Relationship Between Histomonas meleagridis Infection and Cecal Intestinal Microbiota of Chickens
Qiaoguang Chen, Yaxin Liu, Wendi Zhu, HsuPan Aye, Ruting Li, Zhaofeng Hou, Dandan Liu, Yuelan Yin, Jianping Tao, Jinjun Xu

TL;DR
This study shows how infection with Histomonas meleagridis affects chicken growth, causes lesions, and changes gut bacteria in the cecum.
Contribution
The study identifies specific shifts in cecal microbiota composition during H. meleagridis infection in chickens.
Findings
Infected chickens had reduced body weight and severe liver and cecum lesions at 14 days post-infection.
Beneficial bacteria like Verrucomicrobia and Lactobacillus aviarius decreased, while harmful bacteria like Proteobacteria and Fusobacteria increased.
Microbial diversity and structural differences were significantly altered in infected chickens compared to controls.
Abstract
This study evaluated the effects of Histomonas meleagridis infection on growth performance, lesion indicators, and the composition of the cecal microbial community in chickens. The results showed that fourteen days post-infection represented the peak stage of disease development. Compared with the control group, infected chickens exhibited a significant reduction in body weight, along with severe lesions in the liver and cecum. At the same time, the abundance of beneficial bacteria in the cecum, such as Verrucomicrobia and Lactobacillus aviarius, decreased, whereas the abundance of harmful