Gene Expression, Docking and Machine Learning in Malaria Drug Discovery: A Systematic Review
Reuben Samson Dangana, Israel Ehizuelen Ebhohimen, Samson Anjikwi Malgwi, Samuel Chima Ugbaja, Moses Okpeku

TL;DR
This review explores how gene expression, molecular docking, and machine learning are being used to discover new malaria drugs, focusing on herbal compounds and computational methods.
Contribution
The paper systematically reviews the integration of molecular and computational techniques for antimalarial drug discovery from 2014 to 2024.
Findings
Molecular docking was the most used technique, with compounds like isorhamnetin showing strong binding to Plasmodium proteins.
ML models predicted bioactivity and resistance patterns, identifying flavonoids and terpenoids as promising drug candidates.
RNA-seq analysis revealed key genes modulated by herbal treatments, including those involved in apoptosis and inflammation.
Abstract
Malaria remains a significant and worldwide health threat with increasing resistance to current treatments, stimulating the demand for innovative approaches in pursuing drug discovery. This systematic review integrates the progress made from 2014 through 2024 regarding molecular methods like gene expression profiling, molecular docking and machine learning to understand the biology of Plasmodium and identify new drug targets and compounds, focusing on herbal remedies and computational methods. Several studies were found using a PRISMA‐guided search of PubMed, Scopus and Web of Science (64 studies found). The data extracted were gene expression outcomes, docking affinities, ML models and experimental validations (in vitro/in vivo). Molecular docking emerged as the dominant technique (32.37%), followed by in vitro antiplasmodial assays (14.39%), ADMET profiling (10.79%) and gene…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
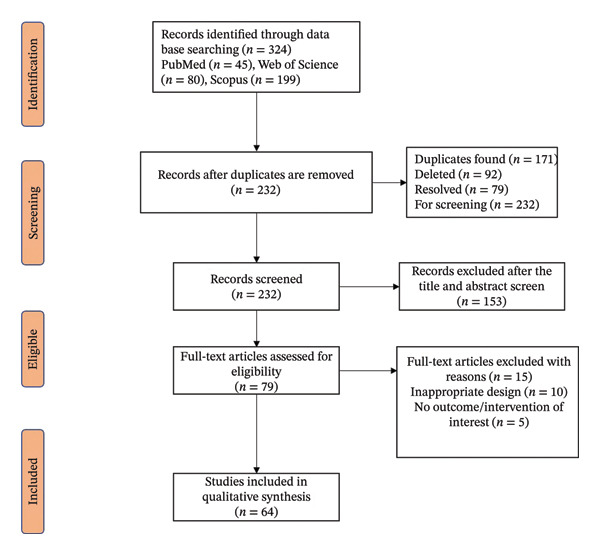 FIGURE 1
FIGURE 1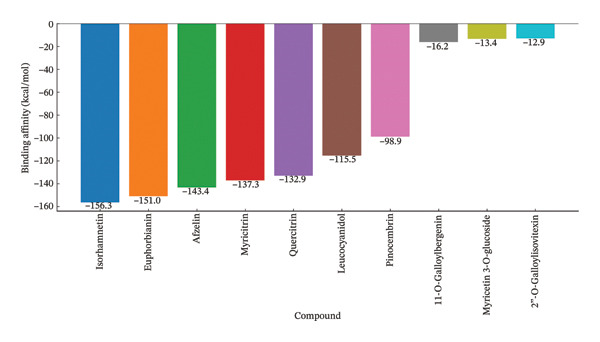 FIGURE 2
FIGURE 2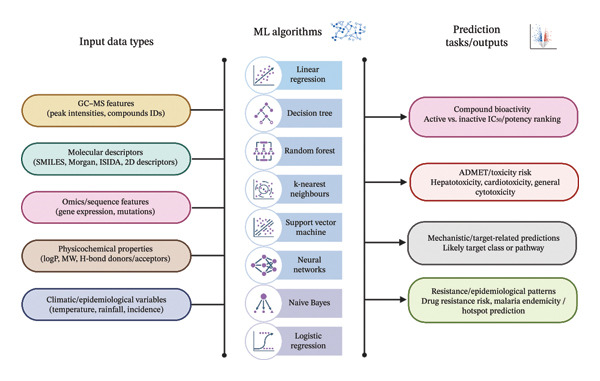 FIGURE 3
FIGURE 3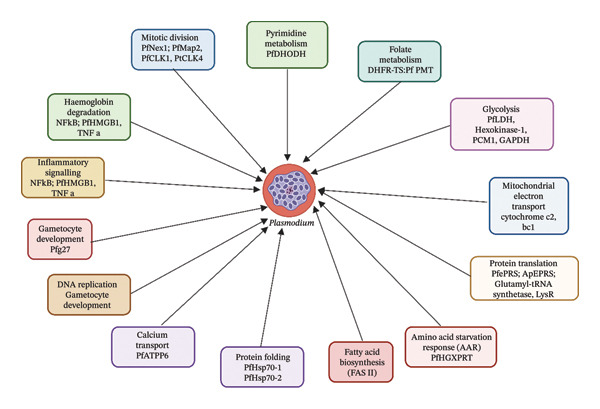 FIGURE 4
FIGURE 4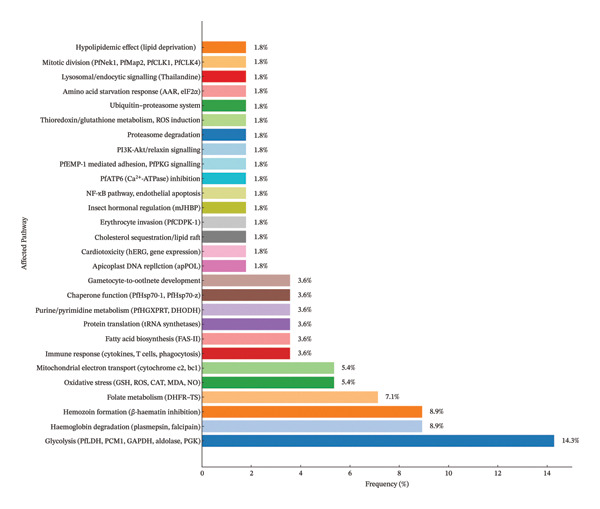 FIGURE 5
FIGURE 5| References | In silico methods | In vitro methods | In vivo methods | Chemical analysis and compound characterisation | Docking software | Validation method |
|---|---|---|---|---|---|---|
| Adelusi et al. [ | 3D homology modelling (I‐TASSER, Rosettafold), ADMET profiling (ADMETlab 2.0), molecular docking, molecular dynamics (GROMACS), MM/GBSA analysis | — | — | — | AutoDock Vina v1.5.7 | Ramachandran plot, ProSA‐web z‐score, RMSD comparison of I‐TASSER and Rosettafold models |
| Onguéné et al. [ | 3D molecular modelling (MOE, LigPrep), ADMET descriptor calculation (QikProp), energy minimisation (MMFF94, OPLS force fields) | — | — | — | — | Comparison with Lipinski’s Rule of Five, Jorgensen’s Rule of Three, ADMET descriptors of known drugs |
| Happi et al. [ | Molecular docking, ADMET prediction (ADMETLab 2.0) | Bioassay‐guided fractionation, antiplasmodial assay (schizont maturation inhibition), cytotoxicity assay (MTT on P388 cells) | — | — | PyRx/Vina, BIOVIA Discovery Studio | IC50 values, selectivity index (SI = Cytotoxicity IC50/Antiplasmodial IC50), docking binding energies (ΔG) |
| Zininga, et al. [ | — | Chaperone activity assay (MDH aggregation), ATPase activity assay, antiplasmodial assay (pLDH), protein expression/purification (PfHsp70‐1, PfHsp70‐z) | — | UPLC–QTOF–MS (phenolic quantification) | — | Chaperone assay normalised to spontaneous MDH aggregation, ATPase assay normalised to basal activity, pLDH assay validated with chloroquine (IC50: 8.5 ng/mL), SDS–PAGE for protein purity |
| Khan et al. [ | Molecular docking | DPPH radical scavenging, reducing power assay, total antioxidant capacity, pLDH assay | — | — | MOE | IC50 (antiplasmodial), EC50 (antioxidant), Moldock score, pKi |
| Elmaidomy et al. [ | PASS virtual screening, molecular docking, MDS (Desmond, 50 ns), free energy perturbation (FEP) | Malstat assay ( | — | 1H, DEPT‐Q, HSQC, HMBC NMR; HRESIMS; UV; IR | AutoDock/Vina | PASS prediction (Pa: 0.892 for cytochrome‐C reductase inhibition), docking/MDS binding to cytochrome bcl (ΔG: −8.33 kcal/mol vs. −14.46 kcal/mol for atovaquone) |
| Knockleby et al. [ | Molecular docking | Cytotoxicity (SRB), anti‐plasmodial (HRP2 ELISA), cell cycle (flow cytometry), apoptosis (Annexin V), protein analysis (Western blot), kinase assay (ADP‐Glo) | — | NMR, HR‐ESIMS | PyRx (0.9.8) with Autodock 4.2 | Spectroscopic comparison with standards (NMR, MS), repeated experiments ( |
| Gomes et al. [ | Molecular docking | Oxidative stress (TEAC, GSH, TBARS) | Antimalarial activity (4‐day suppressive test) | NMR | Molegro Virtual Docker 5.5 | Compound identification via NMR, repeated oxidative stress measurements, statistical significance ( |
| Rawa et al. [ | Molecular docking, MD simulations (AMBER 18), MM–PBSA, computational NMR, ECD (DFT, TD‐DFT) | Antimalarial ( | — | HR‐ESI‐TOF‐MS, HR‐EI‐TOF‐MS, NMR, DEPT, HSQC, HMBC, DQF‐COSY, NOESY | AutoDock 4.2 | Redocking of co‐crystallised ligand (RMSD ∼2.0 Å), MD stability (RMSD, RMSF, Rg, H‐bond profiles), MM–PBSA, ECD spectra comparison |
| Al‐Huqail et al. [ | Molecular docking | IC50 via sigmoidal dose–response curves | Parasitaemia, suppression, survival time | UPLC–ESI–MS/MS (20 compounds), HPLC (rutin: 3.65 mg/g, hesperidin: 36.17 mg/g) | Accelrys Discovery Studio 2.5 | HPLC accuracy (99.70%–100.23%), precision (%RSD 1.38–3.07), LOD/LOQ, docking on PfDHFR–TS (1J3I), Pf‐PMT (2UJ9), Lm‐PTR1 (2BFM), Lm‐FPPS (4JZX) |
| Ikpa et al. [ | Molecular docking, ADMET (admetSAR), drug likeness (OSIRIS), toxicity risk assessment | — | — | FTIR, GC–MS | — | FTIR, GC–MS characterisation, OSIRIS toxicity/drug likeness, admetSAR ADMET, docking binding affinity, 2D/3D interaction visualisation |
| Enyiekere et al. [ | Molecular docking, ADMET profiling | Antiplasmodial assays (suppressive, prophylactic, schizonticidal) | Parasitaemia reduction, chemosuppression, survival time | GC–MS | PyRx, BIOVIA Discovery Studio | Comparison with chloroquine/pyrimethamine, docking/ADMET profiling |
| Olaosebikan et al. [ | Molecular docking, ADMET prediction | — | — | GC–MS | AutoDock Vina | Redocking the native ligand (DSM1, RMSD 0.15 Å), ADMETlab 2.0 predictions |
| Pradhan et al. [ | Molecular docking | In vitro antimalarial (modified WHO MARKIII) | — | GC–MS | Biovia Discovery Studio 2021 | Dose–response curves, microscopic parasite clearance |
| Wiraswati et al. [ | Molecular docking | Antiplasmodial (Giemsa staining, microscopy) | — | GC–MS | PyRx, AutoDock Vina | — |
| Herrera‐Calderon et al. [ | Molecular docking, MD (Desmond 2020.1) | Antioxidant assays (DPPH, ABTS, FRAP) | — | GC–MS | AutoDock 4.2 | RMSD, RMSF, Rg, SASA |
| Kane et al. [ | Molecular docking | Gene expression (qPCR) | — | GC–MS | AutoDock Vina (via PyRx) | qPCR for gene expression, GC–MS compound identification, docking binding affinity |
| Deligianni et al. [ | — | High‐content microscopy (GFP‐tagged parasites) | — | GC–MS | — | Inhibition > 50% ( |
| Samuel et al. [ | High‐throughput virtual screening, molecular docking, pharmacophore modelling, ADME, toxicity profiling | — | — | — | — | Binding affinity scores, molecular interaction analysis (H‐bonds, hydrophobic), pharmacophore modelling, ADMET predictions |
| Shekari et al. [ | Homology modelling, molecular docking, ADME–Tox, PCA, energy minimisation, Ramachandran plot | — | — | — | — | Homology model quality (GMQE, sequence identity, Ramachandran plot), binding affinity, molecular interactions (H‐bonds, pi‐alkyl, pi‐cation/anion), ADME–Tox (SwissADME, OSIRIS), PCA correlations |
| Adeoye et al. [ | Molecular docking | In vitro β‐haematin inhibition | — | HPLC–DAD, HRGC–MS | AutoDock 4.2, AutoDock Vina | — |
| Taranto et al. [ | Molecular docking, comparative homology modelling | — | — | HPLC–MS/MS, NMR | AutoDock Vina | — |
| Chaniad et al. [ | Molecular docking | Hypoxanthine uptake inhibition, MTT (cytotoxicity) | — | — | AutoDock 4.2, UCSF Chimaera 1.14, PLIP | 3H‐hypoxanthine uptake, MTT assay, docking binding interactions |
| Birgit Viira [ | SVM classification, molecular descriptors (ISIDA), virtual screening | Antiplasmodial (SYBR Green I), cytotoxicity (resazurin, MRC‐5) | — | — | — | Cross validation (leave‐1/3‐out, balanced accuracy > 0.7), consensus voting (> 70% active likelihood), IC50 via ICEstimator, selectivity index (CC50/IC50) |
| Rehman [ | Molecular docking, binding pocket prediction (CASTp), visualisation (PyMOL, LigPlot+), network pharmacology, pathway enrichment | Isothermal Titration Calorimetry (ITC) | — | — | AutoDock Vina 4.2, AutoDock Tools 1.5.7 | Binding energy (ΔG), STRING DB PPI ( |
| Konyanee et al. [ | Molecular docking, ADMET (SwissADME, ProTox‐II) | Antiplasmodial (pLDH), cytotoxicity (MTT, Vero cells) | — | NMR spectroscopy | AutoDock Vina v. 1.1.2, PyMOL v. 2.5.2, PLIP | IC50 determination, CC50 and selectivity index (SI), redocking NADH (RMSD = 0.775 Å), NMR/literature comparison |
| Camara et al. [ | — | Antiplasmodial (SYBR Green I), anti‐inflammatory (RT‐qPCR, ELISA), antioxidant (LUCS), brain cell analysis (flow cytometry), gene expression (RT‐qPCR), cytokine levels (ELISA), acute oral toxicity | ECM model (parasitaemia, survival, RMCBS) | UHPLC–HRMS (MzMine, MSDIAL, MS‐FINDER) | — | IC50 via nonlinear regression, LDH assay, EC50 for antioxidant index, parasitaemia suppression, survival curves, RMCBS, GAPDH as qPCR control, HRMS/MS fragmentation vs. DNP database |
| Asanga et al. [ | Molecular docking, ADME (SwissADME) | — | Antiplasmodial (Rane’s curative test) | GC–MS | AutoDock Vina 4.2, Biovia Discovery Studio | Parasite density, % growth inhibition, GC–MS characterisation, docking protein–ligand interactions, ADME analysis |
