Comparative Evaluation of Mucosal Adjuvants for Intranasal Immunization with a Recombinant RSV Prefusion F Protein
Hongqiao Hu, Lei Cao, Jie Jiang, Yuqing Shi, Liang Du, Mengxuan Chu, Hai Li, Yan Zhang

TL;DR
This study compares different adjuvants for intranasal RSV vaccination in mice, finding that moderate doses of CpG-ODN and PEI offer the best immune response and protection.
Contribution
The study provides a systematic comparison of dose-effect profiles across multiple mucosal adjuvants for intranasal RSV vaccine development.
Findings
CpG-ODN at 10 µg improved neutralizing antibodies, IgA, and reduced lung viral load without aggravating pathology.
PEI and IFN-α showed dose-dependent effects, with 10 µg being sufficient for robust immunity.
CpG-ODN and PEI outperformed CTA1-DD in immunogenicity and protection, though PEI induced a Th2-biased response.
Abstract
Background: Respiratory syncytial virus (RSV) remains a major etiologic agent of acute lower respiratory tract infection (ALRTI). Currently licensed RSV vaccines are administered by intramuscular injection and induce limited immunity at the respiratory mucosal interface, underscoring the need for effective mucosal vaccination strategies. Methods: To enhance mucosal immune responses, we used prefusion F protein (Pre-F) as the antigen and performed intranasal immunization in BALB/c mice. Four mucosal adjuvants (CpG-ODN, CTA1-DD, IFN-α, and PEI) were systematically compared across different dose levels to evaluate their immunological and protective efficacy. Results: Both adjuvant type and dose helped shape the magnitude and quality of the immune response and the level of protection. CpG-ODN showed a dose-restricted immunopotentiating effect: an intermediate dose (10 µg) significantly…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
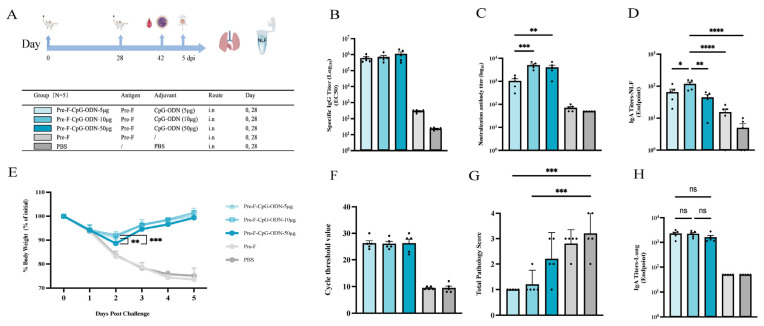 Figure 1
Figure 1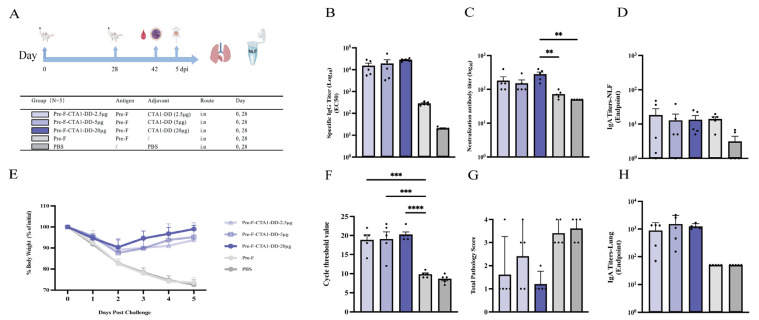 Figure 2
Figure 2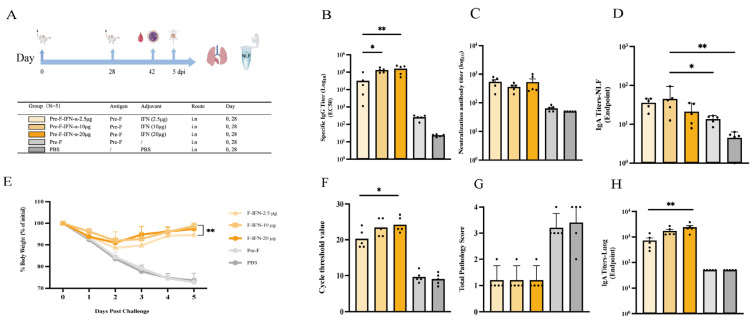 Figure 3
Figure 3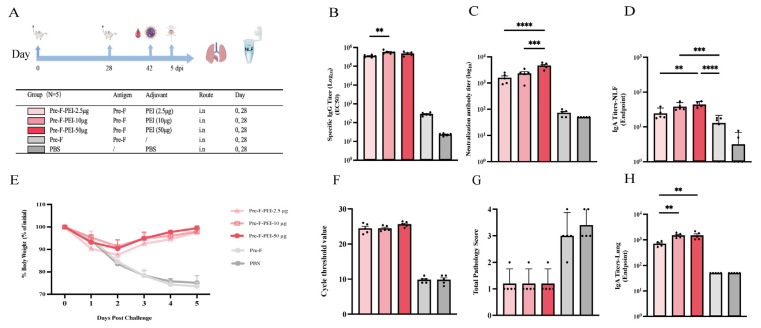 Figure 4
Figure 4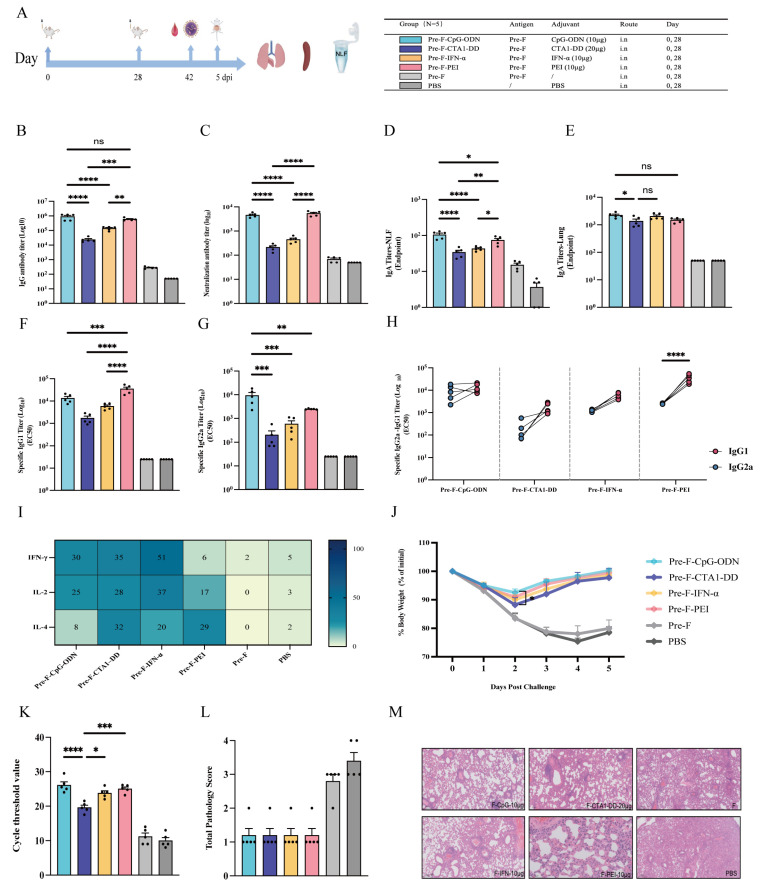 Figure 5
Figure 5- —Beijing Natural Science Foundation and Haidian Original. Innovation Joint Fund
- —Monitoring, prediction, and early-warning technologies for unknown and emerging pathogens
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRespiratory viral infections research · Pediatric health and respiratory diseases · Inhalation and Respiratory Drug Delivery
1. Introduction
