) demonstrate that, even among genetically related isolates, FTIR is capable of detecting relevant phenotypic variations, such as changes in lipid and protein composition and PHA reserves, reflecting adaptive metabolic responses to different ecological niches. These findings reinforce the value of spectral fingerprinting techniques in microbial ecology and environmental adaptation studies, with great potential for functional monitoring of bacterial communities (Sindhu et al. 2011; Fang et al. 2013; Kourmentza et al. 2017).
In the clinical field, Barker et al. (2021) investigated the B. cenocepacia ET12 lineage, a pathogen of high relevance in CF patients due to its associated high morbidity, mortality, and antimicrobial resistance. The study compared the performance of FTIR spectroscopy (IR Biotyper® system) with the MALDI-TOF MS method coupled with ClinProTools software (MALDI-TOFBruker Daltonics, Bremen, Germany, version 2.2) for typing clinical isolates of this strain. FTIR showed a sensitivity of 84.6% and specificity of 83.3%, results comparable to MALDI-TOF MS, which presented 87.5% sensitivity and 80% specificity, respectively (Barker et al. 2021).
The authors noted that the performance of FTIR could be enhanced by expanding the analyzed spectral range to include the region from 900 to 400 cm⁻^1^, which is rich in polysaccharide signals and carbohydrate skeletal vibrations, as well as by improving classification algorithms based on machine learning. These improvements would be especially relevant for discriminating highly related strains, a critical challenge in outbreak investigations at specialized CF treatment centers (Barker et al. 2021; Passaris et al. 2022). The study also reinforced the role of FTIR as a complementary tool, particularly in contexts where high-resolution genotypic methods such as PFGE or WGS are not readily available or economically viable for routine use (Guerrero-Lozano et al. 2022; Yang et al. 2023).
Strategic laboratory setup for Burkholderia identification and typing: optimizing costs, methods, and analytical precision
The establishment of a microbiology laboratory specialized in the analysis of bacteria from the genus Burkholderia requires careful planning, particularly regarding cost estimation and the selection of appropriate laboratory techniques. Considering the diversity of available methodologies, ranging from classical culture-based methods to advanced genomic sequencing approaches, it becomes essential to evaluate the balance between cost, technical complexity, analytical capacity, and practical applicability (Guo et al. 2014; Calderaro and Chezzi 2024).
In an initial phase, traditional isolation and screening techniques using selective culture media (SCM) present the lowest implementation costs, involving basic equipment, low-cost consumables, and reagents specific for selective cultivation. Although they form the basis for preliminary strain screening, these methods have intrinsic limitations in specificity and resolution, which may compromise their effectiveness in differentiating genetically closely related strains (Calderaro and Chezzi 2024) (Supplementary Table 1; Fig. 3).Fig. 3. Cost estimates by category (A and B) and total cost per microbial identification and typing method (C and D), expressed in Brazilian reais (R$) and US dollars (USD), respectively. To enable comparative analysis, values were converted to a logarithmic scale. Cost categories include: Equipment (GEN), Equipment (SPC), Consumables and utensils (GEN), Consumables and utensils (SPC), Reagents (GEN), Reagents (SPC), Software and licenses, and Technical training. The evaluated methods were: selective culture media (SCM), biochemical profile (BCP – VITEK® 2), genomic DNA banding pattern (GDBP – PFGE), sequencing of conserved and variable regions (SCVR – 16S rRNA), multilocus sequence typing (MLST), matrix-assisted laser desorption/ionization–time of flight mass spectrometry (MALDI-TOF MS), Fourier Transform Infrared Spectroscopy (FTIR), and whole-genome sequencing (WGS). Abbreviations: GEN, general; SPC, specific