| Apeh et al. [ | Molecular docking, ADMET (SwissADME) | — | 4‐day suppressive test, haematological analysis | — | PyRx v0.8, AutoDock Vina | Binding affinity, interaction analysis, parasitaemia suppression, haematological indices |
| Nwonuma et al. [ | Molecular docking, molecular dynamics (100 ns) | — | Parasitaemia, biochemical assays (lipid, enzyme), histopathology | — | PyRx 0.8, GROMACS | Binding affinity, MD stability (RMSD, RMSF, Rg), parasitaemia reduction, biochemical parameter analysis |
| Chaurasia and Pandey [ | Molecular docking, MD simulation (100 ns), SWISSADME | β‐Haematin assay | — | GC–MS | AutoDock | Comparison with artemisinin (binding affinities, β‐haematin efficiency: 91.7%), RMSD, RMSF, Rg, SASA, Lipinski’s rule, SWISSADME, GC–MS retention times/mass spectra |
| Soeiro et al. [ | Molecular docking, NMR structural analysis | Antiplasmodial, cytotoxicity (Vero cells) | Parasitaemia clearance ( | Molecular modelling (drug interactions) | — | IC50 vs. chloroquine, parasitaemia clearance vs. chloroquine, NMR (PfHMGB1, GLR‐HsHMGB1), glabridin cytotoxicity (SI = 9.6), docking interaction energies (ΔE, ΔG) |
| Dzouemo et al. [ | Molecular docking, ADMET predictions | Antiplasmodial (SYBR Green I) | — | NMR (1H, 13C), EI‐MS | AutoDock Vina (Version 1.2.0) | Spectroscopic data vs. literature, antiplasmodial assay vs. chloroquine/artemisinin, BIOVIA Discovery Studio visualisation |
| Samuel [ | Molecular docking, ADMET predictions | β‐Haematin inhibition | — | NMR (1H, 13C), FTIR | AutoDock Vina (Vina Dock Wizard) | Spectroscopic data vs. literature, β‐haematin assay vs. chloroquine/artesunate, redocking of the |
| Hidayati et al. [ | Molecular docking | Antimalarial (Giemsa‐stained microscopy) | — | NMR, MS spectra, TLC | Molegro Virtual Docker 5.5 | — |
| Adams et al. [ | Molecular docking, MD simulation, MM–PBSA | — | — | LC‐ESI‐Q‐TOF‐MS | AutoDock Vina v.1.2.0, PyRx 0.8 | ROC curve (AUC = 0.733), RMSD of redocked ligand (0.959 Å) |
| Sikam et al. [ | Molecular docking, ADMET studies | In vitro antiplasmodial (schizont maturation inhibition) | — | LC‐ESI‐Q‐TOF‐MS, NMR | AutoDock Vina® | IC50 vs. chloroquine |
| Kadioglu et al. [ | Molecular docking | Cytotoxicity (AC16), microarray, zebrafish toxicity | — | — | AutoDock 4, AutoDock Tools 1.5.7 | — |
| Muema et al. [ | Molecular docking, ADMET (SWISSADME) | SYBR Green I fluorescence, IEV susceptibility, gametocyte morphology | — | — | AutoDock Vina, PyRx | In vitro and ex vivo susceptibility assays |
| Elmaidomy et al. [ | Inverse docking, molecular docking, MD simulation (Desmond), PPI network (Cytoscape) | Malstat assay (pLDH) | — | NMR (1H, 13C, DEPT‐Q), HRESIMS | AutoDock Vina | Malstat assay validation, Cytoscape PPI network |
| Snider and Weathers [ | — | Microscopy (Giemsa), RT‐qPCR (gametocyte genes) | — | GC–MS (artemisinin quantification) | — | Microscopic quantification of parasitaemia/gametocytaemia, RT‐qPCR for gene expression changes |
| Tjitraresmi et al. [ | Molecular docking, pharmacokinetic/toxicity (PreADMET, Toxtree), MD (AMBER 16), Lipinski’s Rule | — | — | — | AutoDock 4.2.6 | Redocking chloroquine (RMSD = 1.13 Å), MD stability (RMSD, RMSF) |
| Singh et al. [ | Molecular docking | Antimalarial (Giemsa), hemocompatibility, MTT (HEK293) | — | UV–Vis, DLS, Zeta Potential, FTIR, XRD, FESEM | AutoDock (blind docking), Schrödinger (Glide, Sitemap) | Docking scores, glide energy, parasitaemia reduction, IC50, hemocompatibility, MTT, physicochemical characterisation (UV–Vis, DLS, Zeta, FTIR, XRD, FESEM) |
| Murugesan and Kaleeswaran [ | Molecular docking, ADME/Tox (SWISS‐ADME), ProtParam | — | — | — | AutoDock Vina, PyRx | PROCHECK, ERRAT for protein structure, PASS server for toxicity |
| Affected pathway | Pathway description | Key gene (RNA seq) | Specific gene function | References |
|---|---|---|---|---|
| Apoptosis | Programmed cell death eliminates infected cells or regulates immune responses in malaria. | Bax | Pro‐apoptotic protein; promotes mitochondrial membrane permeabilisation | [ |
| Bcl2 | Antiapoptotic protein; inhibits programmed cell death by regulating mitochondrial membrane | [ | ||
| Caspase‐3 | Executioner caspase cleaves cellular proteins to execute apoptosis | [ | ||
| Inflammation/immune response | Immune activation to combat | CD11b | Integrin mediates leucocyte adhesion and migration in immune response | [ |
| IFN‐I3 | Cytokine: activates macrophages, enhances antigen presentation | [ | ||
| IL‐1I2 | Proinflammatory cytokine; activates immune cells and induces fever | [ | ||
| IL‐6 | Cytokine regulates immune response, inflammation and haematopoiesis | [ | ||
| IL‐12 | Cytokine stimulates T‐cell and NK‐cell activity, enhances IFN‐Î3 production | [ | ||
| iNOS | Inducible nitric oxide synthase, produces nitric oxide, enhances pathogen killing | [ | ||
| NFkB | Transcription factor; regulates inflammatory and immune gene expression | [ | ||
| PGES | Prostaglandin E synthase; produces prostaglandins, amplifies inflammation | [ | ||
| TLR2 | Toll‐like receptor; recognises pathogens, initiates inflammatory signalling | [ | ||
| TNF | Proinflammatory cytokine; promotes inflammation and cytotoxicity in infected cells | [ | ||
| TNF‐I | Proinflammatory cytokine; induces inflammation and apoptosis in infected cells | [ | ||
| Immune signalling/activation (upregulated) | Enhanced immune cell recruitment and pathogen recognition in response to malaria infection. | Ccl8 | Chemokine; attracts monocytes and lymphocytes, upregulated in immune response | [ |
| Cd209a | C‐type lectin receptor; mediates pathogen recognition, upregulated in immune response | [ | ||
| Cd209b | C‐type lectin receptor; involved in immune cell adhesion, upregulated | [ | ||
| Saa3 | Serum amyloid A3, an acute‐phase protein, promotes inflammation (upregulated) | [ | ||
| Cellular stress and related processes | Stress responses, cell proliferation or immune modulation induced by antimalarial compounds like artemisinin B. | APP | Amyloid precursor protein; involved in neuronal signalling, upregulated in artemisinin B‐treated cells | [ |
| CCDD1 | Unknown function; likely involved in cellular stress response (upregulated by artemisinin B) | [ | ||
| NT5E | Ecto‐5′‐nucleotidase; hydrolyses extracellular nucleotides, modulates immune response | [ | ||
| PCNA | Proliferating cell nuclear antigen; facilitates DNA replication and repair | [ | ||
| PRNP | Prion protein; involved in cellular signalling, upregulated in stress response | [ | ||
| STK39 | Serine/threonine kinase; regulates cellular stress and ion transport | [ | ||
| TXNIP | Thioredoxin‐interacting protein; inhibits antioxidant pathways, promotes oxidative stress | [ | ||
| Cell signalling, antimalarial activity | Signalling pathways targeted to disrupt | EphA2 | Receptor tyrosine kinase; mediates cell–cell signalling, a potential antimalarial target | [ |
| ephrin‐A | Ligand for EphA2; facilitates cell signalling in immune and endothelial cells | [ | ||
| Fatty acid biosynthesis ( | Essential for | Fab_I (ENR) | Enoyl‐ACP reductase; catalyses fatty acid elongation in | [ |
| Fab_Z | β‐hydroxyacyl‐ACP dehydratase; catalyses dehydration in | [ | ||
| Housekeeping (cellular structure, metabolism) | Baseline cellular are functions used as a reference in gene expression studies. | Actin | Cytoskeletal protein; maintains cell structure, used as a housekeeping gene | [ |
| GAPDH | Glyceraldehyde‐3‐phosphate dehydrogenase; catalyses glycolysis, a housekeeping gene | [ | ||
| Inflammation, apoptosis, oxidative stress | Multifaceted host response to malaria, balancing inflammation, cell death and oxidative stress. | APOE | Apolipoprotein E regulates lipid metabolism, modulates inflammation | [ |
| CASP3 | Executioner caspase activates apoptosis by cleaving cellular substrates | [ | ||
| EGFR | Epidermal growth factor receptor promotes cell proliferation and survival | [ | ||
| HMOX1 | Heme oxygenase‐1; degrades heme, reduces oxidative stress | [ | ||
| IL6 | Cytokine promotes inflammation and immune cell differentiation | [ | ||
| MMP2 | Matrix metalloproteinase‐2; degrades extracellular matrix, involved in tissue remodelling | [ | ||
| MMP9 | Matrix metalloproteinase‐9; degrades extracellular matrix, aids immune cell migration | [ | ||
| NOS3 | Endothelial nitric oxide synthase; produces nitric oxide, regulates vascular tone | [ | ||
| PTGS2 | Cyclooxygenase‐2; synthesises prostaglandins, promotes inflammation | [ | ||
| SRC | Nonreceptor tyrosine kinase; regulates cell signalling and survival | [ | ||
| Malaria parasite development (early gametocyte) | Early gametocyte formation in | PfGEXPS | Exported protein; marker for early‐stage gametocyte development in | [ |
| Malaria parasite development (late gametocyte) | The late gametocyte stage in | Pfs25 | Surface protein; marker for late‐stage gametocyte transmission in | [ |
| Anti‐inflammatory response | Suppression of excessive inflammation to limit host tissue damage in malaria. | CD36 | Scavenger receptor; binds oxidised LDL, modulates immune response | [ |
| HO‐1 | Heme oxygenase‐1; degrades heme, exerts anti‐inflammatory effects | [ | ||
| TGF‐β | Cytokine; suppresses inflammation, promotes immune tolerance | [ | ||
| Endothelial activation | Vascular changes facilitating immune cell adhesion and response in malaria infection. | ICAM‐1 | Intercellular adhesion molecule‐1 facilitates leucocyte‐endothelial interactions | [ |
| VEGF | Vascular endothelial growth factor promotes angiogenesis and vascular permeability | [ | ||
| Cytotoxicity | Direct killing of infected cells by immune effectors during the malaria immune response. | Granzyme B | Serine protease induces apoptosis in target cells during cytotoxic response | [ |
| Malaria parasite detection | Molecular detection of |
| Unknown specific genes; used for detecting | [ |
| Compound | Binding affinity (kcal/mol) | Target protein | Experimental IC50 | References |
|---|---|---|---|---|
| Isorhamnetin | −156.333 | Plasmepsin II (2BJU) | NA | [ |