Respiratory syncytial virus (RSV) belongs to the family Pneumoviridae and the genus Orthopneumovirus. It is an enveloped, single-stranded, negative-sense RNA virus with a genome of approximately 15 kb that contains 10 genes and encodes 11 proteins. Based on sequence variation in the G gene, RSV is classified into two subtypes, RSV-A and RSV-B; globally, the predominant circulating genotypes are currently ON1 and BA9 [1]. RSV is an important respiratory pathogen across all age groups, causing substantial disease burden, particularly severe acute lower respiratory tract infections in children under 5 years of age (especially preterm infants and those with underlying cardiopulmonary diseases) and in adults aged ≥65 years [2]. The fusion (F) protein contains multiple highly conserved antigenic epitopes and is a key target for monoclonal antibody therapies and vaccine design [3]. To date, three monoclonal antibodies (Palivizumab, Nirsevimab, and Clesrovimab) and three vaccines (Abrysvo, Arexvy, and mRESVIA) have been approved [4,5]. However, currently available RSV vaccines are administered intramuscularly and induce relatively limited local mucosal immunity in the respiratory tract, highlighting the potential value of mucosal immunization strategies for RSV prevention.
Mucosal immunity offers clear advantages in preventing respiratory viral infections by rapidly inducing high levels of IgA antibodies and local T cell responses at the respiratory mucosa, the primary site of viral entry, thereby establishing a first-line immune barrier that effectively inhibits viral attachment, entry, and spread. Such localized immune responses can not only reduce viral transmission but also lower disease incidence and severity [6]. In the development of intranasal RSV vaccines, most candidates that have advanced into clinical evaluation are still predominantly live-attenuated vaccines (LAVs). For example, the NIAID live-attenuated RSV/6120/ΔNS2/1030s vaccine showed a degree of protective efficacy in children aged 6–24 months, but local adverse events such as rhinorrhea were also observed. Sanofi’s live-attenuated vaccine, SP0125, progressed the furthest; however, it was discontinued after failing to meet the primary efficacy endpoint in a phase 3 clinical trial [7,8,9]. Overall, intranasal LAVs must achieve a balance between sufficient attenuation and preserved immunogenicity, and the consistency of clinical protection and the safety margin still requires further validation. In addition, intranasal viral-vectored vaccines in adults may be affected by pre-existing immunity (preformed antibodies), which can limit nasal retention and reduce the efficiency of immune induction. By contrast, intranasal recombinant protein vaccines avoid the risk of reversion to virulence and are less susceptible to anti-vector immunity; however, they typically require appropriate mucosal adjuvants and delivery systems to enhance both mucosal and systemic immune responses, thereby achieving more stable protective efficacy [10,11].