Selective media such as BCSA, OFPBLA, and PCA exhibit moderate to high specificity for the genus Burkholderia, with BCSA standing out, reported to have specificity of up to 95%. However, none of these media allow identification at the species level, as non-Bcc microorganisms, such as Ralstonia and Stenotrophomonas, can also grow on these substrates, generating false positives. Therefore, although useful for initial isolation, selective media lack discriminative power and should be complemented by molecular or proteomic methods (Henry et al. 1999; Coenye et al. 2001) (Supplementary Table 9; Fig. 4).Fig. 4. The values represent the percentage sensitivity (SP) of each method at different taxonomic levels: genus, species, and clone. 1. Selective media: BCSA, OFPBLA, and PCA show high sensitivity at the genus level (69.5–92.5%) but do not discriminate species or clones; MacConkey/Abx shows lower genus-level sensitivity (59%) and also fails to identify species or clones. 2. Automated phenotypic identification: BCP (VITEK® 2) reaches good sensitivity at the genus (92.5%) and species (80%) levels but does not distinguish clones. 3. Classical molecular typing: GDBP (PFGE) fails at genus-level identification but is effective for species (60%) and excellent for clones (97.5%); SCVR (16S rRNA) identifies genus (94.5%) and shows moderate performance for species (55%) and clones (59%). 4. Advanced molecular typing: MLST and WGS achieve maximal or near-maximal sensitivity at all levels, with WGS reaching 100% for genus, 99.5% for species, and 100% for clones. 5. Mass spectrometry and spectroscopy: MALDI-TOF MS shows excellent sensitivity at genus (99%) and species (91.5%) levels but does not distinguish clones; FTIR demonstrates good sensitivity for genus (92.5%) and species (77.5%) and moderate sensitivity for clones (60%). These data indicate that sequencing- and molecular typing–based methods provide higher accuracy at all levels, whereas selective media and classical phenotypic techniques are limited, especially for species and clone discrimination. Abbreviations: BCP, Biochemical Profile; BCSA, Burkholderia cepacia selective agar; FTIR, Fourier Transform Infrared Spectroscopy; GDBP, Genomic DNA Banding Pattern; MacConkey/Abx, MacConkey agar supplemented with antibiotics; MALDI-TOF MS, Matrix-Assisted Laser Desorption Ionization – Time of Flight Mass Spectrometry; MLST, Multilocus Sequence Typing; OFPBLA, Oxidation-Fermentation Polymyxin Bacitracin Lactose agar; PCA, Pseudomonas cepacia agar; PFGE, Pulsed-Field Gel Electrophoresis; SCVR, Sequencing of Conserved and Variable Regions; WGS, Whole-Genome Sequencing
For rapid and automated identification, systems such as VITEK® 2 and MALDI-TOF MS represent alternatives with medium and high initial costs, respectively, offering the advantage of significantly reducing identification time and standardizing results. These systems are especially relevant in clinical settings, where the speed and reliability of analyses directly impact therapeutic decisions. However, both depend on the continuous supply of specific consumables and require technical training for operation and maintenance (Deng et al. 2014; Guo et al. 2014) (Supplementary Tables 2 and 6; Fig. 3).
The VITEK® 2 system, widely used in routine practice, offers high specificity at the genus level (90–95%) but only moderate to limited performance at the species level (83–90%). This is due to the phenotypic similarity among Bcc species and the limitations of the system’s biochemical database. Accuracy may improve with database updates, but misclassification errors among closely related species remain frequent, limiting its reliability as a definitive identification tool in sensitive clinical contexts (WHO, 2018; Periaiah et al. 2024) (Supplementary Table 9; Fig. 4).
MALDI-TOF MS has gained prominence for its high specificity at the genus level (> 98%) and moderate to high specificity at the species level (94–98%). However, accuracy critically depends on the quality and updating of the database, as emerging or underrepresented species may still be misclassified (Fehlberg et al. 2013; Chen et al. 2021). Sample preparation also directly influences method performance. Studies report overall accuracy of 99.6% for genus and 93–94% for species; for Bcc, genus-level concordance reaches 100%, while species-level concordance ranges from 53 to 76.9%, depending on score cut-offs and the species involved (Barberis et al. 2014; Wilkendorf et al. 2020) (Supplementary Table 9; Fig. 4).