| Euphorbianin | −151.001 | Plasmepsin II (2BJU) | NA | [ |
| Afzelin | −143.385 | Plasmepsin II (2BJU) | NA | [ |
| Myricitrin | −137.29 | Plasmepsin II (2BJU) | NA | [ |
| Quercitrin | −132.938 | Plasmepsin I (3QRV) | NA | [ |
| Leucocyanidol | −115.458 | Plasmepsin II (2BJU) | NA | [ |
| Pinocembrin | −98.8994 | Plasmepsin II (2BJU) | NA | [ |
| 11‐O‐Galloylbergenin | −16.22 | PfLDH (Not specified) | 7.85 A ± 0.61 AµM | [ |
| Myricetin 3‐O‐glucoside | −13.413 | PfLDH (1T2C) | NA | [ |
| 2″‐O‐Galloylisovitexin | −12.896 | PfLDH (1T2C) | NA | [ |
| Bergenin | −12.13 | PfLDH (Not specified) | 6.92 ± 0.43 µM | [ |
| Sitoglucoside | −11.6 | pfENR (1VRW) | NA | [ |
| Isovitexin | −11.485 | PfDHFR–TS (3UM8) | NA | [ |
| Rutin | −11.25 | AMA1 (3SRI), M18AAP (6PEV), Pf12p (7KJ7), TyrRS (3VGJ) | NA | [ |
| Astragalin | −11.02 | AMA1 (3SRI), M18AAP (6PEV), Pf12p (7KJ7), TyrRS (3VGJ) | NA | [ |
| Ergosterol peroxide | −10.9 | pfENR (1VRW) | NA | [ |
| Friedelin | −10.6 | DHFR (2BL9) | NA | [ |
| Vitexin | −10.601 | PfDHFR–TS (3UM8) | NA | [ |
| Bauerenol | −10.5 | DHFR (2BL9) | NA | [ |
| Cynaroside | −10.5 | LysRS (4YCV) | NA | [ |
| Quercetin 3‐O‐alpha‐L‐rhamnopyranoside | −10.45 | AMA1 (3SRI), M18AAP (6PEV), Pf12p (7KJ7), TyrRS (3VGJ) | NA | [ |
| Î2‐Sitosterol | −10.2 | pfENR (1VRW) | 734.3 ± 1.213 µg/mL | [ |
| Sesamin | −10 | PfDHODH (5TBO) | > 10,000 nM | [ |
| Isoquercitrin | −9.93 | AMA1 (3SRI), M18AAP (6PEV), Pf12p (7KJ7), TyrRS (3VGJ) | NA | [ |
| Sophoroside | −9.711 | PfDHFR–TS (3UM8) | NA | [ |
| Alpha‐muurolene | −9.7 | mJHBP (5V13) | NA | [ |
| Gamma‐cadinene | −9.7 | mJHBP (5V13) | NA | [ |
| Artocarpin | −9.6 | Falcipain‐2 (3BPF) | NA | [ |
| Pyrogallol | −9.5 | Falcipain‐2 (3BPF) | NA | [ |
| Decarine | −9.5 | PfDHODH (5TBO) | 1333 nM (with Trans‐fagaramide) | [ |
| Luteolin | −9.5 | EphA2 (6FNH) | 3.4 µM | [ |
| Epicatechin‐gallate | −9.3 | PfCDPK‐1 (I‐TASSER model) | NA | [ |
| 2‐Ethylacridine | −9.3 | PfDHODH (3165) | NA | [ |
| Rhaponticin | −9.3 | PfDHODH (5TBO) | NA | [ |
| Ascorbic acid | −9.3 | Falcipain‐3 (3BWK) | NA | [ |
| Isocolumbin | −9.3 | EphA2 (6FNH) | NA | [ |
| Pachyrrhizin | −9.9 | Hexkinase‐1 (1CZA) | NA | [ |
| Ursolic acid | −9.7 | PfEMP‐1 (7JGD) | NA | [ |
| Betulinic acid | −9.1 | PfPKG (5DYL) | NA | [ |
| Picracin | −9.1 | PfCDPK‐1 (I‐TASSER model) | NA | [ |
| Biological function | Target protein | PDB ID/status | References |
|---|---|---|---|
| Proteases | Falcipain‐2 (FP2) | 3BPF, 6JW9, 7e10 | [ |
| Falcipain‐3 (FP3) | 3BWK | [ | |
| M18 aspartyl aminopeptidase | 6PEV | [ | |
| Plasmepsin I | 3QS1, 3QRV | [ | |
| Plasmepsin II | 1LF3, 2BJU, 2IGY, 4YA8, 4CKU, 5JOD | [ | |
| Plasmepsin IV | 5JOD | [ | |
| Plasmepsin X (PMX) | 7RY7 | [ | |
| 20S proteasome | 7LXU | [ | |
| Metabolic enzymes | Dihydrofolate reductase (DHFR) | 2BL9 | [ |
| Dihydrofolate reductase–thymidylate synthase (DHFR–TS) | 1J3I, 3UM8, 113K | [ | |
| Dihydroorotate dehydrogenase (PfDHODH) | 5TBO, 1TV5, 3165 | [ | |
| Enoyl acyl‐carrier protein reductase (pfENR) | 1VRW | [ | |
| Fructose‐bisphosphate aldolase | PF3D7_1444800 (no PDB ID) | [ | |
| Hexokinase‐1 | 1CZA | [ | |
| Lactate dehydrogenase (PfLDH) | 1LDG, 1U5A, 1T2C, 1CET, 1U40, no PDB ID | [ | |
| LDH/GAPDH | PF3D7_1320800/PF3D7_1462800 (no PDB ID) | [ | |
| Phosphoethanolamine methyltransferase (PMT) | 3UJB, 2UJ9 | [ | |
| Phosphocholine cytidylyltransferase | 4ZCS, 4ZCR | [ | |
| Spermidine synthase | 4CWA | [ | |
| Choline kinase | 6YXT | [ | |
| Hypoxanthine‐guanine‐xanthine phosphoribosyltransferase (PfHGXPRT) | 1TUX | [ | |
| Glyoxalase‐1 (GLO‐1) | No PDB ID specified | [ | |
| Phosphoglycerate kinase (PGK) | PF3D7_0922500 (no PDB ID) | [ | |
| Transport proteins | Ca2+‐ATPase | 2KNE | [ |
| Calcium transporter CAX | 4K1C | [ | |
| Mosquito juvenile hormone‐binding protein (mJHBP) | 5V13 | [ | |
| PfATP6 | Homology modelled | [ | |
| Signalling proteins | Aurora A | 4O0U | [ |
| Aurora B | 4AF3 | [ | |
| EphA2 receptor | 6FNH, 5NK0 | [ | |
| PfCDPK‐1 | No PDB ID modelled via I‐TASSER | [ | |
| PfPKG (cGMP‐dependent protein kinase) | 5DYL | [ | |
| PfCLK1, PfCLK4, PfMap2, PfNek1 | Homology models | [ | |
| tRNA synthetases | Anopheles gambiae bifunctional glutamyl/prolyl‐tRNA synthetase (AgEPRS) | No PDB ID homology modelled | [ |
| Glutamyl‐tRNA synthetase (ERS) | 7WAJ | [ | |
| Lysyl‐tRNA synthetase | 4YCV | [ | |
| Tyrosyl‐tRNA synthetase | 3VGJ | [ | |
| PfcPRS | PF3D7_1213800 modelled on human PRS (4HVC) | [ | |
| Chaperones/stress response | Anopheles gambiae heat shock protein 70 kDa (AgHSP70KDa) | No PDB ID homology modelled | [ |
| Hsp70‐1 (PfHsp70‐1) | Homology model no PDB ID; PF3D7_0818900 | [ | |
| Hsp70‐z (PfHsp70‐z) | No PDB ID | [ | |
| Antioxidant enzymes | Superoxide dismutase (SOD1) | 2C9V | [ |
| Catalase (CAT) | 1QQW | [ | |
| Glutathione peroxidase (GPx1) | 1GP1 | [ | |
| Glutathione reductase (GR) | 1XAN | [ | |
| DNA/RNA processing | Apicoplast DNA polymerase (apPOL) | 7SXQ | [ |
| DNA topoisomerase II | 6CA8 | [ | |
| Membrane/structural proteins | Apical membrane antigen 1 | 3SRI | [ |
| Erythrocyte membrane protein 1 (PfEMP‐1) | 7JGD, 3C64 | [ | |
|
| 2LOE | [ | |
| Pf12p | 7KJ7 | [ | |
| Pfg27 (gametocyte protein) | No PDB ID | [ | |
| PCM1 | PF3D7_1120100 (no PDB ID) | [ | |
| Electron transport | Cytochrome bcl reductase | 4PD4 | [ |
| Cytochrome c2 (cyt c2) | 7TXE | [ | |
| Cytochrome c2 domain‐swapped dimer (cyt c2 DSD) | 7TXE | [ | |
| Immune/inflammatory response | High mobility group Box 1 (PfHMGB1 HsHMGB1) | 2MRC, 1HME | [ |
| IL6 | No PDB ID | [ | |
| TNF Alpha | No PDB ID | [ | |
| Haemozoin formation | Haemozoin crystal lattice | No PDB ID specified | [ |
| Ion channel | hERG | 5VA2 | [ |
| Machine learning model | Input data type | Prediction output | AUC/accuracy | References |
|---|---|---|---|---|
| KNN, logistic regression, decision tree | GC–MS data (239 components), chemical composition | % inhibition of zygote/ookinete formation | MCC: 0.701–0.840, Best MCC: 0.840 | [ |
| Binary logistic regression | Climatic variables (temperature, humidity, rainfall, elevation) | Endemicity prediction (1 = endemic, 0 = non‐endemic) | Not specified | [ |
| Chemprop, MAIP | Molecular structures (SMILES, Morgan fingerprints) | Antimalarial activity probability | AUC: 0.868590 ± 0.007887 | [ |
| Genetic function algorithm (GFA) | 2D descriptors (ALogP2, ATS1m, ATS7m, ATS0v) | Binding affinity (QSAR model) |
| [ |
| Random forest (RF) | Chemical descriptors (logP, drug likeness, amines, etc.) | Cardiotoxicity, hepatotoxicity, mutagenicity | AUC: 0.830–0.869 | [ |
| Support vector machine (SVM) | Molecular structures (SMILES), ISIDA fragment descriptors (39 sets) | Binary classification (active/inactive) | Balanced accuracy: 0.701–0.953, prospective: 0.62 | [ |
| S/No. | Affected pathway | Validation methods | References |
|---|---|---|---|
| 1 | Immune response (inflammation, T cell recruitment, phagocytosis) | RT‐qPCR (gene expression: TNF, IL‐1β, IL‐6, IL‐12, IFNγ), histological analysis, TUNEL assay (apoptosis), in vivo parasitaemia suppression, LDH assay (cytotoxicity), EC50 (antioxidant), HRMS/MS (phytochemicals) | [ |
| 2 | Oxidative stress (GSH/CAT/MDA/NO, ROS, HO‐1) | Biochemical assays (GSH, CAT, MDA, NO), RT‐qPCR (Bcl2/Bax/caspase‐3), histopathology, TEAC (ABTS assay), GSH (DTNB assay), TBARS (lipid peroxidation), in vivo parasitaemia suppression, microscopy (Giemsa‐stained smears) | [ |
| 3 | Lysyl‐tRNA synthetase (LysRS) | Molecular docking (AutoDock Vina), ADMET prediction (SwissADME), in vivo parasitaemia suppression, haematological indices | [ |
| 4 | Dihydrofolate reductase (DHFR) pathway | Molecular docking (AutoDock Vina), ADMET prediction (SwissADME), in vivo parasitaemia suppression, dose‐response curves (nonlinear regression), microscopic parasite clearance, ROC curve (AUC = 0.733), RMSD (0.959 Å) | [ |
| 5 | Plasmepsin I inhibition (haemoglobin degradation) | In vitro antiplasmodial assay (HRP2 ELISA), molecular docking (AutoDock Vina), Ramachandran plot, ProSA‐web z‐score, RMSD comparison (I‐TASSER, Rosettafold) | [ |
| 6 | Amino acid starvation response (AAR, eIF2 | Whole‐genome sequencing (SNPs via Illumina, SAMtools), yeast complementation, Western blot (eIF2 | [ |
| 7 | Anaerobic glycolysis (PfLDH inhibition, ATP synthesis) | Molecular docking (AutoDock Vina, RMSD ∼2.0 Å), MD simulation (RMSD, RMSF, Rg, H‐bonds), MM–PBSA, pLDH assay, IC50 determination, NMR/HR‐MS, redocking (NADH, RMSD = 0.775 Å), Moldock score, pKi | [ |
| 8 | Antioxidant defence (SOD, CAT, GPx, GR) | Repeated oxidative stress measurements (TEAC, GSH, TBARS), molecular docking (Molegro Virtual Docker), statistical significance ( | [ |
| 9 | Apicoplast DNA replication (apPOL), Protein translation (glutamyl‐tRNA synthetase) | Molecular docking (PyRx), RMSF, Rg, Protein Plus analysis, Lipinski’s Rule of Five | [ |
| 10 | Haemozoin formation (β‐haematin inhibition) | In vitro β‐haematin assay, luciferase assay (gametocyte viability), SYBR Green I assay (ABS parasite growth), molecular docking (Schrödinger, AutoDock 4.2), UPLC–QTOF–MS, HMBC, COSY, comparison with artemisinin | [ |
| 11 | Cardiotoxicity (hERG, gene expression) | In vitro (AC16 cardiomyocytes), in vivo (zebrafish), molecular docking, microarray | [ |
| 12 | Cholesterol sequestration, lipid raft disorganisation, HMGB1, GLO‐1 | In vitro pLDH assay (IC50), in vivo parasitaemia clearance, NMR (PfHMGB1, GLR‐HsHMGB1), molecular docking (ΔE, ΔG), MTT assay (Vero cells, SI = 9.6) | [ |
| 13 | Dihydroorotate dehydrogenase (DHODH) | In vitro schizont maturation inhibition, molecular docking (AutoDock Vina, RMSD = 0.15 Å), ADMETlab 2.0, IC50 values, selectivity index (SI) | [ |
| 14 | Cysteine/methionine‐rich protein functions (M18, Tyrosyl‐tRNA synthetase, Pf12p) | Molecular docking, in vitro parasitaemia reduction (Giemsa smears), IC50, hemocompatibility assay, MTT assay (HEK293), physicochemical analysis (UV–vis, DLS, Zeta, FTIR, XRD, FESEM) | [ |