Respiratory mucosal adjuvants encompass a broad range of types, including polymers, bacterial toxins (and their derivatives), cytokines, pattern-recognition receptor (PRR) agonists, and small peptides [12]. Among these, CpG oligodeoxynucleotides (CpG-ODN), a classical TLR9 agonist, can robustly promote Th1-biased immune responses; TLR9 agonists represented by CpG-ODN have demonstrated clear immunostimulatory effects with acceptable safety profiles in multiple vaccine clinical trials [13,14]. Animal studies further show that intranasal immunization with Pre-F protein in combination with CpG-ODN can elicit high levels of antigen-specific IgA and neutralizing antibodies in both the upper and lower respiratory tracts, significantly reduce lung viral loads, and alleviate histopathological injury after RSV challenge, without evidence that immune enhancement aggravates disease [15,16].CTA1-DD is composed of the A1 subunit of cholera toxin fused to the D domain of staphylococcal protein A, combining non-toxicity with potent mucosal adjuvant activity. When administered intranasally with protein antigens or vectors, CTA1-DD markedly enhances local IgA responses, promotes Th1/Th17 immunity, and facilitates the formation of tissue-resident memory T cells (TRM) [17,18]. Previous studies indicate that CTA1-DD can dose-dependently augment B-cell activation, germinal center formation, and the generation of long-lived plasma cells, thereby supporting the establishment of protective Th1/Th17 responses [19]. Interferons (IFNs) are typically classified into three types: type I (e.g., IFN-α/β), type II (IFN-γ), and type III (IFN-λ). As a key cytokine of mucosal innate immunity, type I interferon (IFN-α), when co-administered intranasally with antigen, can promote dendritic cell recruitment and antigen presentation, enhance TRM and neutralizing antibody responses, and thereby accelerate viral clearance [20,21]. Polyethylenimine (PEI), widely used as a nucleic acid transfection reagent, carries a strong positive charge and can form nanocomplexes with negatively charged molecules to improve cellular delivery. Recent evidence suggests that PEI functions not only as a delivery vehicle but also as an effective mucosal adjuvant: through electrostatic interactions, PEI forms nanocomplexes with antigens, enhancing mucosal adhesion and transepithelial transport and promoting phagocyte uptake and cross-presentation [22]. Collectively, CpG-ODN, CTA1-DD, IFN-α, and PEI represent PRR agonists, toxin-derived adjuvants, cytokine adjuvants, and polymer-based adjuvants, respectively. Although these respiratory mucosal adjuvants offer distinct immunostimulatory features, rational dose selection to ensure both safety and efficacy remains a central challenge in vaccine design.
Unlike intramuscular injection, the nasal mucosa is structurally more delicate and is richly innervated and vascularized; excessive adjuvant dosing is therefore more likely to provoke local inflammation or tissue injury, nonspecific immune activation, and antigen-independent inflammatory responses. Accordingly, in respiratory mucosal vaccine research, adjuvant dose is a key determinant of the balance between safety and immunogenicity. Because mucosal adjuvants differ substantially in mechanism of action, inflammatory potency, and mucosal irritancy, it is difficult to apply a single standardized dose for direct cross-adjuvant comparisons. Based on these considerations, using RSV prefusion F (Pre-F) protein as the antigen in an intranasal immunization model in BALB/c mice, we performed dose escalation screening of four mucosal adjuvants, CpG-ODN, CTA1-DD, IFN-α, and PEI, to first define their safe and effective dose ranges under mucosal administration; based on these ranges, we then systematically compared the immunogenicity and protective efficacy of the different adjuvants. Thus, rational selection and optimization of mucosal adjuvant dosing to achieve an appropriate balance between immune enhancement and safety remains a core issue in mucosal vaccine development, and the present work provides experimental evidence to inform the optimization of future RSV mucosal immunization strategies.
2. Materials and Methods
2.1. Cells and Viruses
HEp-2 cells used in this study were obtained from the American Type Culture Collection (ATCC). The RSV-A long strain was maintained in our laboratory.
2.2. Experimental Animals
Female BALB/c mice (6–8 weeks of age) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. All animal procedures were approved by the Institutional Animal Care and Use Committee of the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention (Approval No. 20231113086).
2.3. Major Reagents
Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H + L) was purchased from Zhongshan Jinqiao (ZSGB-BIO, China). Mouse IgG1, IgG2a, and IgA antibodies were purchased from Abcam (UK). Mouse IFN-γ and Mouse IL-4 precoated ELISPOT kits were obtained from MabTech (Sweden). Branched 25 kDa PEI (Polysciences, Cat. No. 24765) was purchased from Shanghai Faqi Laboratory Reagents Co., Ltd. Type C CpG-ODN (ODN 2395, VacciGrade™), PMA plus ionomycin, and mouse lymphocyte separation medium were purchased from Dakewe Biotech Co., Ltd. Viral nucleic acid extraction kits were purchased from Xi’an Tianlong Science and Technology Co., Ltd. The fluorescent quantitative RT-PCR kit PrimeScript™ One Step RT-PCR Kit was purchased from Takara (Japan).
2.4. Construction of Pre-F Protein, IFN-α Adjuvant, and CTA1-DD Adjuvant
The Pre-F protein used in this study was prepared and expressed by our department in previous work [16]. The sequence of the Pre-F protein was selected based on the ON1 circulating strain (GenBank: KY296733.1), and its construction was optimized based on DS-Cav1 [23]. The IFN-α and CTA1-DD adjuvants were prepared and stored in our laboratory, as previously described [18,20].