bacteria, including Proteobacteria and Fusobacteria, increased. These findings contribute to a better understanding of microbial changes in the chicken cecum following infection with H. meleagridis. Histomonosis, caused by Histomonas meleagridis, leads to economic losses in the poultry and livestock…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
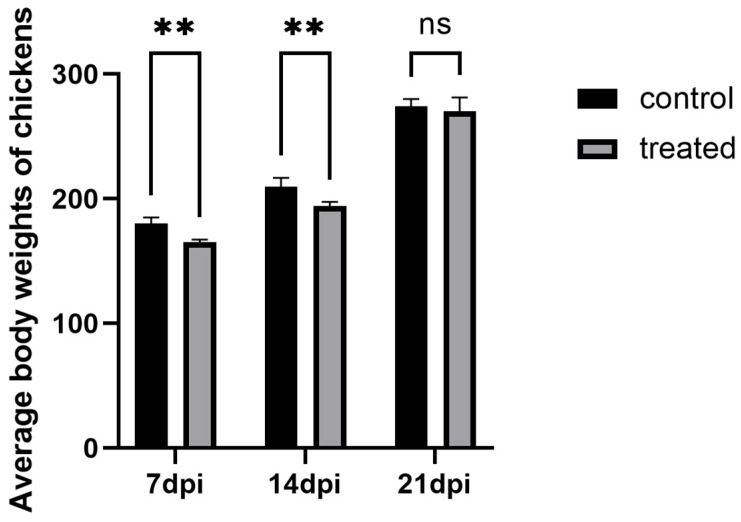 Figure 1
Figure 1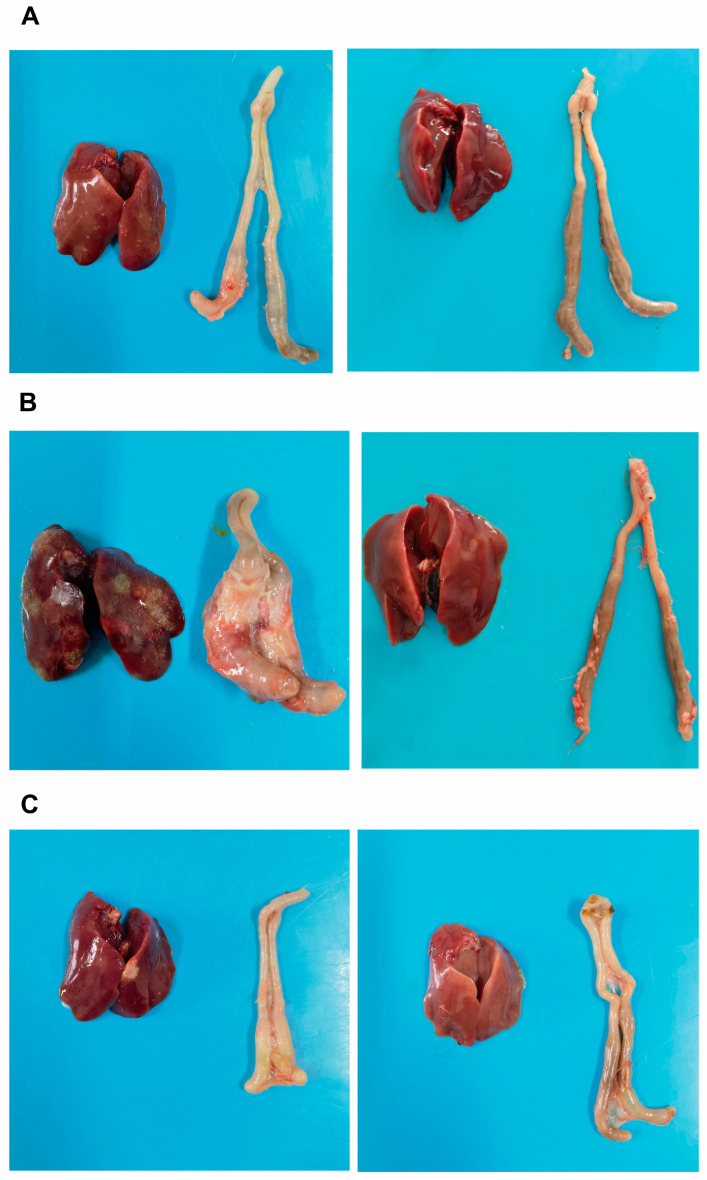 Figure 2
Figure 2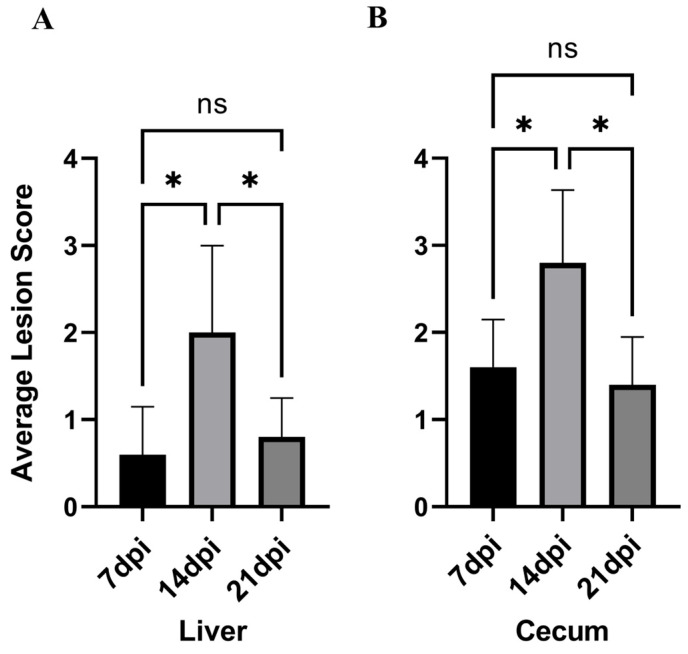 Figure 3
Figure 3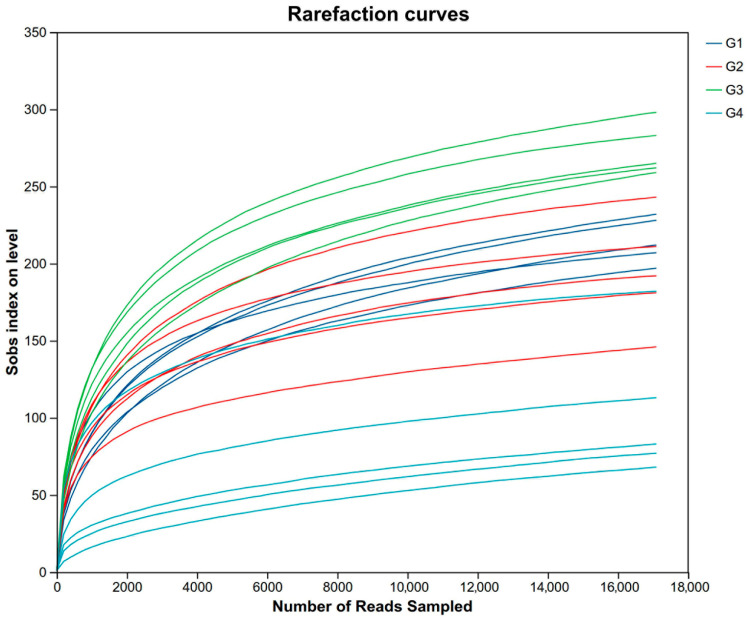 Figure 4
Figure 4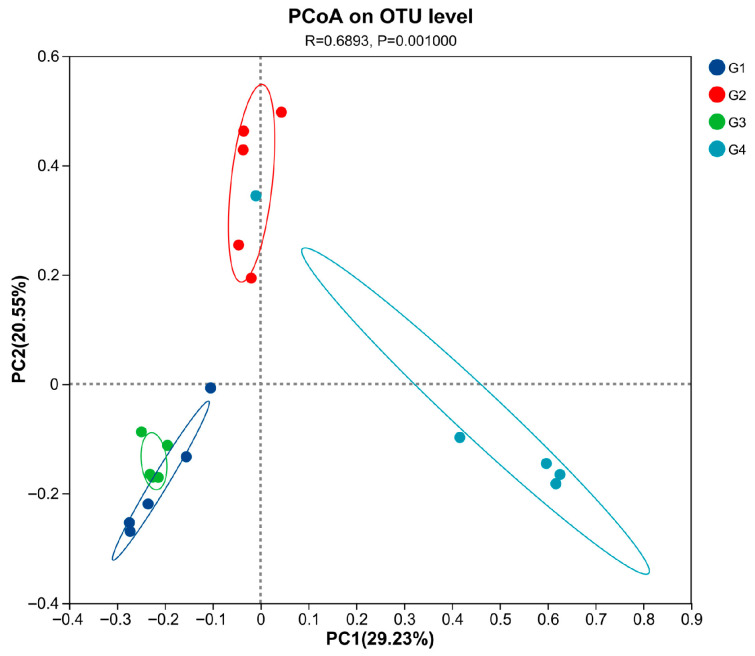 Figure 5
Figure 5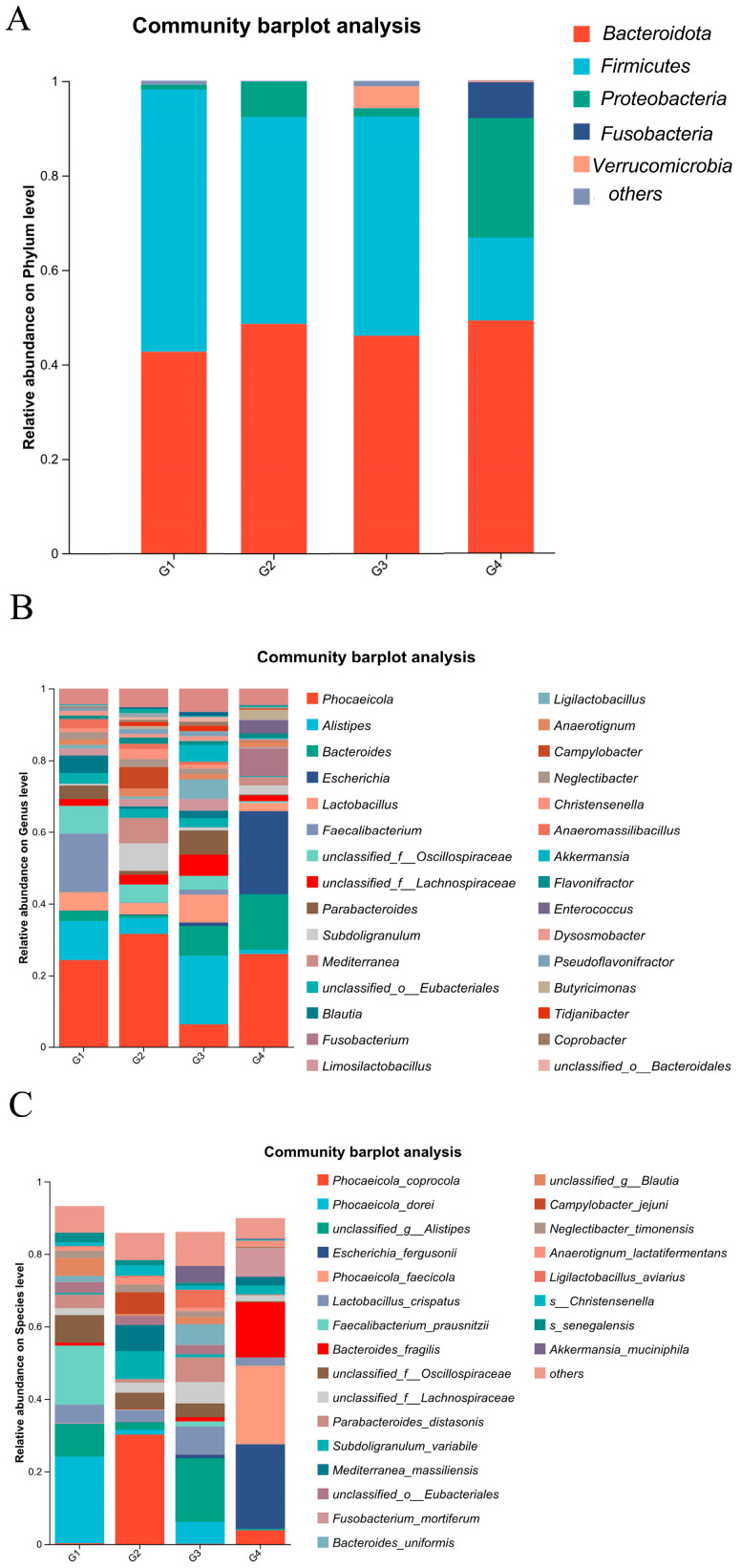 Figure 6
Figure 6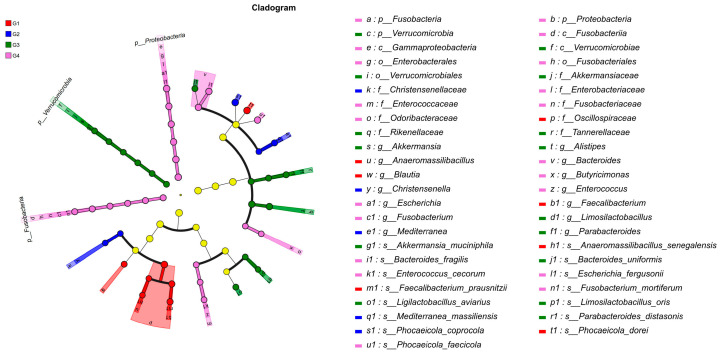 Figure 7
Figure 7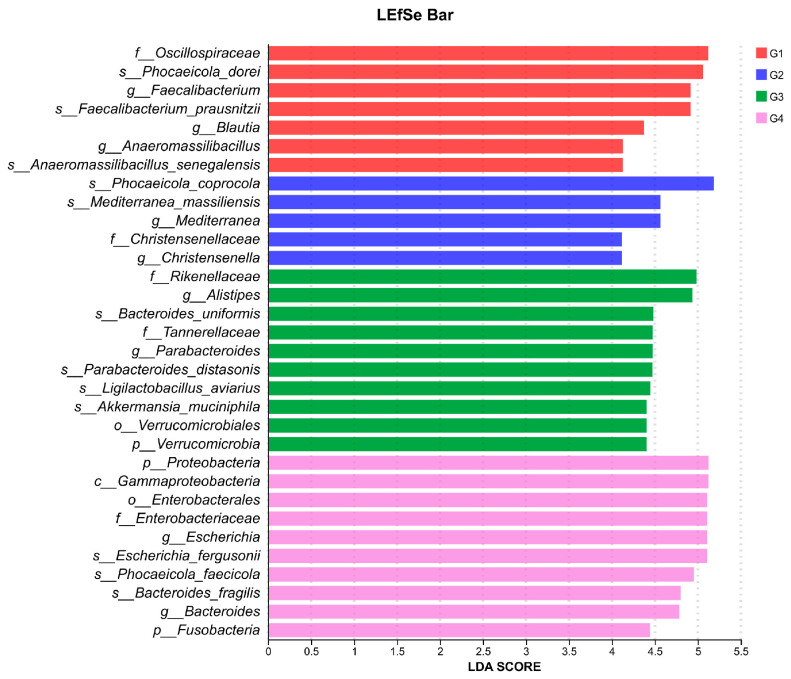 Figure 8
Figure 8- —National Natural Science Foundation of China
- —the 111 Project D18007
- —the Key Laboratory of poultry preventive medicine of the Ministry of Education
- —the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD)
- —Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCoccidia and coccidiosis research · Veterinary medicine and infectious diseases · Aquaculture disease management and microbiota
1. Introduction
Histomonas meleagridis is a protozoan parasite responsible for histomonosis, commonly known as “blackhead disease” or infectious enterohepatitis [1,2]. Characteristic lesions occur in the liver and cecum, typically presenting as caseous cores in the cecum and volcanic crater-like focal necrosis on the liver surface [3]. The disease poses a serious threat to turkeys, with mortality rates reaching up to 100%. In chickens, while the mortality rate is comparatively lower, histomonosis significantly impairs production performance, resulting in decreased egg production, reduced body weight, and an elevated mortality rate that can persist for several weeks [4,5]. In recent years, concerns regarding food safety and public health have led to the progressive ban of nitroimidazole compounds, nitrofuran compounds, and other effective veterinary drugs in the United States, the European Union, and China [6,7,8,9,10]. The live attenuated vaccine obtained through serial in vitro passaging has demonstrated effective immunoprotection but has not yet been successfully commercialized [11,12,13]. This situation has created significant challenges in the prevention and control of histomonosis, necessitating urgent exploration of novel strategies. Previous in vitro studies have shown that Escherichia coli in the gut influences the energy absorption and metabolism of H. meleagridis [14], while in vivo research has revealed that the presence of H. meleagridis increases the relative abundance of E. coli, thereby exacerbating pathological changes [15,16]. Thus, elucidating the interactions between H. meleagridis and intestinal microbiota populations may provide critical insights for developing novel alternative therapies.