| 15 | Gametocyte‐to‐ookinete development | In vitro inhibition (> 50%, | [ |
| 16 | Erythrocyte invasion (PfCDPK‐1) | Molecular docking, Ramachandran plot, ProSA‐web z‐score, RMSD comparison (I‐TASSER, Rosettafold), in vitro assays | [ |
| 17 | Fatty acid biosynthesis (FAS‐II) | qPCR (gene expression), GC–MS, molecular docking (binding affinity) | [ |
| 18 | Folate metabolism (DHFR–TS) | Molecular docking (Glide, PyRx), ROC curve (AUC = 0.733), RMSD (0.959 Å), correlation with pIC50 ( | [ |
| 19 | Glycolysis (hexokinase‐1, PCM1, GAPDH, Aldolase, PGK), purine recycling | In silico validation (PROCHECK, ERRAT, PASS), molecular docking, QSAR ( | [ |
| 20 | Proteasome degradation | Protein spot quantification (≥ 2‐fold up‐regulation), PIQUES™ software, 2‐DE, LC/MS/MS | [ |
| 21 | Mitochondrial electron transport (cytochrome c2, bc1) | Molecular docking (AutoDock Vina), PASS (Pa: 0.892), MD simulation, NMR, HRESIMS, Malstat assay, in vitro parasite growth inhibition, haemolytic activity, cytotoxicity (A549, Vero) | [ |
| 22 | PfHsp70‐1 chaperone function, protein folding | In vitro UV–Vis spectroscopy, MDH aggregation suppression assay, pLDH assay, ATPase assay (normalised to basal activity), SDS–PAGE, comparison with chloroquine (IC50 = 8.5 ng/mL) | [ |
| 23 | Purine salvage pathway (PfHGXPRT) | In vitro pLDH assay, cytotoxicity (MTT), biophysical assays (DSF, UV–vis), molecular docking | [ |
| 24 | Pyrimidine metabolism (UMP biosynthesis, DHODH) | Molecular docking (AutoDock Vina, RMSD = 0.15 Å), ITC (Ka, ΔH), network pharmacology (STRING DB, | [ |
| 25 | NF‐κB pathway, endothelial apoptosis | Molecular docking, binding interactions (hydrogen bonds, hydrophobic), computational physicochemical analysis | [ |
| 26 | PfATP6 (Ca2+‐ATPase) inhibition | Comparative homology modelling, molecular docking, in vitro β‐haematin assay | [ |
| 27 | PfEMP‐1 mediated adhesion, PfPKG Signalling | In vivo parasite density, % growth inhibition, GC–MS, molecular docking, ADME analysis | [ |
| 28 | Insect hormonal regulation (mJHBP) | Molecular docking, MD simulation (RMSD, RMSF, Rg, SASA) | [ |
| 29 | Thioredoxin reductase (TrxR), glutathione metabolism, ROS induction | SVM model (cross‐validation, balanced accuracy > 0.7), IC50 (ICEstimator), cytotoxicity (MRC‐5, SI), in silico consensus voting (> 70% likelihood) | [ |
| 30 | Hypolipidemic effect, metabolic enzymes (PfLDH, Plasmepsin II) | In vivo parasitaemia reduction, biochemical assays, molecular docking, MD simulation stability (RMSD, RMSF) | [ |
| 31 | Lysosomal/endocytic signalling (Thailandine) | NMR, MS, in vitro cytotoxicity ( | [ |
| 32 | Ubiquitin–proteasome, glycerophospholipid metabolism, DNA replication | In vitro Malstat assay, molecular docking (AutoDock Vina), MD simulation (Desmond), PPI network (cytoscape), docking scores, ΔG_binding < −7 kcal/mol | [ |
| 33 | Mitotic division (PfNek1, PfMap2, PfCLK1, PfCLK4), transmembrane transport | In vitro/ex vivo susceptibility assays, molecular docking (AutoDock Vina), IC50, selectivity index | [ |
| 34 | PI3K‐Akt, relaxin signalling | Molecular docking, MD simulation, machine learning (Chemprop, MAIP), binding scores | [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMalaria Research and Control · vaccines and immunoinformatics approaches · Genetics, Bioinformatics, and Biomedical Research
- •Molecular docking identified nonstandard antimalarials like isorhamnetin (−156.33 kcal/mol, Plasmepsin II), targeting haemoglobin degradation.
- •RNA‐seq showed herbal extracts modulating Plasmodium genes (PfGEXPS) and host immunity (IL-6), disrupting parasite development.
- •ML models (KNN, Chemprop) predicted phytochemical bioactivity with AUC 0.87–0.95, enhancing antimalarial screening.
- •Multiomics and network pharmacology prioritised PfDHODH, integrating docking and proteomics to combat resistance.
- •Techniques targeted Plasmodium pathways like glycolysis and folate metabolism, validated in vitro, supporting multistage therapies.
1. Introduction
Malaria remains one of the most pressing global health challenges, with significant socioeconomic impacts, particularly in low‐ and middle‐income countries [1, 2]. According to the World Health Organisation (WHO), an estimated 247 million cases of malaria and 619,000 related deaths occurred in 2021, with the vast majority reported in sub‐Saharan Africa. In 2022, malaria disease increased to 249 million cases in 85 countries, most of which were in Africa (93.6%), the Eastern Mediterranean (3.3%), Southeast Asia (2.1%), the Western Pacific (0.8%) and the Americas (0.2%) [3–6]. The child population aged below 5 years contributes to the deaths by almost 79%, and this shows the incessant vulnerability of the population [2]. The deadliest of the malaria‐causing parasites, Plasmodium falciparum, has evolved resistance to almost all first‐line antimalarial treatments over time [7], including the artemisinin‐based combination therapy (ACT), which is now regarded as the gold standard in malaria treatment. Drug resistance is a current menace that highlights the importance of novel research methods in enhancing malaria control and treatment interventions.
Over the past few years, molecular technologies have turned out to be an invaluable asset in malaria research, helping scientists to unravel the complicated biology of Plasmodium species and providing a way to speed up drug discovery. Among them, one can distinguish gene expression profiling, molecular docking and machine learning (ML) because of their influence and relevance to different fields of research. RNA sequencing (RNA‐seq), especially at the single‐cell scale, has also been used to offer information on the biology of parasites by showing cell–cell variability when it comes to gene expression [8]. This degree of granularity is essential to the comprehension of stage‐specific transcriptional programmes that cannot be identified by bulk transcriptomics. As an example, scRNA‐seq has been applied to dissect the life cycle of Plasmodium and has helped identify genes that mediate the switch between asexual and sexual stages that are critical to its transmission and survival [2, 9]. Another application of RNA‐seq is the evaluation of the changes in gene expression with respect to environmental signals and drug exposure, such as herbal intervention, though this field is not thoroughly studied yet [10].
Molecular docking is used as an adjunct method in that researchers can predict how the small molecules interact with Plasmodium proteins. This is a critical computational approach to determine binding affinities and possible drug‐target interactions and therefore inform experimental validation and rational drug design [11]. The power of docking studies is specifically strong in conjunction with the omics data, and it can be used to explore the protein function and ligand specificity in a more systematic way. The last studies have used deep learning docking methods, that is, the diffusion model (Targetdiff), to generate and screen novel compounds using structural features of essential Plasmodium proteins [2, 9, 12].
Another transformative tool is ML, which has made breakthroughs in the analysis of large‐scale biological data in malaria studies. ML algorithms are effective in the classification of compounds as active or inactive, drug resistance pattern prediction, and the optimal selection of lead compounds to be subjected to experimental testing, based on molecular descriptors [13]. Various experiments have indicated that the random forest (RF), support vector machines (SVMs) and artificial neural network models can be used to predict antimalarial compounds with a high level of accuracy [9, 12, 14, 15]. Notably, feature selection methods help in boosting the performance of these models by selecting the most informative descriptors, thereby improving the performance of the models at the expense of low computational complexity.
The systematic review examines the role of the modern state‐of‐the‐art molecular and computational tools in revolutionising malaria research, and three areas are discussed as follows. First, it discusses the capability of RNA‐seq to trace genetic changes in Plasmodium parasites that have encountered natural therapies, which sheds some light on the way in which traditional therapies can interfere with the process of infections. Second, it also measures the accuracy of molecular docking to study drug‐target interactions and thus its importance in the search for promising compounds among large chemical libraries. Lastly, it evaluates the twofold influence of ML in forecasting the emergent drug resistance trends as well as simplifying the process of identifying new antimalarial compounds. The review highlights the potential of combining laboratory‐based molecular understanding with computational creativity to speed up advances in one of the oldest infectious diseases that has reached humankind by exploring the extent to which these tools can be effective in overcoming critical difficulties, such as treatment modification and bottlenecks in drug development.
2. Methods
2.1. Database Search Process
The Preferred Reporting Items of Systematic Reviews and Meta‐Analyses (PRISMA) guidelines [16] were used to complete this systematic review. Three major electronic databases (PubMed, Scopus and Web of Science) were searched. The search was conducted to find the studies that were applicable to the analysis of applying molecular techniques in malaria research, especially the studies that involved gene expression profiling, molecular docking and ML. A combination of the following search terms was used: (Plasmodium OR malaria) AND (RNA sequencing OR gene expression) AND (herbal treatment), as well as (machine learning OR AI) AND (drug resistance prediction). Peer‐reviewed articles published in English were considered, published between the year 2014 and 2024.
