Comparative Analysis of Next- and Third-Generation Sequencing Platforms for Chikungunya Virus Whole-Genome Sequencing Using a Lineage-Inclusive Primer Set During the 2025 Foshan Outbreak
Penghui Jia, Xiao Cong, Chang Zhang, Zhe Liu, Xiaofang Peng, Juan Su, Qiqi Tan, Shen Huang, Changyun Sun, Xin Zhang, Baisheng Li

TL;DR
This study compares sequencing technologies for tracking the Chikungunya virus during an outbreak and introduces a new primer set that works across virus lineages.
Contribution
A lineage-inclusive primer set and direct comparison of NGS and TGS for CHIKV whole-genome sequencing during an outbreak.
Findings
Illumina NGS showed higher raw read accuracy, while Nanopore TGS provided better UTR coverage and faster processing.
After polishing, variant calls from both sequencing platforms were fully concordant.
Phylogenetic analysis suggested a single introduction event linking the Foshan outbreak to strains from Réunion Island.
Abstract
Chikungunya virus (CHIKV) poses an increasing global public health threat, as evidenced by the significant 2025 Foshan outbreak in China. Rapid, whole-genome sequencing (WGS) is critical for outbreak response but is challenged by primer mismatches across diverse lineages and a lack of direct sequencing platform comparisons. To address this, we developed a novel lineage-inclusive primer set and performed parallel WGS on 24 clinical samples from the outbreak using both Illumina (NGS) and Oxford Nanopore Technologies (TGS) platforms. Our lineage-inclusive primer set successfully amplified full-length CHIKV genomes across all samples. Comparisons revealed that Illumina NGS provided higher raw read accuracy, while Nanopore TGS achieved more complete coverage of terminal UTRs with a faster turnaround time. Crucially, after polishing, variant calls between the two platforms were 100%…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