The intestinal microbiota refers to the microbial community inhabiting the animal gut, including trillions of bacteria, fungi, viruses, archaea, and other microorganisms [17]. The chicken intestine harbors a highly diverse microbiota that interacts with the host to establish a stable and coordinated intestinal ecosystem [18]. Complex interactions between intestinal microorganisms and the host influence physiological balance, health, immunity, and production performance [19,20,21]. Recent studies have demonstrated that parasitic infections can induce gut microbiota dysbiosis, leading to intestinal barrier dysfunction and exacerbation of disease progression. Infection with Trichuris ovis significantly altered the diversity of the cecal microbiota in goats, increasing the abundance of opportunistic pathogens such as Proteobacteria and Bacteroides while reducing beneficial bacteria [22]. Entamoeba histolytica infection induced notable shifts in the gut microbiota of amoebiasis patients, characterized by an increased abundance of Prevotella copri, which was associated with inflammatory diarrhea [23]. Similarly, in broilers infected with Eimeria acervulina, the alpha diversity of the luminal and mucosal microbiota in the duodenum and jejunum was altered, manifested by a reduced abundance of short-chain fatty acid-producing bacteria and an increased abundance of opportunistic pathogens [24]. Infection with Eimeria tenella modified the composition and structure of the cecal microbiota in broilers, leading to a decrease in beneficial commensals such as Lactobacillus spp. and Faecalibacterium prausnitzii, while increasing the relative abundance of Enterococcus spp., Streptococcus spp., Escherichia coli, and Campylobacter jejuni [25,26,27]. However, current research on H. meleagridis infection and changes in the chicken gut microbiota has primarily focused on the phylum and genus levels, with no reports at the specific species level. Based on this background, the present study investigated changes in body weight, liver, and cecal lesions in chickens following H. meleagridis infection, as well as alterations in the cecal microbiota composition at the phylum, genus, and species levels during both the early and peak stages of infection. This approach aims to identify bacterial taxa associated with histomonosis and provide a basis for developing alternative strategies to mitigate the impact of the disease on the poultry industry.
2. Materials and Methods
2.1. Ethics Statement
All animal procedures were performed in accordance with the guidelines and regulations of the Animal Experiment Ethics Committee of Yangzhou University, under license no. SYXK (SU) 2021-0027 for chickens. All chickens were housed and handled in accordance with the principles of humane care and use of laboratory animals.
2.2. Animals
A total of 30 one-day-old white leghorn chickens were purchased from Jiangsu Boehringer Ingelheim Vitong Biotechnology Co., Ltd. (Nantong, China). The chickens were housed in an H. meleagridis-free environment, and their feed was supplemented with multivitamins and trace elements.
2.3. Parasite
The H. meleagridis strain JSYZ-D, isolated from the liver of an infected chicken in Yangzhou, is preserved in liquid nitrogen at the Parasite Laboratory of the School of Veterinary Medicine, Yangzhou University. After resuscitation, it was inoculated into a mixed medium consisting of 9 mL Medium 199 (Gibco, Grand Island, NY, USA) and 1 mL of 10% inactivated horse serum (Gibco), supplemented with 11 mg of sterilized rice starch (Sigma-Aldrich, Shanghai, China), and incubated at 40 °C in an anaerobic incubator [28].
2.4. Experimental Design
At 14 days of age, all chickens were weighed and randomly divided into two groups—the H. meleagridis-infected group and the control group (after adjustment to ensure similar average body weights across groups). Each group consisted of 15 chickens, housed in separate cages. On the same day (14 days of age), each chicken in the experimental group was artificially infected via the cloacal route with 2.0 × 10^5^ H. meleagridis. Prior to artificial infection, the number of H. meleagridis cells/mL was calculated by hemocytometer and trypan blue (Sigma-Aldrich, Shanghai, China) staining to ensure the viability of the parasites at the time of infection. Artificial infection was performed according to the method described by Beer et al. [11].
2.5. Sample Collection and Intestinal Lesion Score
On 7, 14, and 21 days post-infection, five chickens were randomly selected from both the infected group and the control group at each experimental time point. Thus, all chickens were divided into six groups according to the dissection time, with five chickens in each group. At the three experimental time points, the chickens in the infected and control groups were weighed and then euthanized via cervical dislocation, followed by lesion scoring of the cecum and liver. At each experimental time point, cecal contents were collected from each euthanized chicken, and these content samples were stored at −80 °C. Cecal content samples from the infected group and their corresponding control group chickens with severe lesions and high lesion scores were selected for microbial sequencing.
2.6. Lesion Scoring Rules
Cecal scoring criteria [29,30] were as follows: the longitudinal fold of the cecal wall was well-characterized and lacked macroscopical lesions, and the cecal contents were thick with dark feces and no caseous exudate, score 0; cecal wall thickening or presence of scattered petechiae, or both, score 1; moderate thickening of the cecal wall with caseous exudate or contents forming a caseous core, color change of cecal contents or absence of contents and bleeding spots in the cecum, score 2; the cecal wall was thickened, with a prominent caseous core of cecal contents, the cecum had no contents, or the cecal wall appeared petechiae, score 3; the wall of the cecum was significantly thickened, and the cecal mucosal layer appeared fibrotic necrotic and ulcerated, with a caseous core or no contents in the cecum, the presence of a hemorrhagic blind end, or cecal rupture leading to peritonitis, score 4. Liver scoring criteria were as follows [29,30]: no macroscopic round necrotic lesion, score 0; presence of 1–5 small, round necrotic foci (<5 mm in diameter), score 1; many small, round necrotic foci (≥5), or large necrotic foci (≥5 mm in diameter), score 2; many macroscopic small and large necrotic foci, score 3; presented with complex lesions and numerous mixed lesions, score 4. All lesions were scored without knowing the grouping.
2.7. DNA Extraction, Library Construction, and Sequencing
Microbial DNA was extracted from cecum contents using the E.Z.N.A. soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s protocol. The quality and concentration of DNA extract were determined by 1.0% agarose gel electrophoresis and a NanoDrop2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA) and kept at −80 °C prior to further use. The full-length bacterial 16S rRNA gene was amplified with primer pairs 27F (5′-AGRGTTYGATYMTGGCTCAG-3′) and 1492R (5′-RGYTACCTTGTTACGACTT-3′) using a T100 Thermal Cycler PCR thermocycler (BIO-RAD, Hercules, CA, USA). The PCR reaction mixture included 4 μL 5 × Fast Pfu buffer, 2 μL 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL Fast Pfu polymerase, 10 ng of template DNA, and ddH_2_O to reach a final volume of 20 µL. PCR amplification cycling conditions were as follows: initial denaturation at 95 °C for 3 min, followed by 29 cycles of denaturing at 95 °C for 30 s, annealing at 60 °C for 30 s and extension at 72 °C for 45 s, and single extension at 72 °C for 10 min, ending at 4 °C. The PCR product was extracted from 2% agarose gel and purified using the PCR Clean-Up Kit (YuHua, Shanghai, China) according to manufacturer’s instructions and quantified using Qubit 4.0 (Thermo Scientific, Waltham, MA, USA). Purified amplicons were pooled in equimolar amounts and paired-end sequenced on an Illumina Nextseq2000 platform (Illumina, San Diego, CA, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China).