2.2. Criteria for Eligibility
Studies were also included in case they satisfied several predetermined criteria. Only studies that used gene expression profiling, molecular docking or machine learning methods were included in the scope of malaria research. Only the studies that are related to the Plasmodium species, including P. falciparum, P. vivax or P. berghei, were taken into consideration. The research that involved the use of natural products or herbal plants as an intervention was taken into consideration. To be eligible, the studies had to be primary research articles, and they needed to have computational or in silico analyses and have been published in English in the last 10 years, 2014–2024, in any peer‐reviewed journal. In addition, the inclusion criteria required that the molecular procedure should be well outlined and its use in malaria studies should be explicitly stated. Articles were filtered by eliminating those that were not molecular, malaria‐unrelated, non‐Plasmodium‐related, opinion pieces, editors, conference abstracts, reviews or preprints that were not peer reviewed. Articles that were older than the specified range of dates of publications or were not in English and articles that were not clear on the method or technique used were also excluded.
2.3. Data Extraction
The systematic selection of data on eligible articles by using a predesigned template, specific to the objectives of the review, was used to collect crucial information related to gene expression profiling, molecular docking and ML studies on malaria. Fields that were extracted were as follows: study title, year of publication, study design/type, species/strain of Plasmodium, sample size, herbal treatment/intervention, comparator group/standard, technique used, key genes (RNA‐seq), affected pathways, validation method, docking software, target protein (PDB ID), ligands screened, binding affinity (ΔG), half maximal inhibitory concentration (IC_50_), ML model, input data type, prediction output, AUC/accuracy, limitations, key findings, research gaps and reference/author. Data were extracted by two reviewers into an Excel spreadsheet, and differences were resolved by agreement. Missing data were denoted as ‘Not Reported’, and supporting information was used where possible so that a complete and reproducible synthesis of findings was done [17].
2.4. Quality Assessment
The quality of included studies had been assessed with a stringent level so that the reliability and validity of the findings could be considered credible in this systematic review. Articles found in e‐libraries were uploaded to Rayyan [18], a web‐based application, to undergo independent screening. The scientific names of plants used in the studies were picked and cross‐tabulated to ascertain accuracy in the scientific name using the World Flora Online (https://wfoplantlist.org) to ensure taxonomic accuracy. Study integrity assessment criteria were specific to the gene expression profiling, molecular docking and ML articles, which were implemented to test, study design, methodology and reporting to guarantee a strong evidence synthesis of the molecular techniques in malaria research.
3. Results
3.1. Overview of Included Studies
The systematic review identified 324 records through database searches (PubMed: 45, Web of Science: 80, Scopus: 199), with 232 unique records after duplicate removal. Following title and abstract screening, 153 records were excluded, and 79 full‐text articles were assessed for eligibility. Fifteen articles were excluded due to inappropriate design (n = 10) or lack of relevant outcomes/interventions (n = 5), resulting in 64 studies included in the qualitative synthesis; see Figure 1. Among 64 studies analysed for molecular techniques, molecular docking was identified as the most commonly employed method, constituting 32.37% of the 139 total technique findings. In vitro antiplasmodial assays were utilised in 14.39% of the findings, while ADMET profiling was applied in 10.79% of cases. Cytotoxicity assays were documented in 9.35% of the findings, and GC–MS was used in 8.63%. NMR spectroscopy was employed in 7.19% of cases, followed by molecular dynamics (MDs) simulation at 6.47%. Bioassay‐guided fractionation was reported in 5.04% of the findings, and gene expression techniques were noted in 3.60%. ML and UPLC–QTOF–MS were each recorded in 2.16% of the findings, while 3D homology modelling, MM/GBSA or MM/PBSA analysis, 3D molecular modelling and energy minimisation were each implemented in 1.44% of cases. These results highlight the predominant role of docking and profiling methods in advancing molecular research on malaria. Table 1 also provides a comprehensive review of methodologies in antimalarial drug discovery: in silico, in vitro, in vivo, chemical characterisation, docking and validation strategies reported by included studies.
Prisma flowchart for study selection.
3.2. Gene Expression Profiling
The application of RNA‐seq to investigate Plasmodium gene expression has been extensively explored across the included studies. Table 2 gives a summary of key genes grouped by affected pathways in malaria, with functional descriptions, with notable findings related to gene expression changes induced by herbal treatments. In a study by Dkhil et al. [16], female mice upregulation of Ccl8, Saa3, Cd209a and Cd209b was observed in response to Indigofera oblongifolia leaf extract (IOLE), while 24 immune response genes were downregulated, indicating a complex immunomodulatory effect. Similarly, Dkhil et al. [62] reported that IOLE modulated the expression of apoptotic genes (Bcl2, Bax, Caspase-3) and cytokine genes (IL-1β, IL-6, IFN-γ, TNF-α), suggesting its potential to influence both programmed cell death and inflammatory pathways during malaria infection. Another investigation by Kadioglu et al. [55] identified upregulation of APP, CCDD1, NT5E, PCNA, PRNP, STK39 and TXNIP in cells of zebrafish larvae treated with artemisinin derivatives, such as artemisinin B and deoxydihydro‐artemisinin, highlighting their role in cellular proliferation and stress response pathways. Additionally, Miao et al. [14] demonstrated, via in silico studies, that Cordia myxa compounds altered the expression of IL‐6, CASP3, PTGS2, SRC, HMOX1, MMP‐9, APOE, NOS3, EGFR and MMP‐2, thereby affecting inflammatory, oxidative stress and extracellular matrix remodelling pathways. Camara et al. [44] reported in P. berghei‐infected mice modulation of proinflammatory (IL-1β, TNF, IL-12, IFN-γ, CD11b, TLR2, NFkB, PGES, iNOS), anti‐inflammatory (TGFβ, CD36, HO-1), endothelial activation (ICAM-1, VEGF) and cytotoxic (Granzyme B) genes in response to methanolic crude extract of Terminalia albida stem bark, with GAPDH and P. berghei‐specific primers used as controls. Snider and Weathers [58] observed altered expression of gametocyte markers PfGEXPS (early‐stage) and Pfs25 (late‐stage) in response to Artemisia annua and Artemisia afra tea infusions from NF54 P. falciparum asexual parasites, indicating potential interference with parasite sexual development.
Techniques such as single‐cell RNA‐seq and bulk RNA‐seq were commonly employed to capture these gene expression profiles, offering high‐resolution insights into Plasmodium and host responses to herbal interventions. For instance, Kane et al. [35], using P. falciparum cultures (W2 and D6) with a parasitaemia level of 4%, utilised RNA‐seq to assess the impact of A. afra extracts on Fab_I (ENR), Fab_Z (β‐hydroxyacyl‐ACP dehydratase) and Actin (housekeeping) genes, revealing disruptions in fatty acid biosynthesis pathways critical to parasite survival. Challenges in these studies included data variability due to differences in sample quality, RNA extraction methods and sequencing depth, as well as complexities in bioinformatics pipelines for data normalisation and differential expression analysis. Variability in herbal extract composition, as seen with methanolic extracts of T. albida or Artemisia species, further complicates reproducibility across studies. Despite these challenges, the identification of key genes and pathways, such as those involved in apoptosis [15], inflammation [17] and parasite development [18], underscores the potential of gene expression profiling to elucidate the molecular mechanisms of herbal interventions in malaria research. While the reviewed studies highlight the regulatory potential of herbal compounds on both host and parasite gene expression, an important yet underexplored opportunity lies in systematically linking these transcriptional changes to phenotypic outcomes using integrative approaches. For instance, correlating differential expression patterns with parasite clearance rates, cytokine profiles or survival outcomes could validate the functional relevance of these transcriptional shifts. Moreover, single‐cell RNA‐seq offers a promising avenue to resolve intra‐population heterogeneity in Plasmodium responses to natural products, which bulk RNA‐seq may mask. Going forward, coupling gene expression data with metabolomic or proteomic readouts could provide a more holistic understanding of the molecular cascades influenced by herbal interventions, potentially revealing synergistic effects or compensatory mechanisms that underlie treatment efficacy. Such integration would not only enrich mechanistic insight but also enhance the translational value of gene expression profiling in antimalarial research.
3.3. Molecular Docking for Drug‐Target Interactions
Computational tools, including AutoDock, AutoDock Vina, PyRx, Schrödinger and Molegro Virtual Docker, were used to predict interactions between phytochemicals and well‐defined Plasmodium protein targets, primarily P. falciparum enzymes such as PfLDH (1CET, 1LDG, 1T2C, 1U5A), Falcipain-2 (3BPF, 6JW9, 7E10), Plasmepsin II (1LF3, 2BJU, 2IGY, 4CKU, 4YA8), DHFR–TS (1J3I, 2BL9, 3UM8), PfDHODH (1TV5, 5TBO, 3I65) and the 20S proteasome (7LXU), with species, strain and structural identifiers detailed in Tables 3 and 4. Among the most frequently targeted proteins, P. falciparum lactate dehydrogenase (PfLDH, PDB IDs: 1CET, 1LDG, 1T2C, 1U5A) was prominent, with a targeting frequency of approximately 21.43% across the dataset, as evidenced by its mention in 12 studies. In all included studies, docking was performed against structurally defined targets, most commonly P. falciparum proteins such as PfLDH (e.g., PDB IDs 1CET, 1LDG, 1T2C, 1U5A), Falcipain-2 (3BPF, 6JW9, 7E10), Falcipain-3 (3BWK), Plasmepsin I (3QRV, 3QS1), Plasmepsin II (1LF3, 2BJU, 2IGY, 4CKU, 4YA8), DHFR–TS (1J3I, 2BL9, 3UM8), PfDHODH (1TV5, 5TBO, 3I65) and the 20S proteasome (7LXU). P. falciparum 3D7 structures were used as reference models, even when the biological focus included other Plasmodium species such as P. vivax or P. berghei, consistent with the high sequence and functional conservation of these enzymes across species.
Other key targets included Dihydrofolate Reductase–Thymidylate Synthase (DHFR–TS, 1J3I, 2BL9, 3UM8), Dihydroorotate Dehydrogenase (PfDHODH, 1TV5, 5TBO, 3165) and 20S proteasome (7LXU). For example, Samuel et al. [36] reported myricetin 3‐O‐glucoside with a binding affinity of −13.413 kcal/mol against PfLDH (1T2C), validated through in silico pharmacophore modelling and ADMET predictions. Similarly, Elmaidomy et al. [57] identified limonin, luteolin and myricetin with ΔG_binding < −7 kcal/mol for 20S proteasome (7LXU), choline kinase (6YXT) and phosphocholine cytidylyltransferase (4ZCS), confirmed by in vitro Malstat assays and MDs simulations using Desmond. Chaurasia and Pandey [48] demonstrated strong binding of artocarpin (−9.6 kcal/mol) to Falcipain-2 (3BPF) and ascorbic acid (−9.3 kcal/mol) to Falcipain-3 (3BWK) among 50 phytochemicals from Artocarpus species, supported by in vitro β‐haematin assays (91.7% efficiency compared to artemisinin). Singh et al. [60] reported rutin’s exceptional affinity (−11.25 kcal/mol) for Apical Membrane Antigen 1 (3SRI), validated through in vitro parasitaemia reduction and hemocompatibility assays. Dzouemo et al. [50] found sesamin (−10.0 kcal/mol) and decarine (−9.5 kcal/mol) binding strongly to PfDHODH (5TBO), corroborated by in vitro antiplasmodial assays and spectroscopic data. Binding affinities across studies typically ranged from −6.0 to −13.4 kcal/mol, with notable outliers like euphorbianin (−151.001 kcal/mol) against Plasmepsin II (2BJU) in Shah et al. [65], validated via Lipinski’s rule and ADMET profiling. Where P. falciparum structures were used as surrogates for orthologous proteins from other Plasmodium species, this choice was supported by reported high sequence identity and conservation of active‐site residues (e.g., PfLDH, DHFR–TS, PfDHODH), indicating that the docking poses are likely to be biologically relevant across species.
Validation of these docking predictions was robustly conducted using a variety of in vitro and in vivo methods to ensure reliability. Several studies combined docking with in vitro, in vivo and cytotoxicity profiling of nonstandard compounds, providing more translational evidence for their antimalarial potential (e.g., β‐carboline derivatives and plant‐derived scaffolds) [80–82]. In vitro assays, including Malstat, SYBR Green I, pLDH, β‐haematin inhibition and luciferase assays, were frequently utilised, most commonly against P. falciparum laboratory strains such as 3D7 (chloroquine‐sensitive), NF54 and W2 or K1 (chloroquine‐resistant), as well as rodent models like P. berghei ANKA where applicable, as reported by Hidayati et al. [52], Adeoye et al. [38] and Mianda et al. [77]. However, not all studies consistently reported the Plasmodium strain used, which may complicate comparisons given known strain‐dependent differences in drug response. In vivo validations, such as parasitaemia suppression, survival curves and Peters’ tests, were reported by Asanga et al. [45], Herman et al. [75] and Alebachew et al. [68]. Additional methods included spectroscopic analyses (NMR, MS, FTIR), cytotoxicity assays (MTT, LDH) and MD simulations assessing RMSD, RMSF and hydrogen bond profiles, as noted in Rawa et al. [27] and Chaurasia and Pandey [48]. Computational validations, such as redocking with RMSD < 2.0 Å, were reported by Adams et al. [53] (RMSD = 0.959 Å). Ramachandran plots and ADME–Tox profiling were also reported by Shekari et al. [37] to further strengthen the findings. For instance, Konyanee et al. [43] validated rheediachromenoxanthone’s binding (−8.6 kcal/mol) to PfLDH (1LDG) through in vitro IC_50_ determination and redocking (RnMSD = 0.775 Å).