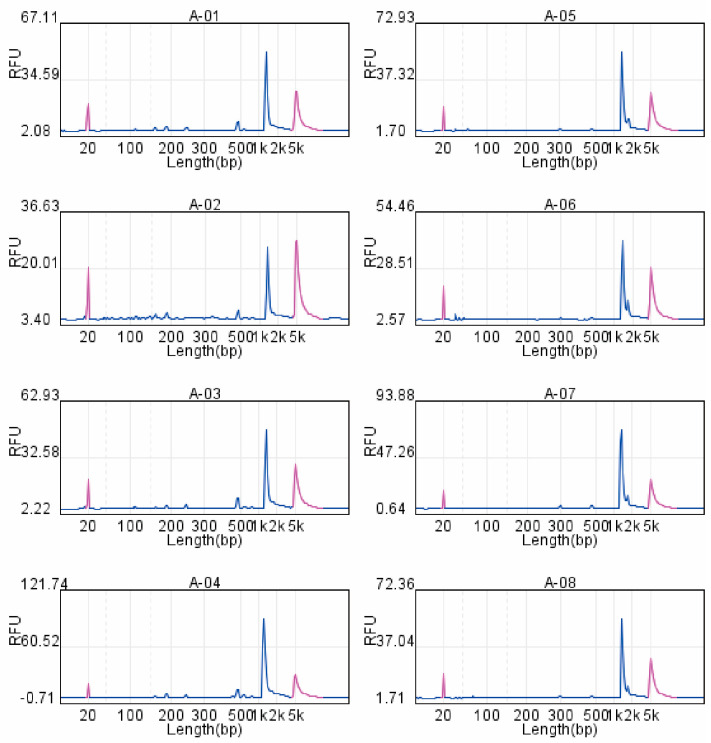 Figure 1
Figure 1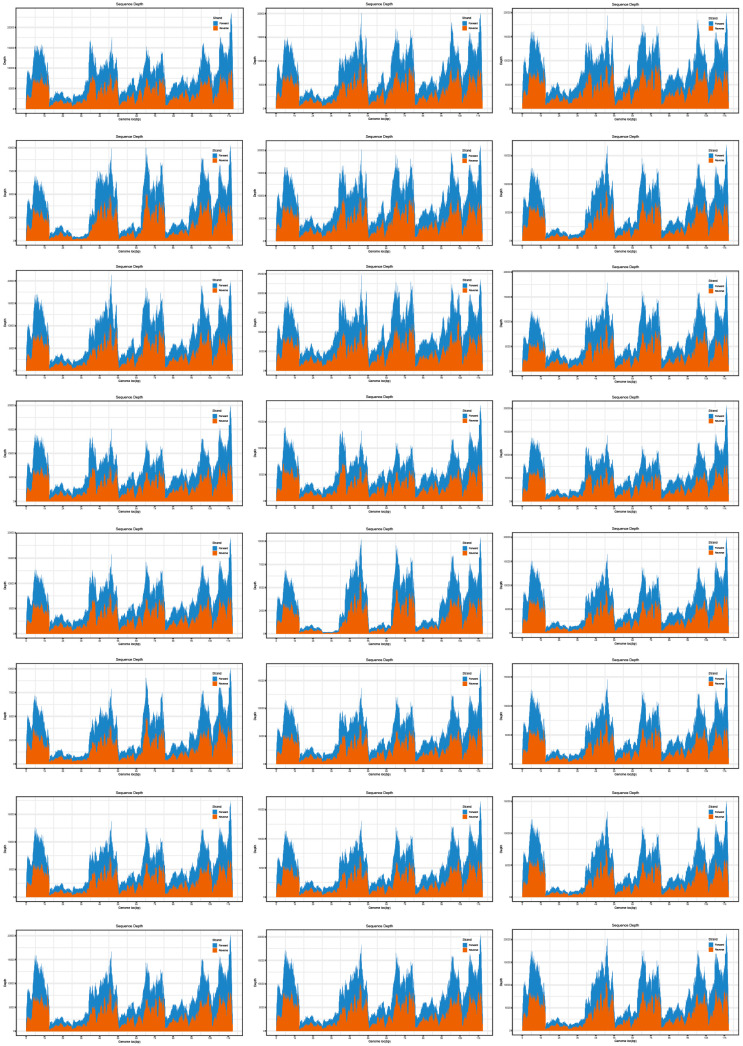 Figure 2
Figure 2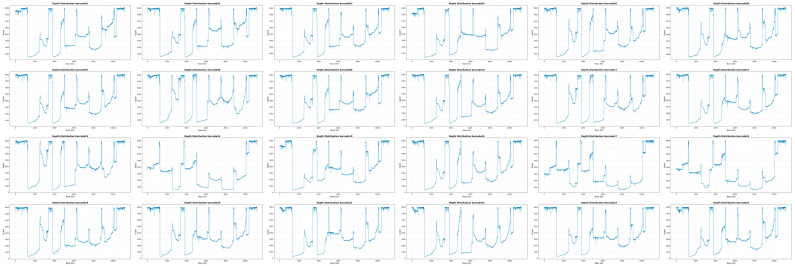 Figure 3
Figure 3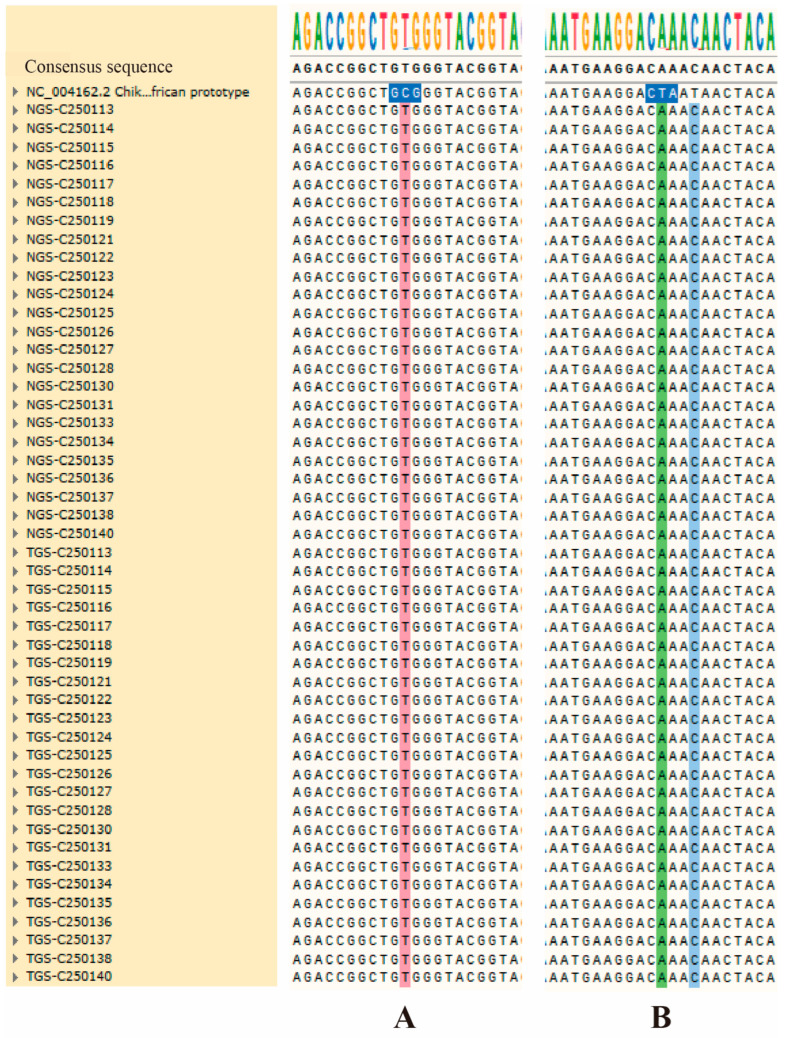 Figure 4
Figure 4 Figure 5
Figure 5- —Guangdong Provincial Medical Science and Technology Research Fund
- —Key Areas R&D Program Project of Guangdong Province
- —Guangdong Provincial Key Laboratory of Pathogen Detection for Emerging Infectious Disease Response
- —Emergency Research Project for the Prevention and Control of Chikungunya Virus Infection in Guangdong Province
- —Guangdong Provincial Center for Disease Control and Prevention Supports Talent Projects
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Genomics and Phylogenetic Studies · Biological Research and Disease Studies
1. Introduction
Chikungunya virus (CHIKV) is a mosquito-borne, positive-sense single-stranded RNA virus belonging to the family Togaviridae and genus Alphavirus [1]. Since its first isolation in Tanzania in 1952, CHIKV has been responsible for recurrent and increasingly frequent outbreaks across Africa, Asia, Europe, and the Americas, facilitated by globalization, climate change, and expanding distributions of the competent vectors Aedes aegypti and Aedes albopictus [2,3]. Clinical manifestations typically include acute fever and severe polyarthralgia; however, up to 40–60% of patients may develop chronic, debilitating joint symptoms that persist for months to years, imposing a substantial long-term socioeconomic burden [4,5,6].