2.8. Bioinformatic Analysis
Raw FASTQ files were de-multiplexed using an in-house perl script, and then quality-filtered by fastp version 0.19.6 and merged by FLASH version 1.2.7 with the following criteria: (i) The reads were truncated at any site receiving an average quality score of <20 over a 50 bp sliding window, and truncated reads shorter than 50 bp or reads containing ambiguous characters were discarded. (ii) Only overlapping sequences longer than 10 bp were assembled according to their overlapped sequence. The maximum mismatch ratio of overlap region was 0.2. Reads that could not be assembled were discarded. (iii) Samples were distinguished according to the barcode and primers, and the sequence direction was adjusted and exact barcode matching was performed, with a maximum 2-nucleotide mismatch in primer matching. Then, the optimized sequences were clustered into operational taxonomic units (OTUs) using UPARSE v7.0.1090 with 97% sequence similarity level. The most abundant sequence for each OTU was selected as a representative sequence. To minimize the effects of sequencing depth on alpha and beta diversity measurements, the number of 16S rRNA gene sequences from each sample was rarefied to 20,000, which still yielded an average Good’s coverage of 99.09%. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier version 2.2 against the 16S rRNA gene database (e.g., Silva v138) using a confidence threshold of 0.7.
2.9. Statistical Analysis
Bioinformatic analysis of gut microbiota was carried out using the Majorbio Cloud platform (https://cloud.majorbio.com, accessed on 28 February 2025). Based on the OTUs information, rarefaction curves and alpha diversity indices including observed OTUs, Chao1 richness, Shannon index, and Good’s coverage were calculated using Mothur v1.30.1. The similarity among the microbial communities in different samples was determined by principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarity using the Vegan v2.5-3 package. The PERMANOVA test was used to assess the percentage of variation explained by the treatment, along with its statistical significance, using Vegan v2.5-3 package. The linear discriminant analysis (LDA) effect size (LEfSe) (http://huttenhower.sph.harvard.edu/LEfSe, accessed on 28 February 2025) was performed to identify the significantly abundant taxa (phylum to genera) of bacteria among the different groups (LDA score > 4, p < 0.05).
IBM SPSS Statistics 22.0 software (version 22.0; IBM SPSS, Inc., Chicago, IL, USA) was used to conduct a one-way ANOVA test and one-dimensional variance descriptive statistical analysis on data such as body weight comparison and lesion scores from the six experimental groups of chickens, with (p < 0.05) as the significance judgment criterion.
3. Results
3.1. Growth Performance
During the infection process, the chickens in each experimental group were weighed to obtain body weight comparison data. The results showed that significant differences in body weight were observed between the 7-day infected group and the control group, as well as between the 14-day infected group and the control group (p < 0.05). However, no significant difference was found in body weight in the 21-day experimental group (p > 0.05), suggesting that the severity of H. meleagridis infection was more pronounced at 7 days post-infection (dpi) and 14 days post-infection (dpi). The results are shown in Figure 1.
3.2. Intestinal Lesion and Intestinal Lesion Score
The chickens in each experimental group were dissected to observe pathological changes. The results showed that, in the 7 dpi group, during necropsy, a small number of chickens had crater-like necrotic foci on the liver, with the diameter of the necrotic foci being less than 5 mm, and an obvious cecal plug (Figure 2A, the left side of the image represents the infected group, while the right side represents the control group; the same applies below). In the 14 dpi group, most chickens exhibited liver enlargement, obvious congestion, and multiple crater-like necrotic foci, with some necrotic foci exceeding 5 mm in diameter; the liver was brittle and fragile. The cecum was significantly swollen, with increased volume, and when cut open, the cecal wall was thickened, adhered, and a cecal plug was visible inside (Figure 2B). In the 21 dpi group, during necropsy, a small number of chickens had necrotic foci on the liver, and the cecum was slightly swollen (Figure 2C); no pathological changes were observed in the control group.
The average liver–cecum lesion scores of chickens in each experimental group are shown in Table 1 and Figure 3. It can be observed that the lesion score at 14 dpi is the highest, with the average liver–cecum lesion scores at 14 dpi significantly higher than those at 7 dpi (p < 0.05). Furthermore, the average liver–cecum lesion scores at 21 dpi were significantly lower than those at 14 dpi (p < 0.05).
Given that there was no significant difference in body weight between the infected and control groups at 21 days, and that the liver and cecal lesion scores were significantly reduced, we selected the following four groups for 16S rRNA sequencing analysis: the 7-day control group, the 7-day infected group, the 14-day control group, and the 14-day infected group.
3.3. Rarefaction Curve and the Change in the Alpha and Beta of Cecal Gut Microbiota
For descriptive convenience, the 7-day control group, the 7-day infected group, the 14-day control group, and the 14-day infected group are, respectively, designated as G1, G2, G3, and G4. After quality filtering and assembly, we obtained a total of 669,538 sequences; the average sequence lengths were 1458 bp per sample. The rarefaction curves of 20 samples stabilized as the data increased, indicating sufficient data sampling and sequencing depth to assess richness and microbial community diversity (Figure 4).
Compared with the control group (Table 2), there were no significant differences in the ACE, Chao, Shannon, and Simpson indices in the 7 dpi group (p > 0.05). The ACE, Chao, Shannon, and Sobs indices in the 14 dpi group were significantly decreased (p < 0.05). When comparing the 7 dpi group with the 14 dpi group, it was found that the ACE, Chao, Shannon, and Sobs indices at 14 dpi were significantly decreased (p < 0.05). The PCoA results (Figure 5) demonstrated significant differences in cecal microbiota diversity among the groups (p = 0.001). The cecal microbial structure distribution of the infected and control groups at 14 dpi showed distinct clustering on opposite sides, with significant separation. At 14 dpi, the microbial structure distribution of the cecal microbiota in the control and infected groups of chickens showed a clear left–right separation with a substantial distance between them. Compared to the control group, the infected group deviated toward the positive PC1 axis, and this deviation increased over time.