Although these developments were made, there has been a constraint on the accuracy of the calculations and experimental validation. Such dependence on homology models as PfCDPK-1 [19] or Anopheles gambiae HSP70KDa [37] created uncertainty because of possible errors in the binding sites predicted. Due to the variation between AutoDock Vina and Glide docking algorithms, it was observed that the binding affinity estimates were not consistent, as reported by Shah et al. [65]. The small sample sizes and the variations in the composition of the herbal extracts [59, 71], as well as the lack of clinical translation, were also among the challenges in the field of experiments, as Onguéné et al. [20] emphasised. Drug development is further complicated by the complexity of targeting proteins that are specific to a particular stage, for example, Pfg27 in gametocytes [23]. These limitations indicate the importance of standardised docking methods, improved homology modelling methods and thorough in vivo experiments in applying computational predictions to effective antimalarial therapies.
Molecular docking has succeeded in establishing the strong binding effects of phytochemicals with the Plasmodium targets, but much more is still unknown about the mechanism that causes effective inhibition in the biological context of the parasite. Numerous experiments aim to only predict the strength of binding, without follow‐up experiments or kinetic modelling to determine the duration of binding or the selectivity of action. Further, the majority of docking studies use constant protein structures, ignoring the situation in living organisms where binding can be affected due to their dynamic and flexible conditions. The accuracy can be enhanced with the use of sophisticated approaches such as ensemble docking, induced‐fit simulations and ML‐based refinement of binding predictions. Moreover, the compounds that interfere with several important parasite processes, including glycolysis and proteasome, should become the priority to increase the treatment efficacy and decrease the chances of resistance. Implementing these improvements would transform molecular docking into a more reliable tool for developing new antimalarial therapies.
3.3.1. Docking Studies of Nonstandard Drug Compounds Against Plasmodium spp. Targets
Molecular docking studies have identified several nonstandard drug compounds with promising binding affinities (ΔG, kcal/mol) against P. falciparum and related targets, offering potential leads for antimalarial drug development. Table 3 summarises the binding affinities, target proteins, experimental IC_50_ values and sample sizes for 40 nonstandard compounds reported in studies from 2014 to 2024, excluding standard drugs such as chloroquine and artemisinin. Binding affinities for the top‐ranked nonstandard compounds ranged from −156.333 kcal/mol for isorhamnetin to −86.0838 kcal/mol for scoparone, with euphorbianin (−151.001 kcal/mol), afzelin (−143.385 kcal/mol), myricitrin (−137.29 kcal/mol), quercitrin (−132.938 kcal/mol), leucocyanidol (−115.458 kcal/mol), artemisinin (−101.408 kcal/mol), pinocembrin (−98.8994 kcal/mol) and scopoletin (−98.4147 kcal/mol) occupying intermediate positions in this high‐affinity range [65]. Figure 2 illustrates the binding affinities of the top 10 compounds, highlighting the dominance of Plasmepsin II and PfLDH as targets. Notably, several of these ligands, particularly isorhamnetin, euphorbianin and afzelin, target Plasmepsin II and exhibit markedly more negative binding energies than most other screened compounds, indicating exceptionally strong predicted interactions but also highlighting the need for cautious interpretation because such extreme values often arise from MM–PBSA‐type rescoring and are sensitive to methodological differences across studies. Other notable compounds include 11‐O‐galloylbergenin (−16.22 kcal/mol, PfLDH) [21], myricetin 3‐O‐glucoside (−13.413 kcal/mol, PfLDH) [36] and isovitexin (−11.485 kcal/mol, PfDHFR–TS) [66]. Flavonoids and glycosides, such as rutin (−11.25 kcal/mol), astragalin (−11.02 kcal/mol) and isoquercitrin (−9.93 kcal/mol), were frequently among the stronger binders, indicating their promise as antimalarial candidates [60].
Binding affinities of top 10 nonstandard drug compounds to antimalarial targets.
Target proteins were quite variable, with the most common being PfLDH, Plasmepsin II and PfDHFR–TS; see Table 4. Compounds based on Plasmepsin II showed maximum affinities [65] and PfLDH‐targeting compounds such as 11‐O‐galloylbergenin and bergenin have also shown promising binding properties and experimental IC_50_ values (7.85 ± 0.61 and 6.92 ± 0.43 µM, respectively), indicating the antiviral potentials [21]. Few works reported IC_50_ values, which makes it difficult to correlate docking scores and experimental efficacy. Sample sizes were reported sparsely, with some notable exceptions for isovitexin (n = 81), rutin (n = 3) [60] and friedelin (n = 5) [46], just to name a few. Figure 2 shows the binding affinities of the top 10 compounds with the dominance of Plasmepsin II and PfLDH as targets. The variability in docking methodologies (AutoDock, MM–PBSA) and nonstandard deviation and nonstandard error data are challenges for direct comparisons. Nevertheless, these findings highlight the potential of flavonoids, glycosides and other nonstandard compounds for further study in antimalarial drug development.
3.4. ML in Drug Resistance and Discovery
ML models, such as RF, SVM, k‐nearest neighbours (KNN), logistic regression (LR), decision trees (DT), Chemprop and genetic function algorithms (GFA), have made significant contributions to the study of malaria by analysing genomic information and determining the resistance of pathogens to drugs, in this case Plasmodium. To show how these approaches are implemented in studies, Figure 3 is a visual summary of the most commonly used ML algorithms found in this review. Table 5 summarises the ML models for bioactivity prediction with input data, output and performance. These models have also helped in the discovery of new antimalarial compounds from herbal sources. For example, Deligianni et al. [15] reported that KNN, LR and DT models were used to predict the bioactivity of the compounds in 239 components of Euphorbia hirta essential oils (EOs); the best results were 0.701–0.840 MCC based on leave‐some‐out cross‐validation classification. Top predictive compounds, including eugenol, eucalyptol and limonene, were identified based on consensus weighted feature importance (WFI) analysis, with the robustness of our models validated by ROC AUC/PR curves (best MCC: 0.840). Similarly, Miao et al. [14] used Chemprop and MAIP models along with SMILES and Morgan fingerprints from Morus alba S1 methanolic leaf extract that yielded 0.868590 +‐ 0.007887 for the extent of probability of antimalarial. Potential based on flavonoids such as quercetin and kaempferol. Oyebamiji et al. [72] used GFA to model QSAR of C. myxa phytochemicals using 2D descriptors (e.g., ALogP2, ATS1m) for the prediction of binding affinities with a good R2 of 0.806 and cross‐validation R2 of 0.612. Viira et al. [41] developed SVM classification consensus models for curcuminoid derivatives using SMILES and ISIDA fragment descriptor resolutions to obtain cross‐validated balanced accuracy at 0.701–0.953, with 100% sensitivity in prospective predictions of 17 active compounds. Pradhan et al. [32] used the binary LR algorithm to predict the endemicity of malaria using climatic variables, and the results demonstrated the versatility of ML for an epidemiological application.
Visual overview of machine learning algorithms in antimalarial research.
In drug discovery, ML models have played a predominant role in discovering novel compounds of herbal interventions. For instance, Mianda et al. [77] combined ML with UPLC–QTOF–MS data from Aloe marlothii root extracts, and the molecular targets, such as DNA topoisomerase II, were predicted (PDB: 6CA8), and aloesaponarin I and aloesaponol IV, which showed the best IC_50_ values and binding affinities, indicated they are strong inhibitors. Rehman [42] applied network pharmacology and ML to the Xanthium cavanillesii ethanolic extracts, by prediction of binding energy and enriched pathways (e.g., pyrimidine metabolism) for artemisinin derivatives against PfDHODH (1TV5), also confirmed by ITC measurements. Olaosebikan et al. [31] and Brahma et al. [67] used ML to assess the Andrographis paniculata and Hibiscus cannabinus/Corchorus capsularis extracts, respectively, based on GC–MS data, such as predicting binding affinities and ADMET properties and identifying compounds, for example, andrographolide and sesamin. Kadioglu et al. [55] used RF to predict the toxicity profile of artemisinin derivatives from Artemisia species, and the achieved AUC values were 0.830–0.869 for cardiotoxicity and hepatotoxicity models. These studies highlight the usefulness of ML in the prioritisation of phytochemicals with high potential for antimalarial application, which are typically confirmed through in vitro assays (i.e., luciferase and SYBR Green I), as well as the use of in silico docking.
Despite these advancements, there are still challenges in the applications of ML. Data availability has also been a major challenge, as data from several studies have been based on a limited or heterogeneous type of data (GC–MS profiles, SMILES strings, etc.), probably insufficient to reflect the complexity of Plasmodium biology [29]. Model interpretability is another issue, especially in the case of complex models such as SVM and RF, where feature importance (WFI) [15] has to be carefully validated, and drivers/importances of features have to be strictly validated to avoid misinterpretation. Overfitting is a recurrent problem, particularly for QSAR models with high‐dimensional descriptors, such as in the case of Oyebamiji et al. [72], where cross validation was very important to make the model generalisable. Additionally, the combination of genomic data for resistance prediction, such as that attempted by Herman et al. [75] for PfcPRS mutations, is constrained by the lack of well collateralisation of resistance data sets. These challenges highlight an urgent need for larger, standardised datasets, better feature selection techniques and hybrid strategies that combine ML with molecular docking and dynamics to better predict and interpret malaria drug discovery.
ML has enhanced the selection of promising compounds and prediction of resistance in research on malaria, but this approach is limited due to a lack of data availability, unclear outcomes and a lack of biological context. Many ML models rely on simplified forms of chemical data, such as SMILES or even 2D fingerprints or computationally derived fingerprints, which do not truly capture biological factors like protein shapes or host–parasite interactions. To mitigate this, future approaches should integrate chemical, genetic and biological information into unified ML systems. For example, advanced models such as graph neural networks and transformer models are better able to model more complicated interactions in molecules; at the same time, explainable AI can help us understand what drives predictions to better validate experimental results. In addition, up‐to‐date training models using a variety of Plasmodium strains and data from other parasite life stages might render predictions more reliable, especially for the emergence of resistance. In the future, the combination of ML and active learning systems that could be adapted according to the new biological data could make ML a dynamic tool for finding new antimalarial treatments.
3.5. Pathways Modulated by Molecular Techniques in Malaria Research
Molecular techniques, including gene expression profiling, molecular docking and ML, have significantly advanced the understanding of Plasmodium pathways targeted by phytochemicals and synthetic compounds, offering insights into antimalarial drug development [83, 84]. Table 6 summarises the affected pathways and their respective validation methods. One of the most frequently targeted pathways is PfLDH inhibition, critical for parasite survival within erythrocytes. Shah et al. [65] demonstrated that euphorbianin (−151.001 kcal/mol) and isorhamnetin (−156.333 kcal/mol) potently inhibit Plasmepsin II (2BJU), disrupting haemoglobin digestion in the parasite’s food vacuole, validated through ADMET profiling and Ramachandran plots. Chaurasia and Pandey [48] reported artocarpin’s strong binding (−9.6 kcal/mol) to Falcipain-2 (3BPF), confirmed by in vitro β‐haematin assays, which also inhibit hemozoin formation, a key detoxification process. Aini et al. [69] identified quercetin’s inhibition of Plasmepsin X (7RY7, −7.8 kcal/mol), further emphasising haemoglobin degradation as a therapeutic target. Mianda et al. [77] showed that aloesaponarin I from Aloe marlothii inhibits β‐haematin polymerisation in the asexual blood stage, validated by luciferase assays, while Wiraswati et al. [33] highlighted Plasmepsin I inhibition by Breynia cernua extracts.
Pyrimidine and folate metabolism, essential for Plasmodium nucleic acid synthesis, are prominent targets. Rehman [42] used network pharmacology to demonstrate artemisinin derivatives’ inhibition of PfDHODH (1TV5), disrupting de novo pyrimidine synthesis, with enriched KEGG pathways (e.g., map00240) validated by ITC measurements. Dzouemo [50] and Sikam et al. [54] reported sesamin (−10.0 kcal/mol) and rhaponticin (−9.3 kcal/mol) targeting PfDHODH (5TBO), supported by in vitro antiplasmodial assays. The folate pathway, mediated by Dihydrofolate Reductase–Thymidylate Synthase (DHFR–TS), was inhibited by isovitexin (−11.485 kcal/mol, [66]), friedelin (−10.6 kcal/mol, [46]) and dimethylmatairesinol (−8.4 kcal/mol, [53]), with validations including in vivo parasitaemia suppression and high r ^2^ correlations (0.8374). Al‐Huqail [28] confirmed rutin’s inhibition of PfDHFR–TS (1J3I), impacting phosphatidylcholine synthesis via PfPMT.