Driven by globalization, urbanization, and climate change, CHIKV infection exhibits a rapid capacity for geographical expansion, posing a significant threat to newly affected regions. The recent 2025 Foshan outbreak in China exemplifies this persistent threat. In July 2025, an unexpected and rapidly expanding chikungunya fever outbreak occurred in Foshan City, Guangdong Province, China—one of the most significant CHIKV epidemics in the region in recent years. Within a few weeks, cases increased sharply and spread across multiple districts [1,5,7]. Prior to this outbreak, China had documented only imported CHIKV cases in 2008, followed by several localized outbreaks in Guangdong [8], Zhejiang [9], and Yunnan Provinces [10] between 2010 and 2019, all linked to imported infections. Therefore, real-time genomic characterization is essential for identifying the viral origin, tracking transmission routes, detecting adaptive mutations, and providing timely evidence to guide public health interventions.
The ~11.8 kb CHIKV genome encodes four nonstructural proteins (nsP1-nsP4) essential for viral replication and five structural proteins (C, E3, E2, 6K, and E1), whose genetic variability contributes to viral adaptation, transmissibility, and epidemic potential [4]. Of particular epidemiological importance is the E1-A226V mutation, which enhances viral fitness in Ae. albopictus and has been implicated in several major global outbreaks [11,12,13]. Despite the recognized value of genomic surveillance, obtaining complete and accurate CHIKV whole-genome sequences remains challenging. Key obstacles include low viral loads in clinical samples, genetic variation among circulating strains, and uneven genome coverage. This is particularly problematic in the 5′ and 3′ untranslated regions (UTRs), which are essential for viral replication and regulation.
Currently, next-generation sequencing (NGS) and third-generation sequencing (TGS) represent the two primary platforms used in viral genomics [14]. NGS platforms, such as Illumina, offer high-throughput sequencing with exceptional base accuracy, making them the gold standard for reliable single-nucleotide variant (SNV) detection [15]. However, their short read length often results in incomplete assembly of repetitive or structurally complex regions, leading to gaps in UTRs and other regulatory elements. By contrast, TGS platforms such as Oxford Nanopore Technologies (ONT) generate long reads capable of spanning entire amplicons or even complete genomes, providing superior assembly continuity and resolving structural features and haplotypes [16]. Yet, TGS typically exhibits higher raw error rates, requiring robust downstream polishing algorithms to ensure variant-calling accuracy. Thus, choosing between NGS and TGS for outbreak response involves trade-offs among accuracy, completeness, speed, and cost.
A frequently overlooked determinant of sequencing success is the quality of genome amplification. Clinical samples often contain insufficient viral RNA for direct sequencing, necessitating targeted amplification using lineage-inclusive primer sets. Existing primer schemes may fail to amplify diverse CHIKV strains uniformly, leading to coverage bias or incomplete genomes that compromise downstream evolutionary analyses. Moreover, although both NGS and TGS have been individually applied to CHIKV sequencing, a systematic head-to-head comparison using a unified, optimized primer set has not been conducted. Consequently, data are lacking on the relative performance of these platforms in real-world outbreak samples—particularly regarding coverage uniformity, variant concordance, and phylogenetic resolution.
To address these gaps, we designed a novel lineage-inclusive CHIKV primer set targeting highly conserved genomic regions and evaluated its amplification efficiency in clinical samples collected during the 2025 Foshan outbreak. Using this unified primer scheme, we performed parallel WGS on 24 confirmed CHIKV-positive serum samples using both Illumina NGS and Oxford Nanopore TGS. We systematically compared platform performance in terms of genome coverage, sequencing depth, accuracy, variant concordance, and phylogenetic reconstruction. Additionally, we analyzed the molecular characteristics and evolutionary relationships of the outbreak strain to infer transmission dynamics and potential origin. This study therefore provides a validated, practical framework for CHIKV genomic surveillance and offers critical insights into the complementary roles of NGS and TGS in outbreak response.