3.4. Analysis of the Composition and Differential Changes in Species Abundance of Cecal Microbiota in Chickens
As illustrated in Figure 6A, at the phylum level, the dominant bacterial phyla in both the control and infected groups were Bacteroidota, Proteobacteria, and Firmicutes, with the additional presence of Fusobacteria and Verrucomicrobia. After H. meleagridis infection, the abundance of Firmicutes decreased, whereas the abundances of Bacteroidota and Proteobacteria increased. Verrucomicrobia was detected in the control group, while Fusobacteria emerged in the infected group. Compared to the 7 dpi group, the 14 dpi group showed a further reduction in Firmicutes abundance and an elevation in Proteobacteria abundance.
As shown in Figure 6B, at the genus level, the dominant bacterial genera in the chicken intestinal microbiota include Phocaeicola spp., Alistipes spp., Bacteroides spp., Escherichia spp., Lactobacillus spp., and Faecalibacterium spp. Following H. meleagridis infection, compared to the control group, the abundances of Phocaeicola spp., Bacteroides spp., Escherichia spp., Subdoligranulum spp., and Fusobacterium spp. increased, whereas the abundances of Alistipes spp., Lactobacillus spp., Parabacteroides spp., Mediterranea spp., Blautia spp., Limosilactobacillus spp., Ligilactobacillus spp., Anaerotignum spp., and Campylobacter spp. decreased. Compared to the 7 dpi group, the 14 dpi group exhibited increased abundances of Bacteroides spp., Escherichia spp., Fusobacterium spp., Enterococcus spp., and Mediterranea spp., while the abundances of Alistipes spp., Lactobacillus spp., Akkermansia spp., and Limosilactobacillus spp. decreased.
At the species level (Figure 6C), the dominant bacterial species in the chicken intestinal microbiota include Bacteroides fragilis, Lactobacillus crispatus, and Faecalibacterium prausnitzii. Following H. meleagridis infection, compared to the control group, the abundance of Phocaeicola coprocola, Escherichia fergusonii, Bacteroides fragilis, Subdoligranulum variabile, Mediterranea massiliensis, Fusobacterium mortiferum, Anaerotignum lactatifermentans, and Campylobacter jejuni increased, while the abundances of Phocaeicola dorei, Phocaeicola faecicola, Lactobacillus crispatus, Faecalibacterium prausnitzii, Parabacteroides distasonis, Bacteroides uniformis, Ligilactobacillus aviravius, and Akkermansia muciniphila decreased. Compared to the 7 dpi group, the 14 dpi group exhibited a marked increase in the abundance of Escherichia fergusonii, Phocaeicola faecicola, Bacteroides fragilis, Mediterranea massiliensis, and Fusobacterium mortiferum, whereas the abundances of Phocaeicola coprocola, Lactobacillus crispatus, Subdoligranulum variabile, and Campylobacter jejuni showed a pronounced decline.
3.5. LEfSe-Based Discriminant Analysis of Multi-Level Species Differences in Cecal Microbiota Across Experimental Groups
To gain a comprehensive understanding of the impact of H. meleagridis infection on the gut microbiota, we conducted LEfSe analysis. The taxonomic cladogram and LDA scores obtained from the LEfSe analysis confirmed and visualized the effects of infection (Figure 7 and Figure 8). Filtered by LEfSe analysis (LDA threshold of 4), it was found that at 7 dpi, the control group was enriched with Oscillospiraceae spp., Blautia spp., Phocaeicola dorei, and Faecalibacterium prausnitzii, while the infected group was enriched with Christensenella, Phocaeicola coprocola, and Mediterranea massiliensis; at 14 dpi, the control group showed enrichment of Verrucomicrobia, Rikenellaceae, Tannerellaceae, Akkermansiaceae, Alistipes spp., Bacteroides spp., Bacteroides uniformis, Ligilactobacillus aviarius, and Limosilactobacillus oris, whereas the infected group exhibited enrichment of Proteobacteria, Fusobacteria, Escherichia spp., Fusobacterium spp., Enterococcus spp., Escherichia fergusonii, Phocaeicola faecicola, and Bacteroides fragilis.
4. Discussion
Histomonosis, a parasitic disease primarily affecting poultry, such as chickens and turkeys, is globally distributed and presents significant economic challenges to the poultry industry. The absence of prophylactic drugs and commercially available vaccines for treatment or prevention has led to increased efforts to control this disease [31]. Recent advancements in high-throughput sequencing technologies have enabled a more comprehensive understanding of the gut microbiota, which comprise trillions of microorganisms residing in the animal intestine [32]. A stable gut microbiota community is closely linked to host health, with the microbiota playing a pivotal role in modulating the immune system and enhancing production performance [18,33,34].
In this study, experimental infection of white leghorns with H. meleagridis revealed the most pronounced differences in body weight and lesion scores at 7 dpi and 14 dpi compared to controls, whereas lesions alleviated and body weight gradually recovered by 21 dpi, consistent with previous findings [35,36]. We hypothesized that these changes in body weight and pathology might be associated with alterations in the cecal microbiota induced by H. meleagridis infection. Therefore, cecal contents from infected chickens at 7 and 14 dpi were selected for microbiota diversity analysis. The observed weight recovery may be attributed to the resolution of infection and restoration of physiological functions. However, it should be noted that this study involved a limited sample size, which may introduce potential survivor bias or selection bias, warranting further investigation.
Analysis of microbiota diversity indicated that H. meleagridis infection induced marked changes in the cecal microbiota, which were likely associated with the severity of pathological lesions. We therefore hypothesize that reduced diversity of the cecal microbiota is accompanied by more severe cecal lesions and greater body weight loss, suggesting a potential negative correlation between cecal microbiota diversity and pathological damage. This indicates that dysbiosis of the cecal microbiota plays an important role in the pathophysiological changes induced by H. meleagridis infection.