Glycolysis, crucial for parasite energy production, was disrupted through PfLDH inhibition. Samuel et al. [36] reported myricetin 3‐O‐glucoside’s binding (−13.413 kcal/mol) to PfLDH (1T2C), validated by pharmacophore modelling, while Konyanee et al. [43] and Chaniad et al. [40, 73] identified rheediachromenoxanthone (−8.6 kcal/mol) and 2,4,3′,5′‐tetrahydroxybibenzyl (−8.91 kcal/mol), supported by in vitro IC_50_ data. Chaijaroenkul et al. [70] detected upregulated glycolytic enzymes (e.g., PCM1, LDH, GAPDH) in response to Garcinia mangostana extracts, indicating compensatory mechanisms. Mitochondrial electron transport was targeted by quercetin’s inhibition of Cytochrome c2 (7TXE, −8.2 kcal/mol) [69] and compound 2’s binding to Cytochrome bc1 (4PD4, −8.33 kcal/mol) [24] validated by Malstat assays. Murugesan and Kaleeswaran [61] also noted pachyrrhizin’s inhibition of hexokinase‐1 (−9.9 kcal/mol), further disrupting glycolysis.
Protein translation and amino acid metabolism were disrupted by targeting synthetases. Herman et al. [75] showed halofuginone’s inhibition of PfcPRS, triggering the amino acid starvation response (AAR) via eIF2α phosphorylation, validated by yeast complementation (K_i_ = 71.1 nM). Shekari et al. [37] reported sesamin’s inhibition of AgEPRS (−7.4 kcal/mol), disrupting Anopheles gambiae protein synthesis, validated by ADME–Tox profiling. Aini et al. [69] and Apeh et al. [46] identified quercetin (−7.5 kcal/mol) and cynaroside (−10.5 kcal/mol) targeting Glutamyl-tRNA Synthetase (7WAJ) and LysRS (4YCV), respectively, impacting apicoplast translation. Chaperone‐mediated protein folding via PfHsp70-1 and PfHsp70-z was inhibited by Ziziphus mucronata phenolics ([22, 76]), validated by pLDH assays. The purine salvage pathway was targeted by lupeol (−7.6 kcal/mol) against PfHGXPRT (1TUX) [74].
Immune, inflammatory and oxidative stress pathways were also modulated. Soeiro [49] showed glycyrrhizin’s disruption of PfHMGB1‐mediated inflammation, validated by NMR. Camara et al. [44] and Dkhil et al. [62] reported T. albida and I. oblongifolia extracts inducing NFκB signalling, ROS production and cytokine modulation (TNF, IL-6), enhancing T‐cell recruitment, confirmed by in vivo survival studies. Gomes [26] identified eleutherol’s targeting of antioxidant defences (SOD, CAT, GPx, −69.97 kcal/mol for GPx1), validated by oxidative stress assays. Fatty acid biosynthesis (FAS‐II) was disrupted by A. afra extracts targeting Fab I and Fab Z (Kane et al., 2023), while calcium transport via PfATP6 was inhibited by caffeoylquinic acids [39]. Despite robust validations (e.g., RMSD < 2.0 Å, SYBR Green I assay), challenges such as homology model inaccuracies (e.g., PfCDPK-1) [19] and data heterogeneity underscore the need for standardised protocols to translate these findings into effective therapies.
The various research works collectively point to a wide variety of Plasmodium pathways that are targeted by molecular techniques (see Figure 4), such as haemoglobin degradation (Plasmepsin I, II, IV, X, Falcipain-2, Falcipain-3), hemozoin formation, pyrimidine metabolism (PfDHODH), folate metabolism (DHFR–TS, PfPMT), glycolysis (PfLDH, Hexokinase-1, PCM1, GAPDH), mitochondrial electron transport (Cytochrome c2, bc1), protein translation (PfcPRS, AgEPRS, Glutamyl-tRNA Synthetase, LysRS), amino AAR, purine salvage (PfHGXPRT), fatty acid biosynthesis (FAS-II), calcium transport (PfATP6), protein folding (PfHsp70-1, PfHsp70-z), ubiquitin–proteasome system, DNA replication (DNA topoisomerase II, apPOL), gametocyte development (Pfg27), erythrocyte invasion (PfCDPK-1), mitotic division (PfNek1, PfMap2, PfCLK1, PfCLK4), inflammatory signalling (NFκB, PfHMGB1, TNF-α), oxidative stress (SOD, CAT, GPx, GSH) and insect hormonal regulation (mJHBP). These pathways range from metabolic, structural and immunological processes that are important for parasite survival and transmission, illustrating the scope of molecular control. Figure 5 shows what percentage of the pathways are affected in the studies included. The diversity of Plasmodium pathways that are influenced by molecular treatments highlights the importance of moving away from single‐target approaches towards a systems pharmacology approach. Studies performed by docking and gene expression have revealed key pathways such as glycolysis, pyrimidine biosynthesis and haemoglobin degradation; however, these pathways do not act independently in the complex life cycle of the parasite. Future research will have to investigate how these pathways interact and adapt, particularly in the presence of drug or immune pressure. For instance, stress on glycolysis could increase antioxidant defences or alternative energy sources with consequences for the success of a drug. Combining the data set from RNA‐seq, protein structures and metabolites with ML network models may allow prediction of resistance patterns or identify combined weaknesses of the parasite. Moving from a single‐target approach to drug design to a dynamic, networked approach would help improve the selection of effective compounds and be more appropriate for herbal treatments, which affect many pathways in both parasite and host. Ultimately, interactions among pathways instead of just identification of affected pathways are essential to developing effective, drug‐resistant antimalarial treatments.
Molecular pathways targeted in Plasmodium by antimalarial strategies.
Percentage of pathways affected in malaria studies.
3.5.1. Strengths and Limitations
The strength of these findings lies in their good coverage of the pathway with strong validations such as in vitro assays (Malstat, SYBR Green I, β‐haematin), in vivo assays (Peters’ tests, parasitaemia suppression) and in silico metrics (RMSD < 2.0 Angstroms, r ^2^ = 0.8374). Molecular docking and ML have provided us with a high‐throughput screening procedure and precise identification of targets, whereas gene expression profiling has given us insight into compensatory mechanisms (upregulation of glycolytic enzymes). However, shortcomings include the use of homology models (PfCDPK-1) [19], which might lead to binding site inaccuracies. Data heterogeneity (variable herbal extract compositions [59, 71]) and discrepancies in docking algorithms (AutoDock Vina vs. Glide [65]) are some of the challenges to reproducibility. Limited clinical translation, according to Onguéne et al. [20], and small sample sizes in experimental validations also make therapeutic development more difficult. Addressing these limitations requires the use of standardised docking protocols, better homology modelling and larger and well‐curated data sets, which will increase the reliability and translatability of molecular results in malaria research.
4. Discussion
4.1. Integrating Multiomics and Network Pharmacology for Antimalarial Target Prioritisation
The integration of multiomics approaches, including proteomics, metabolomics and transcriptomics, with network pharmacology has emerged as a powerful strategy to prioritise antimalarial targets, addressing the complexity of Plasmodium biology and drug resistance [86]. Proteomics has been instrumental in validating molecular docking predictions by identifying upregulated proteins in response to phytochemical interventions. For instance, Chaijaroenkul et al. [70] used proteomics to detect elevated levels of glycolytic enzymes (PCM1, LDH, GAPDH) in P. falciparum treated with G. mangostana extracts, suggesting compensatory metabolic adaptations that align with docking studies targeting PfLDH [36, 43]. Metabolomics, particularly through GC–MS profiling, has enabled the identification of bioactive compounds and their metabolic impacts. Deligianni et al. [15] analysed 239 components from Euphorbia hirta EOs, using GC–MS data to inform ML models (KNN, LR, DT) that predicted bioactivity with an MCC of 0.701–0.840, highlighting compounds like eugenol and limonene. Although transcriptomics data are less prevalent in the provided studies, the potential for single‐cell RNA‐seq to reveal stage‐specific gene expression as observed in gametocyte‐specific Pfg27 [23] could complement docking and ML predictions, enhancing target specificity.
Network pharmacology has further advanced target prioritisation by mapping protein–protein interactions (PPIs) and enriched pathways, integrating docking and omics data to identify functional partners and therapeutic targets. Rehman [42] employed network pharmacology to demonstrate artemisinin derivatives’ inhibition of PfDHODH (1TV5), with enriched KEGG pathways (pyrimidine metabolism, map00240) validated by isothermal titration calorimetry (ITC). Similarly, Mianda et al. [77] integrated UPLC–QTOF–MS, molecular docking and STRING DB‐based PPI networks to identify PfDHODH functional partners (PfOPRT, PF14_0697), with enriched GO terms like pyrimidine nucleobase biosynthesis (GO:0019856) supporting PfDHODH as a priority target. Elmaidomy et al. [57] combined docking, MDs and cytoscape‐based PPI networks to prioritise 20S proteasome (7LXU) and choline kinase (6YXT), with compounds like limonin and luteolin achieving ΔG‐binding < −7 kcal/mol, validated by in vitro Malstat assays. These studies illustrate how network pharmacology bridges molecular and systems‐level insights, enhancing the identification of multitarget strategies.
ML complements multiomics and network pharmacology by predicting bioactivity and refining target prioritisation. Miao et al. [14] utilised Chemprop and MAIP models with SMILES and Morgan fingerprints from Morus alba extracts, achieving an AUC of 0.868590 ± 0.007887 for antimalarial activity, identifying flavonoids like quercetin as key inhibitors of PfDHFR–TS. Deligianni et al. [15] applied KNN and LR models to GC–MS‐derived descriptors, using consensus WFI to prioritise compounds disrupting gametocyte‐to‐ookinete development. These ML models integrate omics data with docking results, enabling high‐throughput screening of phytochemicals and synthetic derivatives. For example, Oyebamiji et al. [72] used GFA with 2D descriptors (ALogP2, ATS1m) to predict binding affinities for C. myxa compounds, achieving an R ^2^ of 0.806, validated by cross‐validated QSAR models.
The strength of integrating multiomics and network pharmacology lies in its ability to provide a holistic view of Plasmodium biology, enabling the identification of novel targets like PfDHODH, Plasmepsin II and Falcipain-2 through synergistic computational and experimental approaches. Robust validations, including in vitro assays (Malstat, SYBR Green I), in vivo studies (parasitaemia suppression) [42] and in silico metrics (RMSD < 2.0 Å) [53], enhance the reliability of these findings. However, limitations include challenges in data integration due to heterogeneous omics datasets, such as variable GC–MS profiles [71] or limited transcriptomic data for Plasmodium. Computational complexity and model interpretability, particularly in ML‐driven PPI networks, pose additional hurdles, as seen in the need for careful WFI validation [15]. The reliance on homology models (PfCDPK-1) [19] introduces potential inaccuracies in target prediction.
Collectively, the combination of molecular docking, gene expression profiling and ML is a multidimensional framework for antimalarial lead discovery. Docking prefers phytochemicals and synthetic compounds that have favourable binding to essential Plasmodium targets but is limited in itself by inaccuracies in its scoring functions and by homology model uncertainty and poor correlation to in vitro potency. Gene expression studies provide a deeper mechanistic insight, showing not only pathway modulation on target but also different compensatory responses (e.g., upregulation of glycolytic enzymes or stress pathways), as well as host–parasite interaction; however, heterogeneous experimental designs, small sample sizes and variable extract composition typically occur. ML plays a role in scalability and predictive power, for example, virtual screening, QSAR modelling and resistance prediction using large descriptor/omics‐based datasets; however, model interpretability, overfitting and reliance on biased or sparse training data are major constraints. When combined, these methods are not additive, that is, docking‐guided ML models to refine chemical space and improve the hit rates and transcriptomic readouts and pathway enrichment analyses to validate the hypotheses derived from the docking and to inform the selection of ML features, to link the predicted binding to biologically relevant phenotypes. At the same time, divergences between docking scores, ML predictions and experimental gene expression or in vitro efficacy expose context‐specific limitations and highlight where target choice, protein structure selection or training data need refinement. From a translational perspective, studies that couple in silico prioritisation with in vitro, in vivo and cytotoxicity assays offer the most credible leads, yet such fully integrated pipelines remain relatively rare and are unevenly standardised across laboratories. Future work should therefore emphasise prospective, pipeline‐based designs that intentionally combine docking, ML and multiomics readouts under harmonised protocols to generate robust, mechanism‐anchored antimalarial candidates suitable for preclinical development.