2.5. Animal Immunization and Challenge
2.5.1. Evaluation of Immunogenicity and Protective Efficacy of Four Mucosal Adjuvants at Different Doses in BALB/c Mice
A total of 100 female BALB/c mice (6–8 weeks old) were randomly assigned to 20 groups (n = 5 per group). For each mucosal adjuvant, three representative dose levels (low/intermediate/high) were selected based on commonly used intranasal ranges reported in prior studies and mucosal tolerability considerations; detailed literature context is provided in the Discussion. Mice were bled and immunized on days 0 and 28 (50 µL per mouse). The detailed immunization regimens are shown in the Results section. For the RSV BALB/c mouse challenge dose and challenge model, our laboratory has previously explored and established a lethal model [24]. Therefore, based on prior research, we selected a moderate, non-lethal challenge dose (5 × 10^5^ PFU/mice). The specific challenge protocol is as follows: On day 42, mice were anesthetized with isoflurane and intranasally challenged with live RSV-Long at a dose of 5 × 10^5^ PFU/Mice. Body weight was monitored and recorded daily after challenge. Mice were euthanized on day 5 post-challenge, and lung and spleen tissues were collected for subsequent experiments.
2.5.2. Comparative Evaluation of Immunogenicity and Protective Efficacy of Four Mucosal Adjuvants in BALB/c Mice
A total of 30 female BALB/c mice (6–8 weeks old) were randomly assigned to six groups (n = 5 per group). The adjuvant doses used in this comparative experiment were selected from the dose-screening study (Section 2.5.1). Mice were bled and immunized on days 0 and 28 (50 µL per mouse). The detailed immunization regimens are shown in the Results section. On day 42, mice were anesthetized with isoflurane and intranasally challenged with live RSV-Long at a dose of 5 × 10^5^ PFU/Mice. Body weight was monitored and recorded daily after challenge. Mice were euthanized on day 5 post-challenge, and lung and spleen tissues were collected for subsequent experiments.
2.6. Collection of Mouse Nasal Lavage Fluid (NLF) and Lung Homogenate
Mouse NLF was collected as follows: After euthanasia, mice were placed in the supine position, and the head and maxillary regions were fully exposed. The mandible was cut to expose the pharyngeal outlet. Using a pipette, slowly drip pre-cooled sterile PBS (200 µL) into the posterior nares, and collect the lavage fluid flowing from the anterior nares. This process is repeated twice. Collected NLF was transferred to microcentrifuge tubes and centrifuged at 4 °C, 300–500 g for 5–10 min. The supernatants were used for antibody measurements.
Mouse lung homogenate was collected as follows: An appropriate amount of lung tissue was obtained, with 0.5 mL of PBS added for every 0.1 g of tissue. The tissue was placed in a homogenization tube containing precooled PBS and grinding beads. The tissue was then homogenized intermittently at 6000 rpm using a tissue grinder (30 s × 3 times) until it was completely broken. The homogenate was centrifuged at 8000 rpm for 10 min at 4 °C, and the supernatant was collected and stored at −80 °C for future use.
2.7. Detection of Pre-F-Specific Antibody Titers by Enzyme-Linked Immunosorbent Assay (ELISA)
Coating: 96-well plates were coated with Pre-F protein (50 µg/well) and incubated overnight at 4 °C. Washing: Plates were washed four times with PBST and tapped dry. Blocking: Each well was blocked with PBST containing 10% FBS for 2 h at 37 °C, followed by washing as above. Sample incubation: Serum was added to the first well at a 1:100 dilution and serially diluted in 5-fold steps; nasal lavage fluid was added to the first well at a 1:8 dilution and serially diluted in 2-fold steps; and lung homogenates were added to the first well at a 1:50 dilution and serially diluted in 2-fold steps. Plates were incubated for 1 h at 37 °C and washed as above. Secondary antibody incubation: HRP-conjugated goat anti-mouse IgG (1:5000) or HRP-conjugated antibodies specific for IgG1, IgG2a, or IgA (1:20,000) were added and incubated for 30 min at 37 °C, followed by washing as above. Development and reading: TMB substrate was added, and the plate was incubated for 15 min at 37 °C. The reaction was stopped with the stop solution, and absorbance was measured at 450 nm. EC50 values were calculated by four-parameter logistic regression in GraphPad Prism 10 after subtracting background readings from each well prior to curve fitting.
2.8. Neutralizing Antibody Assay
Mice sera were heat-inactivated at 56 °C for 30 min. Sera were initially diluted 1:25 and then serially 2-fold diluted. Each dilution was mixed at an equal volume with RSV-Long (1000 PFU/mL) and incubated at 37 °C for 2 h. The mixtures were added to HEp-2 cell monolayers. After 1 h of adsorption, the inoculum was removed, and cells were overlaid with maintenance medium containing 1.2% microcrystalline cellulose and incubated for 48 h. The overlay medium was removed, and the cells were fixed with 4% paraformaldehyde for 20 min. Cells were then incubated with palivizumab (1:1000) for 1 h at 37 °C and washed four times with PBST, followed by incubation with HRP-conjugated goat anti-human IgG (1:2000) for 30 min at 37 °C and washing four times with PBST. TrueBlue substrate was added for plaque visualization, and plaques were counted. Neutralizing titers were calculated using the Karber method and defined as the highest serum dilution resulting in a 50% reduction in plaque number relative to the positive control.