2. Materials and Methods
2.1. Sample Collection and Clinical Information
A total of 24 laboratory-confirmed CHIKV-positive serum samples were collected during the chikungunya fever outbreak in July 2025 in Foshan City, Guangdong Province, China. Samples were obtained from three hospitals in Beijiao Town, Shunde District: Chencun Hospital (n = 12), Beijiao Hospital (n = 7), and Heyou Hospital (n = 5). All patients presented with typical symptoms, including acute fever and arthralgia. For each case, demographic and clinical data—such as age, sex, date of symptom onset, and sampling date—were recorded. The collected serum samples were residual specimens from routine clinical tests such as complete blood counts, with associated clinical information provided by the hospitals. We extracted RNA from the serum and performed RT-PCR to confirm CHIKV infection, recording the corresponding cycle threshold (Ct) values as indicators of viral load. Samples spanning a wide range of Ct values were intentionally included to evaluate sequencing performance across different viral titers.
2.2. Design of a Lineage-Inclusive CHIKV Primer Set
Complete CHIKV genomes representing the three major phylogenetic lineages—East/Central/South African (ECSA), West African, and Asian (Accessions: PV685524.1, PV685664.1, PV685706.1)—were retrieved from the NCBI GenBank database. Genomes containing >1% ambiguous bases or incomplete 5′/3′ untranslated regions (UTRs) were excluded. Conserved regions across lineages were identified and used as the template for primer design. A set of 10 overlapping primer pairs was designed using PrimalScheme v3.0.2 (https://primalscheme.com/ (accessed on 16 July 2025)), generating amplicons of approximately 1200 bp with ~200 bp overlap between adjacent fragments to ensure robust whole-genome coverage (~11.8 kb). Primer sequences and genomic positions are listed in Table A1.
2.3. RT-PCR Amplification and Amplicon Generation
Viral RNA was extracted from serum samples. cDNA was synthesized from the extracted RNA using the LunaScript RT SuperMix Kit (NEB, Ipswich, MA, USA) under the following conditions: 25 °C for 2 min, 50 °C for 10 min, and 95 °C for 1 min for enzyme inactivation. The resulting cDNA was used for multiplex long-range PCR to amplify the complete CHIKV genome. A tiling-amplicon strategy employing two non-overlapping primer pools was used to generate ~1.2 kb overlapping fragments. PCR amplification was performed with Q5 High-Fidelity 2× Master Mix using the following cycling program: 98 °C for 30 s; 35 cycles of 95 °C for 15 s and 65 °C for 5 min. PCR products were purified using 0.5× AMPure XP beads (Beckman Coulter, Brea, CA, USA) and eluted in nuclease-free water prior to quantification.
2.4. Illumina Library Preparation and Sequencing
Illumina libraries were constructed from purified amplicons using the Illumina DNA Prep Kit (Illumina, San Diego, CA, USA). The workflow included enzymatic fragmentation and size selection, end-repair and A-tailing, and ligation of unique dual indexes (UD Indexes Set A) during a limited-cycle PCR. The indexed libraries were then purified, quantified, normalized, and pooled at equimolar concentrations. Pooled libraries were denatured and diluted to the recommended loading concentration and sequenced on the Illumina MiniSeq platform (Illumina, San Diego, CA, USA) using a single-end configuration (1 × 150 bp). Base calling was performed through the iterative incorporation and imaging of fluorescently labeled nucleotides during bridge amplification on the flow cell.
2.5. Oxford Nanopore Library Preparation and Sequencing
For Oxford Nanopore sequencing, ~1.2 kb amplicons were prepared using the Native Barcoding Kit 24 (SQK-NBD114.24, Oxford Nanopore Technologies plc, Oxford, UK). The workflow consisted of three enzymatic steps: (1) end-repair and dA-tailing, (2) ligation of unique native barcodes using Blunt/TA Ligase Master Mix (NEB, Ipswich, MA, USA), and (3) pooling and adapter ligation with Quick T4 DNA Ligase (NEB, Ipswich, MA, USA). The final barcoded library was purified with AMPure XP beads (Beckman Coulter, Brea, CA, USA) and loaded onto a primed R10.4.1 flow cell for sequencing on the GridION or MinION platform.