Following H. meleagridis infection, the alpha diversity of the cecal microbiota in chickens decreased throughout the course of infection, indicating a reduction in both microbial richness and evenness. This phenomenon is consistent with findings from studies on multiple intestinal parasites. Infection with E. tenella and E. necatrix have been shown to reduce the diversity of the cecal microbiota in chickens [37,38]. Similarly, Cryptosporidium infection causing diarrhea in goats and calves led to decreased gut microbiota diversity [39,40]. To elucidate the dynamic distribution of bacterial species, this study further analyzed bacterial composition at the phylum, genus, and species levels. The most frequently identified bacteria in the cecal microbiota of control group chickens were Firmicutes, Bacteroidetes, Proteobacteria, Verrucomicrobia, and Fusobacteria, consistent with previous reports by Huang et al. [41]. This study revealed that following H. meleagridis infection, the abundances of Firmicutes and Verrucomicrobia decreased, while those of Proteobacteria, Bacteroidetes, and Fusobacteria increased. A higher ratio of Firmicutes to Bacteroidota helps reduce pathogen colonization [42]. In this case, the ratio decreased, while the levels of inflammatory bacteria, such as Proteobacteria and Fusobacteria, increased [43,44,45]. Based on the aforementioned changes, we propose that H. meleagridis infection alters the abundance of beneficial and pathogenic bacteria in the gut, which may influence the progression of histomonosis.
Following H. meleagridis infection, alterations were observed in the cecal microbiota at both the genus and species levels. Based on the cecal species composition, the abundances of L. crispatus, L. aviarius, Limosilactobacillus vaginalis, L. oris, and Limosilactobacillus reuteri were observed to decrease post-infection. Certain species of Lactobacillus spp. are well-established probiotics widely used in food and feed additives for disease prevention and treatment [46,47]. They ferment carbohydrates to produce lactic acid and short-chain fatty acids (SCFAs), secrete antimicrobial factors [48], promote mucus and sIgA secretion [49], modulate immune cytokine expression [50,51,52], and alter gut microbiota composition, thereby exerting antibacterial, anti-inflammatory, and intestinal barrier-enhancing effects [53]. L. crispatus belongs to the genus Lactobacillus, is commonly found in the female urogenital tract, and helps combat colonization by uropathogens [54]. In livestock and poultry, L. crispatus produces lactic acid and lowers intestinal pH to inhibit the growth of harmful bacteria such as C. jejuni and Salmonella [55,56]. Additionally, L. aviarius, L. vaginalis, L. oris, and L. reuteri belong to the genera Ligilactobacillus spp. and Limosilactobacillus spp. within the phylum Firmicutes. The combined application of L. salivarius and L. reuteri in broilers has been shown to enhance intestinal morphology and increase the expression of the anti-inflammatory cytokine IL-10 [57]. Furthermore, L. reuteri and L. vaginalis isolated from chicken ceca produce extracellular polysaccharides (EPSs), which facilitate intestinal colonization of beneficial bacteria and exhibit strong antibacterial activity against E. coli and Salmonella Typhimurium in vitro [58]. Based on these findings, we hypothesize that H. meleagridis infection may lead to a relative reduction in the abundance of these species, thereby decreasing the secretion of anti-inflammatory factors and lactic acid, weakening the intestinal barrier. This could create opportunities for the colonization of opportunistic pathogens, consequently exacerbating pathological lesions.
In addition to the genus Lactobacillus, beneficial bacterial species such as Alistipes, F. prausnitzii, and A. muciniphila were also notably present in the control group. Similarly to Lactobacilli, Alistipes is a propionate producer capable of expressing acetyl-CoA carboxylase. It also expresses glutamate decarboxylase, influencing SCFA levels and the production of the metabolite γ-aminobutyric acid (GABA) [59]. F. prausnitzii produces butyrate with anti-inflammatory properties, increases the secretion of the anti-inflammatory cytokine IL-10 to reduce intestinal inflammation, and exerts anti-inflammatory effects by inhibiting the Th17 pathway [60,61]. A. muciniphila, belonging to the phylum Verrucomicrobia, degrades mucin, enhances SCFA levels, and contributes to intestinal barrier protection and immune regulation [62]. A significant reduction in the abundance of these bacterial species was observed at 14 dpi. We hypothesize that the intestinal inflammation caused by H. meleagridis infection may be related to the decreased abundance of beneficial bacteria possessing anti-inflammatory functions.
In addition to the beneficial bacteria mentioned above, changes in certain pathogenic bacteria are also noteworthy. As the infection progressed, the abundance of Campylobacter increased at 7 dpi. C. jejuni, the predominant species within this genus and a zoonotic pathogen, employs capsular polysaccharide (CPS) production to evade the host immune system [63]. However, existing studies have demonstrated that lactobacilli possess anti-Campylobacter properties and can reduce C. jejuni loads [62]. By 14 dpi, the abundances of Escherichia spp. and Fusobacterium spp. increased, specifically manifested as a rise in E. fergusonii and F. mortiferum. Certain species of Fusobacterium can invade colonic epithelial cells and activate intracellular inflammatory signaling pathways [64]. A research indicated that E. fergusonii carries genes encoding the virulence factor heat-labile enterotoxin (LT), which is also present in enterotoxigenic E. coli (ETEC) [65]. This toxin penetrates cells and induces apoptosis in intestinal epithelial cells, although its activity can be suppressed by SCFAs [66]. In this study, we observed that C. jejuni increased significantly at 7 dpi but markedly decreased at 14 dpi, which we hypothesize may be suppressed by lactobacilli—particularly L. crispatus—consistent with earlier findings. Conversely, the notable increase in E. fergusonii and F. mortiferum at 14 dpi may have resulted from the reduction in beneficial bacterial populations. Therefore, we propose that the decline of these beneficial species likely impaired microbial metabolite levels and cytokine-mediated immune pathways, thereby leading to compromised intestinal barrier function and exacerbated inflammatory responses. As a consequence, more severe pathological manifestations were observed during the later stages of H. meleagridis infection. Although certain functional correlations of the aforementioned bacterial species have been described, this study lacks supporting functional data, such as metagenomics analyses, SCFA quantification, and transcriptomic profiling. Hence, the observed associations between these bacteria and histomonosis should be regarded as preliminary inferences based on current observations, and their functional relevance warrants further investigation.