2.9. Enzyme-Linked Immunospot (ELISpot) Assay
Splenocytes were isolated from BALB/c mice using mouse lymphocyte separation medium according to the manufacturer’s instructions (Dakewe) and counted. Cytokine-secreting cells (IFN-γ, IL-4, and IL-2) were quantified using precoated ELISpot kits (MabTech) following the manufacturer’s protocol. Briefly, plates were washed five times with sterile PBS, then preincubated with RPMI 1640 containing 10% FBS for at least 30 min at room temperature. After removal of medium, cells were added at 3 × 10^5^ cells/100 μL per well, followed by addition of stimulants (10 µL per well). Plates were incubated for 48 h at 37 °C with 5% CO_2_. After incubation, plates were washed five times, and detection antibodies diluted in PBS containing 0.1% BSA were added (100 µL/well) and incubated for 2 h at room temperature. Plates were washed as above, and fluorescent conjugates diluted in PBS containing 0.1% BSA were added (100 µL/well) and incubated for 1 h at room temperature in the dark. Plates were washed again and incubated with fluorescence enhancer (50 µL/well) for 15 min at room temperature in the dark. Plates were then emptied, air-dried in the dark, and spots were counted using an ELISpot reader.
2.10. Lung Viral Load and Histopathology Scoring
The right lung was aseptically collected, homogenized in PBS, and centrifuged; 200 µL of supernatant was used for nucleic acid extraction. RSV viral load in lung tissue was quantified by real-time RT-PCR. The left lung was fixed in 4% paraformaldehyde for 24 h, embedded, sectioned, and stained with hematoxylin and eosin (H&E). Histopathological features were evaluated, including alveolitis, bronchiolitis, and the extent of inflammatory cell infiltration in the perivascular and interstitial regions [25]. Lung sections from individual mice were scored in a blinded manner based on alveolar wall thickening, interstitial pneumonitis, alveolitis, and bronchiolitis, as well as septal widening and perivascular/peribronchiolar inflammatory infiltrates (e.g., mononuclear cells, neutrophils). Overall lung lesion severity was graded as follows: 0 (normal, no lesions), 1 (mild, <1/4 of lung section involved), 2 (moderate, 1/4–2/4 involved), 3 (severe, 2/4–3/4 involved), and 4 (very severe, >3/4 involved).
2.11. Statistical Analysis
GraphPad Prism 10 was used for statistical analyses and graphing. Normally distributed data are presented as mean ± SD and were analyzed by one-way ANOVA followed by Tukey’s multiple-comparisons test. Non-normally distributed data are presented as median (Q1, Q3) and were analyzed by the Kruskal–Wallis test followed by Dunn’s multiple-comparisons test. Statistical significance was defined as * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001; “ns” indicates no significant difference.
3. Results
3.1. Evaluation of the Immunogenicity and Protective Efficacy of CpG-ODN at Different Doses in BALB/c Mice
The results showed that Pre-F-specific serum-binding IgG did not differ significantly among the CpG-ODN dose groups; however, neutralizing antibody titers were higher in the 10 µg and 50 µg groups than in the 5 µg group (p < 0.001 and p < 0.01, respectively) (Figure 1B,C). Mucosal IgA in nasal lavage fluid was significantly higher in the 10 µg group than in the 5 µg and 50 µg groups (p < 0.05 and p < 0.01, respectively) (Figure 1D). In addition, two days after challenge, mice in the 10 µg and 5 µg CpG-ODN groups recovered body weight faster than those in the 50 µg CpG-ODN group (p < 0.001 and p < 0.01, respectively). (Figure 1E). RT-qPCR analysis indicated no significant differences in lung viral loads among the CpG-ODN dose groups (Figure 1F). Nevertheless, lung histopathology scoring suggested more pronounced lung injury in the 50 µg group, with no significant improvement compared with the control group (Figure 1G). Collectively, these findings indicate that low-to-moderate CpG-ODN doses (particularly 10 µg) confer more favorable immunogenicity and protection than the high dose (50 µg).
3.2. Evaluation of the Immunogenicity and Protective Efficacy of CTA1-DD at Different Doses in BALB/c Mice
The results showed no significant differences in Pre-F-specific serum IgG among the CTA1-DD dose groups; however, the 20 µg CTA1-DD group elicited higher serum neutralizing antibody levels than the control groups (Pre-F and PBS) (p < 0.01) (Figure 2B,C). In terms of mucosal immunity, antigen-specific IgA levels in both nasal lavage fluid and lung homogenates did not differ significantly among the CTA1-DD dose groups (Figure 2D,H). Challenge assessments indicated that on day 3 after challenge, mice in the 20 µg CTA1-DD group tended to exhibit a faster body weight recovery than those in the 2.5 µg and 5 µg groups, although the differences were not statistically significant (Figure 2E). Lung viral load measured by RT-PCR showed that the 20 µg CTA1-DD group had significantly lower viral loads than the control group (p < 0.0001), and the 2.5 µg and 5 µg groups also showed reduced viral loads compared with the control group (p < 0.001) (Figure 2F). Overall, these data suggest that a higher dose of CTA1-DD (20 µg) provides more favorable immunogenicity and protective efficacy in this model.