2.6. Bioinformatic Analysis of Illumina NGS and Nanopore TGS Data
Sequencing data from both Illumina NGS and Oxford Nanopore TGS platforms were processed using standardized reference-guided workflows. Initial taxonomic confirmation of CHIKV reads was performed using Kraken2 (Illumina: v2.1.2; Nanopore: v2.1.6). Raw sequencing data of Illumina NGS was processed using fastp (v0.23.2) with the following key filtering steps: (i) automatic adapter trimming; (ii) removal of reads containing more than 10% unidentified bases (N); (iii) truncation of low-quality bases from the 5′ and 3′ ends (quality threshold < 20); and (iv) discarding of reads shorter than 50 bp after trimming. Only reads passing these filters were used for subsequent alignment and variant calling. Illumina reads were aligned to the CHIKV reference genome (strain S27b03, GenBank: PV685524.1) using BWA v0.7.17, whereas Nanopore long reads were aligned using minimap2 v2.17. Variant calling was conducted using FreeBayes v1.3.5 for Illumina data and Medaka v1.7.3 for Nanopore data, followed by low-quality variant filtering with bcftools (Illumina: v1.12; Nanopore: v1.19). Nanopore consensus sequences underwent an additional polishing step using Racon to correct residual base-calling errors. For both platforms, final consensus genomes were reconstructed through reference-guided assembly and subsequently genotyped using the Chikungunya Typing Tool (https://www.genomedetective.com (accessed on 23 December 2025)). Multiple sequence alignment was performed with MAFFT v7.453, and Neighbor-Joining phylogenetic trees were inferred using MEGA11 software, with bootstrap support assessed from 1000 replicates. Phylogenies were visualized using the Interactive Tree of Life (iTOL, https://itol.embl.de (accessed on 23 December 2025)).
2.7. Comparative Evaluation of Sequencing Platform Performance
To directly compare the performance of Illumina NGS and Oxford Nanopore TGS, sequencing data from the same 24 samples were processed in parallel using unified reference genomes and standardized evaluation criteria. Genome coverage, per-base depth, and coverage uniformity were calculated from aligned BAM files. Read quality and length distributions were assessed using Illumina Q20/Q30 scores and Nanopore median Q-scores and N50 values. A site-by-site comparison revealed that the set of high-confidence SNV positions and the genotypes at those positions were identical between platforms for all samples. Phylogenetic consistency was evaluated by comparing Neighbor-Joining trees generated independently from NGS and TGS consensus genomes. Together, these metrics provided an integrated evaluation of the strengths and limitations of both sequencing technologies for CHIKV whole-genome analysis.
3. Results
3.1. Clinical Characteristics of CHIKV-Positive Samples
A total of 24 CHIKV-positive serum samples were collected during the July 2025 Foshan outbreak. All patients presented with acute febrile illness accompanied by arthralgia. Ct values ranged from 20 to 29, representing a wide viral-load distribution suitable for evaluating sequencing performance across titers. Table 1 summarizes demographic and clinical information for all cases.
3.2. Validation of Lineage-Inclusive Amplicon Amplification
To validate the performance of the newly designed lineage-inclusive CHIKV primer set, amplicons generated from four representative clinical samples (Samples 1–4) using two primer pools were first quantified by Qubit fluorometry, yielding DNA concentrations of 30–70 ng/μL. Subsequently, capillary electrophoresis was performed. The 20 individual primers were divided into two multiplex pools. Primer Pool 1 (P1) contained the 10 forward primers, and Primer Pool 2 (P2) contained the 10 reverse primers corresponding to each of the 10 tiling amplicons. All amplification reactions produced a single, dominant peak at the expected size of approximately 1.2 kb (indicated by the dashed vertical line). The absence of secondary peaks confirms the specific and efficient amplification of the target tiling fragments, with no detectable primer-dimer artifacts or non-specific products (Figure 1).
3.3. Illumina NGS Achieves High-Depth and Uniform Coverage of the CHIKV Genome
Illumina sequencing generated a median of 0.90 million high-quality reads per sample (mean: 0.91 million; interquartile range: 0.31 million), with high base quality (mean Q20: 95.77%; Q30: 93.40%). Alignment to the reference genome (S27b03, including both coding and non-coding UTRs) yielded a mean depth of >6000× across all 24 samples, with every sample achieving complete 100% coverage and no gaps in any genomic region. The unequivocally confirms the robustness of the sequencing and the primer set’s comprehensive representation of the viral genome. The sequencing depth profile, illustrated in Figure 2, demonstrates exceptionally uniform read enrichment across the entire coding sequence (CDS), underscoring the platform’s reliability for achieving comprehensive genomic coverage. Detailed per-sample sequencing metrics are provided in Table A2.
3.4. Sequencing Performance of Oxford Nanopore TGS
Nanopore GridION sequencing (20 h run time) generated 200–500 Mb per sample, with a median read length of ~1000 bp and median Q-score ~Q16. Coverage heatmaps (Figure 3) demonstrated complete coverage (100%) with depths ranging from 500× to 8000× across samples. Barcode-specific depth plots confirmed uniform long-read tiling coverage without terminal drop-outs.