4.2. Discovering Antimalarial Leads via Integrated Approaches
The studies reviewed demonstrate a robust pipeline for discovering novel antimalarial leads by combining computational tools, such as molecular docking and ML, with experimental validation, including in vitro and in vivo assays. Molecular docking has been pivotal in identifying compounds with high binding affinities to critical Plasmodium targets, such as PfLDH, PfDHODH, Plasmepsin II and PfEMP-1. For instance, Apeh et al. [46] reported that friedelin exhibited a binding affinity of −10.6 kcal/mol to PfDHFR, outperforming artesunate, with in vivo validation showing significant parasitaemia reduction at 400 mg/kg. Similarly, Shah et al. [65] identified isorhamnetin as a potent Plasmepsin II inhibitor with an exceptionally high binding affinity of −156.33 kcal/mol, supported by favourable absorption, distribution, metabolism, excretion and toxicity (ADMET) profiles and compliance with Lipinski’s Rule of Five. These findings underscore the power of docking to prioritise compounds like 11‐O‐galloylbergenin (−16.22 kcal/mol, PfLDH) [23] and epicatechin‐gallate (−9.3 kcal/mol, PfCDPK-1) [19], which demonstrated stability in MDs simulations (RMSD < 0.4 nm) and strong in vitro antiplasmodial activity (IC_50_ < 8 µM). The integration of docking with MD simulations, as seen by Chaurasia and Pandey [48] for artocarpin (−9.6 kcal/mol, Falcipain-2), ensures reliable prediction of binding stability, enhancing the confidence in lead selection.
Experimental validation by in vitro and in vivo experimentation has played a huge role in proving the antiplasmodial activity of computationally found lead strategies, fortifying the relationship between in silico calculations and drug potential. Studies such as Elmaidomy et al. [57] were performed that validated Limonin’s activity against the 20S proteasome (IC_50_ = 0.2 μM) based on stable MD simulations and Malstat assays associated with ursolic acid (binding energy of the molecular complex: −9.7 kcal/mol), which correlated with 83.4% growth inhibition in vitro [45], comparable to artesunate. In vivo studies also supported these results; Camara et al. [44], for instance, reported that T. albida extract at 100 mg/kg completely suppressed parasitaemia by Day 4–6 and was anti‐inflammatory with a reduction in TNF and IL-6 expression. Similarly, Alebachew et al. [68] revealed the ability of Knipholone (200 mg/kg) to suppress parasitaemia by 60.16% in P. berghei‐infected mice, with docking being able to confirm strong PfLDH binding (−6.96 kcal/mol). These results have demonstrated the necessity of experimental assays in corroborating computational predictions, especially in the case of compounds affecting glycolysis (PfLDH) [27] and hemozoin formation (Falcipain-2/3) [48], which play a vital role in parasite survival.
ADMET profiling and drug‐likeness calculations have also improved the lead compound selection to guarantee the potential of these to be brought into clinical development. Most of the studies reported a good oral bioavailability in accordance with Lipinski’s Rule of Five, as was seen with rhaponticin (−9.3 kcal/mol, PfDHODH) [54] and sesamin (−10.0 kcal/mol, PfDHODH) [50]. However, problems such as low Caco‐2 permeability (epicatechin‐gallate) [19] and possible toxicity (purfalcamine’s hepatotoxicity) [19] indicate the need for structural optimisation. ML has been used in complementing and going hand in hand with these efforts in order to predict bioactivity and prioritise compounds, as Miao et al. [14] showed Cosmosiin’s binding to IL6 (−7.7 kcal/mol) was validated by ML models with high AUC scores. In addition, network pharmacology approaches, such as Rehman [42], identified PfDHODH as an important target for artemisinin stereoisomer C6 (−8.7 kcal/mol) with validation of ITC binding specificity. These integrative approaches have improved the strength of lead identification by addressing in advance the limitations in discovery due to the pharmacokinetic and toxicological aspects.
Despite these advancements, limitations in the current studies need to be considered for future studies. Variability in docking tools (AutoDock Vina vs. Glide) and binding affinity metric (MM–PBSA) [59] can lead to difficulties when trying to directly compare between studies, and therefore, standardised protocols are needed. Additionally, while in vitro IC_50_ and inhibition curve data (aloesaponarin I, IC_50_ = 1.6 micrograms/mL) [77] as well as in vivo plasmodia suppression (T. albida) [44] are promising, in the absence of clinical trial data, the translation of these is limited. Compounds such as myricetin 3‐O‐glucoside (−13.413 kcal/mol, PfLDH) [36] offer high potential for resistance validation against resistant Plasmodium strains. Future research should include all types of multiomics data, like proteomics and metabolomics, to understand the mechanism of actions, such as in Chaijaroenkul et al. [70], where the upregulation of glycolytic enzymes was associated with PfLDH inhibition. Moreover, the extension of ML models to predict resistance profiles and the combination of single‐cell transcriptomics could be used to increase target specificity, especially for gametocyte‐specific proteins, such as Pfg27 [23]. By helping to fill in these gaps, the integrated computational–experimental pipeline can accelerate the development of new types of antimalarial treatments that can be less prone to resistance.
4.3. Future Directions
Future research should prioritise in vivo validation and clinical trials to confirm the efficacy and safety of promising compounds like 11‐O‐galloylbergenin, isorhamnetin and rheediachromenoxanthone, which lack in vivo data despite potent in vitro activity [21, 43, 65]. Mechanistic studies, including knockout assays and enzyme inhibition assays, are needed to validate docking predictions (e.g., PfLDH inhibition by quercetin, PfHsp70-1 disruption by betulinic acid) and elucidate molecular targets [40, 76]. Synergy testing of compound mixtures, such as EOs or plant extracts, could enhance efficacy and overcome resistance, particularly for transmission‐blocking agents like α‐pinene [7, 15]. ML models require larger, standardised datasets to improve specificity and predict resistance mechanisms, building on studies like Viira et al. [41]. Structural optimisation of compounds with high molecular weight or toxicity risks, for example, dimeric NIQs and pachyrrhizin, is critical to improve pharmacokinetics and selectivity [61, 85]. Additionally, exploring non‐oral delivery routes such as vapour sprays for EOs and multitarget strategies could broaden therapeutic applications [24, 34, 57].
4.4. Limitations
The reviewed studies are primarily limited by their reliance on in silico and in vitro data, with minimal in vivo validation, restricting translation to clinical settings [45, 65, 85]. Variability in docking methodologies, such as AutoDock versus MM–PBSA, and the absence of standard deviation or IC_50_ data for many compounds, hinder direct comparisons or a meta‐analysis [65, 66]. Another limitation of the docking literature is that many studies rely on a single P. falciparum structure as a template for other Plasmodium species, without systematically exploring species‐specific structural differences; although core catalytic residues are generally conserved, future work should explicitly assess orthologue conservation and, where possible, use species‐specific models. ML models suffer from low specificity due to heterogeneous training data and sparse mechanistic insights, as seen in curcuminoid predictions [41]. Most studies tested single P. falciparum strains (e.g., 3D7, K1), limiting insights into activity against resistant isolates [27, 40]. Toxicity concerns, such as carcinogenicity risks for compounds like afzelin and hepatotoxicity for pachyrrhizin, remain unaddressed experimentally [61, 65]. Finally, the lack of detailed pharmacokinetic profiles and synergistic effect studies for plant extracts limits their practical application [44, 67].
5. Conclusion
This review shows that molecular docking, gene expression profiling and ML are no longer standalone techniques but interlocking components of a coherent antimalarial discovery pipeline. Together, they have shifted the field from empirical screening towards mechanism‐informed, data‐driven prioritisation of leads from both herbal and synthetic sources. By linking target structure, pathway perturbation and predictive modelling, these approaches enable a more rational selection of compounds with genuine potential to overcome emerging resistance.
NomenclatureADMETAbsorption, distribution, metabolism, excretion, toxicity (used in drug‐likeness prediction)MM/GBSAMolecular mechanics/generalised Born surface area (binding energy estimation)PfLDH Plasmodium falciparum lactate dehydrogenase (drug target in glycolysis)Plasmepsin IIAspartic protease involved in haemoglobin degradationpLDHParasite lactate dehydrogenase (antimalarial assay target)HRP2 ELISAHistidine‐rich protein 2 enzyme‐linked immunosorbent assay (used in malaria diagnostics)ECMExperimental cerebral malaria (murine model)RMCBSRapid Murine Coma and Behaviour Scale (neurological scoring system for ECM)PK/PDPharmacokinetics/pharmacodynamics (used for dose–response and drug action relationship)PASSPrediction of Activity Spectra for Substances (bioactivity prediction tool)GMQEGlobal model quality estimate (used in protein homology modelling)UPLC–QTOF–MSUltra‐performance liquid chromatography–quadrupole time‐of‐flight mass spectrometrySTRING DBSearch Tool for the Retrieval of Interacting Genes/Proteins (for protein–protein interaction networks)Pfg27Gametocyte‐specific gene used as a transmission‐blocking markerPfDHODH Plasmodium falciparum dihydroorotate dehydrogenase (pyrimidine metabolism target)PfCDPK‐1 Plasmodium falciparum calcium‐dependent protein kinase 1 (involved in erythrocyte invasion)PfHsp70‐1/ PfHsp70‐zHeat shock proteins involved in protein folding and parasite stress responsesFalcipain‐2/3Cysteine proteases essential for haemoglobin digestionPfDHFR–TSDihydrofolate reductase–thymidylate synthase (folate metabolism target)PfPMT Plasmodium falciparum phosphoethanolamine methyltransferase (lipid biosynthesis)PfHGXPRTHypoxanthine‐guaninexanthine phosphoribosyltransferase (purine salvage pathway)PfATP6Sarco/endoplasmic reticulum Ca^2+^‐ATPase (targeted by artemisinin)FAS‐IIFatty acid synthesis pathway II (apicoplast‐localised)PfcPRSCytoplasmic prolyl‐tRNA synthetase (translation machinery target)AgEPRS Anopheles gambiae glutamyl‐tRNA synthetase (vector protein synthesis target)mJHBPMosquito juvenile hormone‐binding protein (vector‐based transmission target)
Author Contributions
Reuben Samson Dangana: writing–review and editing, writing–original draft, methodology, investigation, formal analysis, data curation. Israel Ehizuelen Ebhohimen: writing–review and editing, conceptualisation. Samson Anjikwi Malgwi: writing–review and editing, proofreading. Samuel Chima Ugbaja: Moses Okpeku: writing–review and editing, proofreading, review and editing, supervision, resources, funding acquisition.
Funding
This review did not receive any specific grant from funding agencies in the public, commercial or not‐for‐profit sectors.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sarpong E. , Acheampong D. O. , Fordjour G. N. R. et al., Zero Malaria: A Mirage or Reality for Populations of Sub-Saharan Africa in Health Transition, Malaria Journal. (2022) 21, no. 1, 10.1186/s 12936-022-04340-1.PMC 963676636333802 · doi ↗ · pubmed ↗
- 2Choudhuri S. and Ghosh B. , Computational Approach for Decoding Malaria Drug Targets From Single-Cell Transcriptomics and Finding Potential Drug Molecule, Biorxiv Bioinformatics. (2024) 14, 1–18, 10.1038/s 41598-024-72427-7.PMC 1147382639402081 · doi ↗ · pubmed ↗
- 3Shin H.-I. , Ku B. , Park E. , Ju J.-W. , and Lee H.-I. , Status of World Malaria in 2021 (2022 World Malaria Report), 2023.10.56786/PHWR.2023.16.22.2PMC 1247992741332930 · doi ↗ · pubmed ↗
- 4Lawal L. , Buhari A. O. , Jaji T. A. et al., Lingering Challenges in Malaria Elimination Efforts in Sub‐Saharan Africa: Insights and Potential Solutions, Health Science Reports. (2024) 7, no. 6, 10.1002/hsr 2.2122.PMC 1114459638831778 · doi ↗ · pubmed ↗
- 5Shin H.-I. , Ku B. , Jung H. et al., 2023 World Malaria Report (Status of World Malaria in 2022), 2024.10.56786/PHWR.2024.17.32.1PMC 1248038141333952 · doi ↗ · pubmed ↗
- 6Organization W. H. , World Malaria Report 2022, 2022, World Health Organization.
- 7Alghamdi J. M. , Al-Qahtani A. A. , Alhamlan F. S. , and Al-Qahtani A. A. , Recent Advances in the Treatment of Malaria, Pharmaceutics. (2024) 16, no. 11, 10.3390/pharmaceutics 16111416.PMC 1159722739598540 · doi ↗ · pubmed ↗
- 8Ruberto A. A. , Maher S. P. , Vantaux A. et al., Single-Cell RNA Profiling of Plasmodium Vivax-Infected Hepatocytes Reveals Parasite-And Host-Specific Transcriptomic Signatures and Therapeutic Targets, Frontiers in Cellular and Infection Microbiology. (2022) 12, 10.3389/fcimb.2022.986314.PMC 945320136093191 · doi ↗ · pubmed ↗