3.3. Evaluation of the Immunogenicity and Protective Efficacy of IFN-α at Different Doses in BALB/c Mice
The results showed that both the 20 µg and 10 µg IFN-α groups induced significantly higher Pre-F-specific serum IgG levels than the 2.5 µg IFN-α group (p < 0.01 and p < 0.05, respectively) (Figure 3B). In contrast, neutralizing antibody titers did not differ significantly among the IFN-α dose groups (Figure 3C). Regarding mucosal immunity, the 10 µg IFN-α group exhibited higher antigen-specific IgA levels in nasal lavage fluid than the PBS control (p < 0.01). In lung homogenates, the 20 µg IFN-α group showed higher antigen-specific IgA levels than the 2.5 µg IFN-α group (p < 0.01), whereas no significant difference was observed between the 10 µg and 20 µg IFN-α groups (Figure 3D,H). Challenge assessments indicated that on day 3 after challenge, mice in the 10 µg and 20 µg IFN-α groups recovered body weight faster than those in the 2.5 µg IFN-α group, with no significant difference between the 10 µg and 20 µg IFN-α groups (Figure 3E). On day 5 after challenge, the 10 µg IFN-α group maintained a significantly higher body weight recovery rate than the 2.5 µg IFN-α group (p < 0.01) (Figure 3E). Lung viral load measured by RT-PCR showed that the 20 µg IFN-α group had significantly lower viral loads than the 2.5 µg IFN-α group (p < 0.05), while no significant difference was observed between the 20 µg and 10 µg IFN-α groups (Figure 3F). Moreover, lung histopathology scores did not differ significantly among the IFN-α dose groups (Figure 3G). Collectively, these data suggest that intermediate-to-higher doses of IFN-α (particularly 10 µg) confer improved immunogenicity and post-challenge outcomes compared with the low dose (2.5 µg) in this model.
3.4. Evaluation of the Immunogenicity and Protective Efficacy of PEI at Different Doses in BALB/c Mice
The results showed that the 10 µg PEI group induced significantly higher Pre-F-specific serum IgG levels than the 2.5 µg PEI group (p < 0.01). In addition, the 50 µg PEI group elicited significantly higher neutralizing antibody titers than both the 10 µg and 2.5 µg PEI groups (p < 0.001 and p < 0.0001, respectively) (Figure 4B,C). Regarding mucosal immunity, antigen-specific IgA levels in nasal lavage fluid were significantly higher in both the 50 µg and 10 µg PEI groups than in the Pre-F control group (p < 0.0001 and p < 0.001, respectively). In lung homogenates, antigen-specific IgA levels in the 10 µg and 50 µg PEI groups were significantly higher than those in the 2.5 µg group (p < 0.01) (Figure 4D,H). Challenge assessments indicated that on days 2–3 after challenge, mice in the 10 µg and 50 µg PEI groups showed a faster body weight recovery rate than those in the 2.5 µg PEI group, although the differences did not reach statistical significance. Lung viral loads and histopathology scores did not differ significantly among the PEI dose groups (Figure 4E–G). Collectively, these findings suggest that an intermediate PEI dose (10 µg) elicits more robust immune responses and overall protection than the low dose (2.5 µg), whereas the high dose (50 µg), despite further increasing neutralizing antibody titers, does not translate into a significant improvement in post-challenge body weight recovery, lung viral burden, or lung pathology.
3.5. Immunogenicity and Protective Efficacy of Four Mucosal Adjuvants in BALB/c Mice
Based on the above findings, a dose–effect relationship was observed between mucosal adjuvants (and their respective dose levels) and immune potentiation. To further compare the adjuvant-dependent enhancement of intranasal Pre-F immunization, we integrated antibody responses, body weight changes after challenge, and lung histopathology across dose levels to define an empirically appropriate dose for each adjuvant. Accordingly, 10 µg CpG-ODN, 20 µg CTA1-DD, 10 µg IFN-α, and 10 µg PEI were formulated with Pre-F for a head-to-head comparison of immunogenicity and protection (Figure 5A–M). The results showed that both the CpG-ODN and PEI groups elicited higher Pre-F-specific serum IgG and neutralizing antibody titers than the CTA1-DD and IFN-α groups, with statistical significance, whereas no significant differences were observed between the CpG-ODN and PEI groups (Figure 5B,C). In addition, antigen-specific IgA levels in nasal lavage fluid were higher in the CpG-ODN and PEI groups than in the CTA1-DD and IFN-α groups, with statistical significance; notably, the CpG-ODN group induced significantly higher nasal IgA than the PEI group (p < 0.05). Moreover, antigen-specific IgA levels in lung homogenates were higher in the CpG-ODN group than in the CTA1-DD group (p < 0.05) (Figure 5D,E). Analysis of IgG1 and IgG2a titers indicated a more balanced Th1/Th2-associated humoral profile in the CpG-ODN group, whereas the CTA1-DD, IFN-α, and PEI groups exhibited a more Th2-biased humoral pattern (Figure 5F–H). Cellular responses were further assessed by ELISpot for cytokine-secreting splenocytes (IFN-γ, IL-2, and IL-4). The CpG-ODN and IFN-α groups induced higher numbers of IFN-γ and IL-2 secreting cells, consistent with a Th1-biased cellular response, whereas the PEI group showed a Th2-biased cellular profile. The CTA1-DD group exhibited a more balanced Th1/Th2 cellular response (Figure 5I). Challenge evaluation showed that on day 2 after challenge, mice in the CpG-ODN group recovered body weight faster than those in the CTA1-DD group (p < 0.05) (Figure 5J). Quantification of lung viral loads by RT-PCR revealed no significant difference between the CpG-ODN and PEI groups, whereas the IFN-α group showed lower lung viral loads than the CTA1-DD group (p < 0.05) (Figure 5K). Lung histopathology indicated that none of the four adjuvants caused severe lung injury after challenge, and no significant differences in pathology scores were observed among groups (Figure 5L–M). Overall, CpG-ODN and PEI more effectively enhanced systemic and mucosal antibody responses, and CpG-ODN showed a greater capacity to induce nasal mucosal IgA and a Th1-biased response profile.