3.5. NGS and TGS Demonstrate Complementary Performance Characteristics in Coverage and Accuracy
A comparative evaluation of Illumina NGS and Oxford Nanopore TGS demonstrated complementary performance characteristics across the 24 CHIKV genomes. Illumina sequencing yielded more uniform per-base depth, reflected by lower coverage variation, whereas Nanopore achieved more complete coverage of the terminal UTRs and other repetitive regions. Read-quality metrics showed a mean Illumina Q30 exceeding 93%, while Nanopore achieved a median Q-score of approximately Q16 with a read-length N50 of ~1.1 kb. High-confidence SNVs (depth > 100×; allele frequency > 5%) were consistently detected in all samples, and notably, the variant sets derived from both platforms were fully concordant after polishing of Nanopore reads with Medaka and Racon, confirming that consensus-level accuracy was equivalent across platforms. Error-profile analysis indicated fewer indels in Illumina data, whereas Nanopore raw reads exhibited higher insertion and deletion frequencies; however, these errors were largely corrected during polishing, resulting in comparable substitution patterns between platforms. Workflow comparisons showed that Nanopore offered a shorter overall turnaround time (~12–15 h from amplicon to consensus genome), whereas Illumina required ~25–30 h but provided higher intrinsic base accuracy. Together, these metrics highlight the complementary strengths of both technologies and underscore their combined value for rapid and accurate CHIKV whole-genome sequencing.
3.6. Concordance in Variant Calling and Identification of Key Mutations
High-confidence single-nucleotide variants (SNVs) (depth > 100×, AF > 5%) were jointly analyzed across the 24 CHIKV genomes sequenced using Illumina NGS and Oxford Nanopore TGS. Variant calls were fully concordant between the two platforms after polishing of Nanopore reads, and no platform-specific discrepancies were observed for true biological variants. Two lineage-associated adaptive mutations—E1-A226V and E2-L210Q—were detected in 100% (n = 24) of the samples. Multiple sequence alignments confirmed that all consensus genomes shared the characteristic alanine-to-valine substitution at residue 226 of the E1 protein (Figure 4A) as well as the leucine-to-glutamine substitution at E2 position 210 (Figure 4B), both of which have been associated with enhanced transmission by Ae. albopictus and increased epidemic potential.
Beyond these genotype-defining mutations, several additional SNVs were identified across the dataset, including 3879 C → T (24/24 samples), 9874 C → T (8/24), and 2959 G → A (2/24). These lower-frequency variants were not located in known adaptation-associated regions but were consistently detected in both NGS and polished TGS data. Conversely, the 9099 A → T substitution was observed in three Nanopore raw assemblies but was determined to be an artifact upon depth inspection: coverage at this position exceeded 4900× and 7200× in representative samples C250136 and C250137, respectively, with negligible support for the alternate allele. Therefore, 9099 A → T was excluded as a true biological variant. The concordance statement pertained exclusively to SNVs; no high-confidence indels were identified in any sample, consistent with the close evolutionary relationship to the reference strain S27b03 and the acute nature of the outbreak. A complete summary of all detected SNVs is provided in Table 2. Table 2 provides a summary of mutation loci identified by both NGS and TGS. In this table, the columns represent the mutation positions within the reference genome. The notation “C/T”, for example, indicates a substitution where the reference base C is altered to T in the sample. The rows, conversely, correspond to the sample ID.
This combined analysis highlights the strong cross-platform robustness in variant detection and confirms that the Foshan 2025 outbreak strain carries the canonical E1-A226V and E2-L210Q mutations characteristic of the epidemic ECSA lineage.
3.7. Phylogenetic Analysis and Outbreak Origin
To determine the genetic relationship and potential origin of the Foshan outbreak strains, a phylogenetic analysis was conducted using neighbor-joining methods based on the complete genome sequences of the 24 isolates obtained in this study, alongside a curated set of globally representative CHIKV reference sequences encompassing all major genotypes (ECSA, Asian, and West African) and contemporary strains from the 2025 Réunion Island outbreak (Accessions: PV685524.1, PV685664.1, PV685706.1). The neighbor-joining trees reconstructed from both NGS- and TGS-derived consensus sequences yielded identical topologies with strong nodal support (bootstrap values > 90%), demonstrating the robustness and platform-independence of our phylogenetic inferences. The analysis revealed that all 24 Foshan isolates formed a distinct, well-supported monophyletic clade (bootstrap value = 100%) with no discernible genetic diversity, suggesting a scenario consistent with a single introduction event followed by clonal expansion within the local population (Figure 5). Crucially, this Foshan clade robustly nested within the East/Central/South African (ECSA) genotype, and demonstrated the closest evolutionary relationship with the contemporaneous CHIKV sequences from Réunion Island, forming a shared subclade with strong statistical support (bootstrap value = 100%). This high-degree genetic relatedness, evidenced by the minimal number of nucleotide substitutions between the Foshan and Réunion sequences, provides molecular evidence compatible with a recent transmission chain. The phylogenetic data are consistent with the hypothesis that the outbreak may have originated from a single introduction event linked to the ongoing epidemic in Réunion Island.