Through LEfSe analysis, we observed that at 7 dpi, the control group was predominantly enriched with bacterial taxa possessing potential probiotic functions, including key butyrate- and acetate-producing genera such as Blautia and F. prausnitzii. The latter is recognized as a crucial marker of intestinal health, renowned for its anti-inflammatory properties, such as inducing regulatory T cells and generating anti-inflammatory metabolites [67]. Additionally, P. dorei and L. reuteri have also been associated with the maintenance of gut homeostasis [68,69]. In contrast, the 7 dpi group showed enrichment of Christensenellaceae. While Christensenellaceae is often linked to host leanness and is known to influence gut microbiota composition [70], with some studies indicate that its abundance may decrease under disease conditions [71]. Therefore, we hypothesize that early-stage H. meleagridis infection impairs the enrichment of beneficial bacteria in the gut, which may lead to impaired intestinal health and body weight loss in chickens.
As the infection progressed to 14 days, the control group exhibited enrichment in various taxa associated with mucosal health and complex carbohydrate metabolism. These included Akkermansiaceae (particularly A. muciniphila), a representative of the Verrucomicrobia phylum, which plays a key role in maintaining intestinal barrier function by degrading and promoting regeneration of the mucus layer [72,73], as well as members of the Bacteroides spp., which help prevent intestinal inflammation through production of IL-10 and acetate [74]. Conversely, the 14 dpi group demonstrated a marked increase in pathogenic bacteria. Enrichment of Proteobacteria is a classic signature of gut dysbiosis [75]. Among its members, the genus Escherichia (including the species E. fergusonii) represents an emerging zoonotic pathogen that has been isolated from chicken meat in certain regions and from human patients with diarrhea. This bacterium can express the heat-labile enterotoxin (LT) gene [76,77] and participates in carbon source metabolism, playing a significant role in host energy accumulation [78,79]. More notably, the enrichment of Fusobacteria and its representative Fusobacterium spp. is particularly significant, as this phylum has been associated with colorectal cancer in humans [80] and can elevate levels of cytotoxic (CD8^+^) T cells to enhance anti-tumor responses. Additionally, the enrichment of Bacteroides fragilis warrants attention, as certain subtypes (such as enterotoxigenic ETBF) are established pathogenic factors [81]. Based on the above research findings, we hypothesize that under H. meleagridis infection, the abundance of pathogenic bacteria in the chicken gut increases, while the populations of metabolic and beneficial bacterial species decrease. This may adversely affect the growth performance and intestinal barrier function of chickens, leading to aggravated pathological severity.
The LEfSe analysis clearly delineates the structural changes in the gut microbiota induced by H. meleagridis infection: the early stage is characterized by a relative reduction in commensal/beneficial bacteria and an increase in specific bacterial families, while the later stage transitions to a dysbiotic state marked by a significant increase in Proteobacteria (particularly Escherichia), Fusobacteria, and opportunistic pathogens such as B. fragilis. We propose that this microbial shift may be linked to the pathological progression of H. meleagridis infection and the exacerbation of intestinal inflammation. However, our studies have indicated that LEfSe analysis requires a substantial volume of sequencing data to yield accurate results [82,83]. In contrast, the data sample size utilized in this study is relatively limited, which imposes constraints on identifying biomarker species affected by H. meleagridis. Therefore, further investigation is warranted.
5. Conclusions
In summary, this study demonstrates that Histomonas meleagridis infection results in reduced body weight, as well as cecal and hepatic lesions in chickens, leading to decreased species richness in the cecal microbiota, a reduced alpha diversity index of the gut microbial community, and alterations in the composition and structure of the cecal microbiome. Notably, the abundance of non-pathogenic bacteria significantly diminished. Conversely, the abundance of opportunistic pathogens increased. This research provides a more comprehensive theoretical foundation for understanding the interactions between parasites and gut microbiota. Furthermore, these findings may offer new strategic insights for preventing and controlling histomoniasis through microbial intervention strategies. However, the findings of this study still require validation with larger sample sizes and more functional data.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Beer L.C. Petrone-Garcia V.M. Graham B.D. Hargis B.M. Tellez-Isaias G. Vuong C.N. Histomonosis in Poultry: A Comprehensive Review Front. Vet. Sci.2022988073810.3389/fvets.2022.88073835601402 PMC 9120919 · doi ↗ · pubmed ↗
- 2Hess M. Liebhart D. Bilic I. Ganas P. Histomonas meleagridis—New Insights into an Old Pathogen Vet. Parasitol.2015208677610.1016/j.vetpar.2014.12.01825576442 · doi ↗ · pubmed ↗
- 3Liu D. Kong L. Tao J. Xu J. An Outbreak of Histomoniasis in Backyard Sanhuang Chickens Korean J. Parasitol.20185659760210.3347/kjp.2018.56.6.59730630281 PMC 6327192 · doi ↗ · pubmed ↗
- 4Liebhart D. Sulejmanovic T. Grafl B. Tichy A. Hess M. Vaccination against Histomonosis Prevents a Drop in Egg Production in Layers Following Challenge Avian Pathol.201342798410.1080/03079457.2012.76084123391185 · doi ↗ · pubmed ↗
- 5Liebhart D. Hess M. Spotlight on Histomonosis (Blackhead Disease): A Re-Emerging Disease in Turkeys and Chickens Avian Pathol.2020491410.1080/03079457.2019.165408731393162 · doi ↗ · pubmed ↗
- 6Chen Q.G. Wang S. Rong J. Chen C. Hou Z.F. Liu D.D. Tao J.P. Xu J.J. Inhibitory Effects of Different Plant Extracts on Histomonas meleagridis In Vitro and In Vivo in Chickens Vet. Parasitol.202533711048710.1016/j.vetpar.2025.11048740294477 · doi ↗ · pubmed ↗
- 7Dubey J.P. Paker C. Graham D. Hargis B.M. Jenkins M.C. Histomonas meleagridis Infections in Turkeys in the USA: A Century of Progress, Resurgence, and Tribute to Its Early Investigators, Theobald Smith, Ernst Tyzzer, and Everett Lund J. Parasitol.202411026327510.1645/24-838982636 · doi ↗ · pubmed ↗
- 8Liebhart D. Ganas P. Sulejmanovic T. Hess M. Histomonosis in Poultry: Previous and Current Strategies for Prevention and Therapy Avian Pathol.20174611810.1080/03079457.2016.122945827624771 · doi ↗ · pubmed ↗