4. Discussion
Overall, both the type of adjuvant and the dose played an important role in immune quality and protective outcomes following intranasal administration. In this study, we systematically compared the effects of four mucosal adjuvants combined with RSV Pre-F protein at different dose levels, evaluating the induced humoral and cellular immune responses and analyzing post-challenge protection and safety.
CpG-ODN is a synthetic oligodeoxynucleotide containing unmethylated CpG motifs that activate immune responses via Toll-like receptor 9 (TLR9), enhancing B cell and dendritic cell activity and promoting IgA and IgG production, which are crucial for respiratory and intestinal immunity [26]. In animal studies, CpG-ODN is typically tested within a dose range of 5–50 µg, balancing immunogenicity and adverse effects. While lower doses (e.g., 5 µg) enhance antibody responses and survival, higher doses (e.g., 50 µg) may increase immune activation but also exacerbate inflammatory adverse effects [27,28]. In this study, we evaluated three doses of CpG-ODN (5/10/50 µg) combined with Pre-F in BALB/c mice. Our results show that increasing the dose from 5 to 10 µg significantly boosted neutralizing antibodies and mucosal IgA levels, but 50 µg did not further enhance IgA, was associated with slower weight recovery, and worsened lung histopathology. This suggests that protection does not increase linearly with dose, potentially reflecting a ceiling effect in TLR9/MyD88 signaling and inflammation-induced regulatory mechanisms [29]. Additionally, higher doses may compromise the nasal mucosal barrier, increasing cellular infiltration and amplifying immunopathology, thus impairing IgA maintenance [30]. Therefore, considering both immunogenicity and safety, a low-to-moderate CpG-ODN dose, particularly 10 µg, appears to be the most effective dose range in this study.
Previous studies have used CTA1-DD, a bacterial toxin-derived mucosal adjuvant, in intranasal immunization, commonly at doses of 5–10 µg per administration for influenza vaccines [31]. In this study, we compared three doses (2.5/5/20 µg) of CTA1-DD combined with Pre-F in BALB/c mice to determine an appropriate dose range for RSV vaccination. Serum antigen-specific binding IgG increased modestly across doses, with 20 µg demonstrating relatively effective overall immunogenicity and protection, as reflected by higher neutralizing titers, a trend toward faster weight recovery, and significantly reduced lung viral loads. Notably, 2.5–5 µg also lowered viral loads, suggesting partial protection at lower doses. Mucosal IgA responses in nasal lavage fluid and lung homogenates did not differ significantly among doses, indicating that CTA1-DD may preferentially enhance antibody quality and viral clearance rather than further increasing IgA magnitude. This aligns with the known mechanism of CTA1-DD as a B cell-targeted adjuvant that enhances germinal center responses [32] and its established safety profile for intranasal administration [33].
IFN-α is an antiviral cytokine that enhances immune responses and has been explored as a vaccine adjuvant for mucosal immunization [34]. It promotes dendritic cell and B cell maturation and favors Th1 and mucosal IgA responses. Lower doses are more effective for eliciting mucosal immunity, while higher doses may induce excessive inflammation or immune suppression, reducing efficacy [25]. Thus, dose optimization is essential. In this study, BALB/c mice were immunized with Pre-F and IFN-α at 2.5, 10, and 20 µg. IFN-α enhanced immunogenicity but had a modest impact on neutralizing activity. While 10 and 20 µg increased binding antibody titers significantly, neutralizing titers were similar across doses, suggesting a limited role of IFN-α in functional antibody maturation. For mucosal immunity, 10 µg significantly increased nasal IgA, with no difference between the 10 and 20 µg groups in lung IgA, indicating a plateau. After challenge, the 10 µg group showed faster weight recovery than the 2.5 µg group, with no significant difference compared to the 20 µg group. Lung viral loads were lower in the 20 µg group than in the 2.5 µg group but similar between 10 and 20 µg, suggesting antiviral clearance reaches a ceiling at 10–20 µg. The above results support 10 µg as a reasonable dose range for subsequent studies.
As a polymer-based intranasal adjuvant, 25 kDa branched PEI enhances both humoral and mucosal immunity in a dose-dependent manner. However, higher doses may increase local irritation and systemic reactogenicity. In studies with gp140 or HA antigen systems, 20 µg PEI per dose has been commonly used and reported to substantially increase serum and airway secretory antibodies, but it is also associated with higher reactogenicity, including weight loss, and a shift toward Th2 responses. PEI has also been used intranasally before infection as a short-course stimulant for nonspecific protection, highlighting its “double-edged” nature through local stimulation and innate activation [35,36,37]. In our study, we selected three doses of PEI (2.5, 10, 50 µg) combined with Pre-F protein and immunized BALB/c mice. Moreover, 10 µg PEI significantly increased serum IgG and enhanced IgA responses in both nasal lavage fluid and lung homogenates compared to 2.5 µg. Although 50 µg further increased neutralizing titers, it did not lead to additional improvements in post-challenge weight recovery, lung viral loads, or lung pathology scores. Overall, both published studies and our data support a dose-dependent immunostimulatory profile for PEI, with 10 µg providing a favorable balance between immune enhancement and safety, making it a suitable dose for subsequent studies.