4. Discussion
Our study successfully addresses the need for a unified sequencing approach to characterize CHIKV outbreaks by developing a lineage-inclusive primer set and conducting a systematic comparison of NGS and TGS platforms during the 2025 Foshan outbreak. By integrating Illumina NGS and Oxford Nanopore TGS workflows under a unified amplicon-based strategy, we directly compared the performance of these platforms for whole-genome sequencing of CHIKV. Through comprehensive evaluation of genome coverage, sequencing depth, mutation profiling, and phylogenetic resolution, we characterized the strengths and limitations of each method, providing meaningful insights for CHIKV genomic surveillance and outbreak response. Our study directly addresses these gaps by not only delivering a validated lineage-inclusive primer set but also providing a definitive, head-to-head comparison of NGS and TGS platforms using a unified amplification strategy.
A significant contribution of this study is the development of a 10-pair primer scheme that uniformly amplified all genomic regions, including conserved and variable segments across ECSA, Asian, and West African lineages. Capillary electrophoresis verification confirmed clean, monodisperse ~1.2 kb peaks without detectable non-specific amplification, while Qubit quantification demonstrated consistently high DNA yields suitable for downstream library preparation. This finding is significant because primer bias and lineage-restricted amplification remain key obstacles in CHIKV genomic research, particularly during outbreaks in newly affected regions where circulating strains may differ from those used in earlier primer designs [17,18,19,20]. These results highlight the primer set’s strong lineage inclusivity and amplification robustness even at higher Ct values, addressing a common technical barrier in CHIKV sequencing where primer mismatches or low viral loads often lead to incomplete genomes. The development of this primer set therefore provides a valuable tool for both routine molecular surveillance and rapid outbreak investigation.
Parallel sequencing using Illumina and Nanopore platforms clearly demonstrated complementary strengths. Illumina produced highly accurate reads with exceptional base quality (mean Q30 >93%), uniform per-base depth, and minimal indel noise, reaffirming its status as the gold standard for precise SNV detection [21]. In contrast, Nanopore sequencing provided near-complete coverage of the genome—including 5′ and 3′ UTRs that typically exhibit drop-offs in NGS—while maintaining rapid turnaround times (~12–15 h from amplicon to consensus). The ability of Nanopore long reads to bridge structurally complex regions enabled gap-free genome assemblies that are particularly advantageous in outbreak settings requiring rapid molecular characterization [22,23]. Notably, the variant concordance analysis revealed 100% agreement between polished TGS consensus genomes and NGS variant calls, underscoring the effectiveness of Nanopore polishing pipelines in mitigating raw error rates and supporting its reliability when appropriate analytical workflows are applied.
Mutation analysis further highlighted the genomic features associated with the 2025 Foshan outbreak strain. All 24 samples uniformly carried the epidemiologically significant E1-A226V and E2-L210Q mutations, which are known to enhance viral fitness and transmissibility in Ae. albopictus [7,24]. This result aligns with CHIKV evolutionary patterns observed in previous large-scale outbreaks across the Indian Ocean region and Southeast Asia [25,26]. Together, these observations demonstrate both the precision of the analytical pipeline and the value of integrating multiple sequencing platforms for robust mutation detection.
Phylogenetic analyses based on both Illumina and Nanopore consensus sequences yielded identical topologies, reflecting strong platform consistency. All Foshan isolates clustered tightly into a monophyletic clade nested within the ECSA lineage, showing the closest evolutionary affinity to contemporaneous strains from the Réunion Island epidemic [11,27,28]. The minimal genetic diversity observed among Foshan genomes is highly consistent with a single introduction event followed by rapid local transmission, mirroring the epidemiological characteristics of the outbreak. These findings reinforce the critical role of genomic surveillance in tracing viral introductions and reconstructing transmission chains, especially in regions like southern China where Ae. albopictus is abundant and repeated arboviral introductions have been documented over the last decade [8].
This study has several strengths, including a comprehensive cross-platform evaluation, robust primer design, and high-quality genomic data. Nevertheless, some limitations should be acknowledged. First, while the tiling-amplicon approach employed here ensures high genome completeness, it inherently limits the detection of structural variants or recombination events that fall outside the primer binding sites. For a comprehensive view of such genomic alterations, metagenomic or hybrid-capture sequencing would be required in future studies. Second, while Nanopore sequencing offers rapid results, its accuracy remains dependent on coverage depth and subsequent polishing; improvements in base-calling algorithms and pore chemistry may further enhance reliability in future investigations. Finally, the study focused on a single outbreak; integrating vector-derived sequences and broader temporal sampling could provide deeper insights into the ecological and evolutionary dynamics of CHIKV transmission in Guangdong. Additionally, while the primer set demonstrated robust ‘lineage-inclusive’ amplification of the major CHIKV lineages in this study, its comprehensive applicability can be further enhanced. Future work will involve designing and validating primers against a broader geographical and temporal diversity of genomes to progress towards a truly ‘pan-lineage’ assay.