FTIR spectroscopy emerges as a rapid and non-destructive tool, with intermediate implementation costs, positioning itself between proteomic and genomic technologies in terms of investment. According to experimental data, it exhibits high specificity at the genus level (96.5%), moderate specificity at the species level (87.5%), and high clonal differentiation capability (90.0%). It is useful for species discrimination and clonal typing through multivariate analysis of biochemical profiles, being dependent on a reference database. Despite its potential, definitive identification at the species or clone level preferably requires complementation with molecular or proteomic methods (Wenning and Scherer 2013; Uribe et al. 2023) (Supplementary Tables 7 and 9; Figs. 3 and 4).
In contrast, molecular techniques such as PFGE and MLST provide high resolution for epidemiological and phylogenetic analyses but require substantial investments in equipment, reagents, and bioinformatics infrastructure. These methodologies are essential in laboratories dedicated to advanced research and microbiological surveillance, where precision in clonal typing and outbreak traceability is critical (Deng et al. 2014) (Supplementary Tables 3 and 5; Fig. 3).
PFGE is not intended for taxonomic identification, exhibiting low specificity for genus and species but very high specificity for clonal differentiation (> 90%). It is particularly indicated for epidemiological investigations and outbreak tracking, allowing the distinction of clonal strains even within the same species. However, its interpretation requires expertise; it is a labor-intensive technique with high interlaboratory variability (Dallal et al. 2014; WHO, 2018) (Supplementary Table 9; Fig. 4).
Among molecular methods, MLST stands out for offering very high specificity at the genus, species, and clone levels (97–100%). It is one of the most robust methods for epidemiological surveillance and monitoring of patients with CF, allowing the comparison of profiles across laboratories and international databases. Its limitations lie in cost and the need for infrastructure, but its taxonomic accuracy and discriminatory power are unquestionable (Baldwin et al. 2005; Spilker et al. 2009) (Supplementary Table 9; Fig. 4).
Sequencing of the 16S rRNA gene, although exhibiting high specificity at the genus level (90–99%), has limitations in differentiating closely related Bcc species, with moderate to low specificity (50–70%). The high sequence conservation within the complex reduces its usefulness as a standalone tool in more refined clinical and epidemiological studies (Scoffone et al. 2021) (Supplementary Tables 4 and 9; Figs. 3 and 4).
Among the available methodologies, WGS is the most comprehensive and technically advanced approach for microorganism characterization, providing, on a single platform, data on identification, antimicrobial resistance, virulence factors, and clonal typing. Implementing WGS requires the highest financial investment among the described techniques, as well as a qualified team and robust computational infrastructure, factors that may limit its adoption by laboratories with restricted budgets (Calderaro and Chezzi 2024) (Supplementary Table 7; Fig. 3).
WGS represents the current gold standard, exhibiting 100% specificity for genus, species, and clones, and enabling comprehensive and multifunctional genomic analyses. Its main practical limitations lie in the high cost, the need for specialized infrastructure, and bioinformatic processing, factors that still restrict its application to research and reference centers (De Maio et al. 2019) (Supplementary Tables 8 and 9; Figs. 3 and 4).
Comparative analysis of cost estimates demonstrates that the choice of methodologies must fully consider the laboratory’s operational profile, the volume and complexity of samples, as well as specific clinical or investigative demands. In this context, the strategic definition of the set of techniques to be adopted should reflect a balance between economic feasibility, analytical capacity, and impact on the diagnosis and control of Burkholderia infections, thereby contributing to advances in clinical and environmental microbiology (Deng et al. 2014; Guo et al. 2014).
The adoption of a stepped structure is recommended: initially incorporating more accessible methods and progressively integrating advanced technologies in line with the expansion of technical capacity and available resources. The selection of the most appropriate method directly depends on operational objectives, supporting infrastructure, and budget. Laboratories focused on initial screening and basic surveillance may benefit from SCM and the VITEK® 2 system, whereas research centers and genomic surveillance units should prioritize higher-resolution approaches, such as MLST and WGS, which, although more costly, provide superior returns in scientific, epidemiological, and strategic value (Calderaro and Chezzi 2024).