Based on the integrated analysis of the detection indicators across the three dose levels for each adjuvant, we selected representative doses for subsequent head-to-head comparison, namely 10 µg CpG-ODN, 20 µg CTA1-DD, 10 µg IFN-α, and 10 µg PEI. These selections should be interpreted as model-specific representative doses selected from the three dose levels, rather than definitive optimal doses. We then further compared the humoral, cellular, and mucosal immune responses elicited by these formulations and evaluated their protective efficacy and safety. Overall, CpG-ODN and PEI showed a comparatively favorable balance between immune efficacy and safety: both induced higher levels of antigen-specific IgG and neutralizing antibody titers. CpG-ODN also demonstrated a response profile biased toward Th1 (balanced IgG1/IgG2a and increased IFN-γ and IL-2 secretion), which may be advantageous for clearance of intracellular pathogens, whereas PEI exhibited a pronounced Th2 bias at both humoral and cellular levels. CTA1-DD and IFN-α enhanced humoral and cellular responses to some extent, but their overall protective effects were less pronounced than those of CpG-ODN and PEI under the conditions tested. With respect to mucosal immunity, CpG-ODN significantly increased antigen-specific IgA in nasal lavage fluid, consistent with reduced lung viral loads and faster weight recovery, suggesting that appropriately dosed CpG-ODN facilitates the establishment of effective mucosal immune barriers in both the upper and lower respiratory tracts.
5. Conclusions
In summary, integrating immunogenicity, viral clearance, and lung pathology outcomes, low-to-moderate CpG-ODN (particularly 10 µg) and an appropriate dose of PEI appear to be promising adjuvant candidates for RSV mucosal immunization. In contrast, IFN-α and CTA1-DD may require further optimization in terms of dosing and delivery to maximize their benefits while minimizing the risk of inflammation. This study provides an initial comparative analysis of dose–effect relationships for four representative mucosal adjuvants in BALB/c mice. Despite this, further work should focus on validating these findings in additional animal models and immunization regimens that more closely mirror clinical applications.
A limitation of this study is that each adjuvant was evaluated at only three dose levels, and several outcomes approached a plateau within these ranges. Therefore, the data do not establish definitive optimal doses. Formal dose optimization would require broader dose ranges and finer gradients to capture transitions from suboptimal to maximal effects and tolerability. Additional limitations include the use of a single animal species, a limited observation period, and the lack of in-depth analysis of tissue-resident memory T cells in mucosal sites. Nonetheless, the findings provide valuable experimental support for rational adjuvant and dose selection, guiding further optimization for intranasal RSV vaccination and the development of safe, effective, and scalable mucosal immunization strategies against respiratory viruses.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Asseri A.A. Respiratory Syncytial Virus: A Narrative Review of Updates and Recent Advances in Epidemiology, Pathogenesis, Diagnosis, Management and Prevention J. Clin. Med.202514388010.3390/jcm 1411388040507642 PMC 12156131 · doi ↗ · pubmed ↗
- 2Duan Q. Pan J. Zheng L. Jin Z. Li S. Zhang H. Fu X. Chen R. Yu Z. Qin S. Global outbreaks of respiratory syncytial virus infections from 1960 to 2025: A systematic review and meta-analysis E Clinical Medicine 20258610335210.1016/j.eclinm.2025.10335240666171 PMC 12257031 · doi ↗ · pubmed ↗
- 3Wilkins D. Langedijk A.C. Lebbink R.J. Morehouse C. Abram M.E. Ahani B. Aksyuk A.A. Baraldi E. Brady T. Chen A.T. Nirsevimab binding-site conservation in respiratory syncytial virus fusion glycoprotein worldwide between 1956 and 2021: An analysis of observational study sequencing data Lancet Infect. Dis.20232385686610.1016/S 1473-3099(23)00062-236940703 · doi ↗ · pubmed ↗
- 4Kelleher K. Subramaniam N. Drysdale S.B. The recent landscape of RSV vaccine research Ther. Adv. Vaccines Immunother.2025132515135524131060110.1177/2515135524131060139802673 PMC 11724408 · doi ↗ · pubmed ↗
- 5Sanchez-Martinez A. Moore T. Freitas T.S. Benzaken T.R. O’Hagan S. Millar E. Groves H.E. Drysdale S.B. Broadbent L. Recent advances in the prevention and treatment of respiratory syncytial virus disease J. Gen. Virol.202510600209510.1099/jgv.0.00209540202895 PMC 12282282 · doi ↗ · pubmed ↗
- 6Oh J.E. Song E. Moriyama M. Wong P. Zhang S. Jiang R. Strohmeier S. Kleinstein S.H. Krammer F. Iwasaki A. Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral Ig A Sci. Immunol.20216 eabj 512910.1126/sciimmunol.abj 512934890255 PMC 8762609 · doi ↗ · pubmed ↗
- 7Karron R.A. Luongo C. Woods S. Oliva J. Collins P.L. Buchholz U.J. RSV Ped Team Evaluation of the Live-Attenuated Intranasal Respiratory Syncytial Virus (RSV) Vaccine RSV/6120/ΔNS 2/1030 s in RSV-Seronegative Young Children J. Infect. Dis.202422934635410.1093/infdis/jiad 28137493269 PMC 10873187 · doi ↗ · pubmed ↗
- 8Human Challenge Study to Evaluate the Efficacy of MV-012-968 Vaccine Available online: https://clinicaltrials.gov/study/NCT 04690335/(accessed on 4 August 2022)