Overall, this study underscores the value of integrating an optimized primer system with both second- and third-generation sequencing platforms to achieve rapid, high-quality genomic surveillance during an active outbreak. The methodological framework developed here integrates robust amplification, cross-platform sequence evaluation, and comprehensive phylogenetic analysis. This approach can be readily implemented in public health laboratories to facilitate timely molecular characterization of emerging arboviral pathogens. As CHIKV continues to expand into new geographic regions and adapt to diverse ecological environments, the availability of reliable genomic tools such as those described in this study will be essential for early detection, risk assessment, and the effective management of future outbreaks.
5. Conclusions
In conclusion, our study successfully addresses critical challenges in the genomic surveillance of CHIKV outbreaks. We have developed and validated a novel lineage-inclusive, primer set that enables robust and unbiased whole-genome amplification of diverse CHIKV strains, thereby overcoming a significant technical barrier in frontline molecular epidemiology. Through a systematic head-to-head comparison, we demonstrated that Illumina NGS and Oxford Nanopore TGS, when used within this unified amplicon-based framework, offer complementary strengths. NGS remains the gold standard for base-level accuracy, while TGS provides superior speed and completeness, particularly in genomic regions problematic for short reads. Importantly, after rigorous bioinformatic polishing, consensus sequences from both platforms achieved 100% variant concordance, establishing the reliability of TGS for rapid outbreak investigation. Applied to the 2025 Foshan outbreak, this integrated approach precisely characterized the outbreak strain as a monophyletic cluster of the ECSA lineage bearing key adaptive mutations, and pinpointed its origin to the contemporaneous Réunion Island epidemic. The methodological framework and comparative insights provided here offer a practical, scalable, and effective solution for public health laboratories worldwide to conduct real-time genomic surveillance of CHIKV, thereby enhancing preparedness and response capabilities against this re-emerging arboviral threat.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wan S. Zhang X. Cong X. Liu Y. Huang S. Zhou M. Sun C. Peng X. Zhang H. Yu Y. Viral Load Dynamics of Chikungunya Virus in Human Specimens—Foshan City, Guangdong Province, China, 2025 China CDC Wkly.202571067107210.46234/ccdcw 2025.18240843135 PMC 12365650 · doi ↗ · pubmed ↗
- 2Weaver S.C. Chen R. Diallo M. Chikungunya Virus: Role of Vectors in Emergence from Enzootic Cycles Annu. Rev. Entomol.20206531333210.1146/annurev-ento-011019-02520731594410 · doi ↗ · pubmed ↗
- 3de Souza W.M. Lecuit M. Weaver S.C. Chikungunya virus and other emerging arthritogenic alphaviruses Nat. Rev. Microbiol.20252358560110.1038/s 41579-025-01177-840335675 · doi ↗ · pubmed ↗
- 4Freppel W. Silva L.A. Stapleford K.A. Herrero L.J. Pathogenicity and virulence of chikungunya virus Virulence 202415239648410.1080/21505594.2024.239648439193780 PMC 11370967 · doi ↗ · pubmed ↗
- 5Li Y. Jiang S. Zhang M. Li Y. He J. Yang Z. Huang X. Guan Q. Li Z. Xie J. An Outbreak of Chikungunya Fever in China—Foshan City, Guangdong Province, China, July 2025 China CDC Wkly.20257106410654083713910.46234/ccdcw 2025.172PMC 12361914 · doi ↗ · pubmed ↗
- 6Bartholomeeusen K. Daniel M. La Beaud D.A. Gasque P. Peeling R.W. Stephenson K.E. Ng L.F.P. Ariën K.K. Chikungunya fever Nat. Rev. Dis. Primers 202391710.1038/s 41572-023-00429-237024497 PMC 11126297 · doi ↗ · pubmed ↗
- 7Zhou X. Xie X. Wang W. Gao H. Wang K. Liu X. Chen X. Jiang Y. Yu H. Xing D. First whole-genome chikungunya virus sequence detected in mosquitoes during the 2025 Foshan outbreak: Evidence of field vector infection and transmission potential in China Infect. Dis. Poverty 20251410810.1186/s 40249-025-01383-941131572 PMC 12548186 · doi ↗ · pubmed ↗
- 8Wu D. Wu J. Zhang Q. Zhong H. Ke C. Deng X. Guan D. Li H. Zhang Y. Zhou H. Chikungunya outbreak in Guangdong Province, China, 2010 Emerg. Infect. Dis.20121849349510.3201/eid 1803.11003422377135 PMC 3309566 · doi ↗ · pubmed ↗