Conclusion
The identification of species within the genus Burkholderia, particularly those belonging to the Bcc, is hindered by their high phenotypic similarity and extensive genomic diversity. Traditional methods remain useful for initial screening but do not ensure species-level resolution. Proteomic and molecular techniques, such as MALDI-TOF MS, MLST, and especially WGS, provide greater accuracy, albeit with cost and infrastructure limitations.
The adoption of a tiered strategy, combining accessible methods for initial detection with molecular and genomic approaches for confirmation and surveillance, is the most effective way to improve diagnosis and epidemiological control. The standardization of protocols, continuous updating of reference databases, and cooperation among research centers are essential to address the challenges posed by this bacterial genus of significant clinical and epidemiological relevance.
Strengthening integrated and collaborative approaches, together with the continuous advancement of identification technologies, will be decisive in transforming the diagnostic challenge of Burkholderia into an opportunity to enhance surveillance and the global response to emerging pathogens.
Supplementary Information
Below is the link to the electronic supplementary material.Supplementary file1 (PDF 623 kb)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aguiar AR, Sales FLS, Miranda RVSL, Costa LV, Brandão MLL, Santana GS (2025) Use of rec A gene sequencing as a complementary tool for identification of Burkholderia spp. isolated from an immunobiological industry facility. In: International Symposium on Immunobiological, Rio de Janeiro. Annals of the symposium. Instituto de Tecnologia de Imunobiológicos, Rio de Janeiro, vol 9, p 145. 10.35259/isi.2025_70112
- 2List of Prokaryotic names with Standing in Nomenclature (LPSN) (2025) Genus: Burkholderia. DSMZ – Leibniz Institute, Düsseldorf. https://lpsn.dsmz.de/genus/burkholderia. Accessed 5 June 2025
- 3Santana GS de, Aguiar AR, Sales FLS, Miranda RVSL de, Valadão TB, Costa LV da, Brandão MLL (2024) Polyphasic characterization of Burkholderia cepacia complex strains isolated from a pharmaceutical industry facility. In: International Symposium on Immunobiological, Rio de Janeiro. Annals of the symposium. Bio-Manguinhos, Rio de Janeiro, p 144. 10.35259/isi.biomang.2024_63868
- 4Souza PA, Miranda RVSL, Santos MCS, Costa LV, Silva RP, Miranda CAC, Villas Bôas MHS, Brandão MLL (2024) Evaluation of Fourier-Transform Infrared Spectroscopy as a rapid method to type Stenotrophomonas maltophilia strains isolated from pharmaceutical industry. In: International Symposium on Immunobiological, Rio de Janeiro. Annals of the symposium. Bio-Manguinhos, Rio de Janeiro, p 123. 10.35259/isi.biomang.2024_63778
- 5Uni Prot Consortium (2025) ATP synthase subunit beta (atp D) - Escherichia coli (strain K 12). Uni Prot KB: P 0ABB 4. https://www.uniprot.org/uniprotkb/P 0ABB 4. Accessed 10 June 2025
- 6VITEK® 2 (2023) BIOMÉRIEUX. VITEK® 2 Systems: Manual do Usuário. bio Mérieux SA, Marcy-l'Étoile. https://www.biomerieux.com/. Accessed 6 June 2025
- 7VITEK® 2 (2023) Mérieux. VITEK® 2 Systems: GN Card Product Information Sheet. bio Mérieux SA, Marcy-l'Étoile. https://www.biomerieux.com/. Accessed 6 June 2025
- 8VITEK® 2 (2025) VITEK® 2 System Brochure: Detalha o processo de atualização contínua da base de dados. bio Mérieux, Marcy l'Étoile. https://www.biomerieux.com/. Accessed 11 June 2025