Indole-3-Acetic Acid as a Putative Selective AhR Modulator Counteracts Skatole-Induced Dual-Hit Toxicity in Colorectal Cancer Cells
Chihiro Takei, Hidehisa Shimizu

TL;DR
This study shows that indole-3-acetic acid can counteract the harmful effects of skatole on colorectal cancer cells, suggesting a potential dietary strategy for cancer prevention.
Contribution
The study identifies indole-3-acetic acid as a functional antagonist of skatole-induced toxicity through a novel mechanism involving AhR signaling modulation.
Findings
Skatole promotes CRC cell proliferation via AhR-dependent and ERK MAPK pathway activation.
Indole-3-acetic acid counteracts skatole-induced proliferation without affecting MAPK phosphorylation.
IAA acts as a putative selective AhR modulator, reprogramming AhR signaling to block cell cycle progression.
Abstract
The rising incidence of colorectal cancer (CRC) in modernized societies is linked to diet-induced dysbiosis, characterized by a critical metabolic divergence: the depletion of protective indole-3-acetic acid (IAA) concurrent with the accumulation of toxic skatole (3-methylindole). However, the molecular mechanisms by which high concentrations of skatole drive malignancy—and whether IAA can counteract this toxicity—remain elusive. Here, we demonstrate that physiologically relevant concentrations of skatole (500 µM) significantly promote the proliferation of HCT-116 CRC cells through a “dual-hit” mechanism involving both aryl hydrocarbon receptor (AhR)-dependent genomic activity and AhR-independent activation of the ERK MAPK pathway. Notably, co-treatment with IAA (250 µM) effectively abrogated skatole-induced proliferation, restoring cell growth to baseline levels while sparing upstream…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
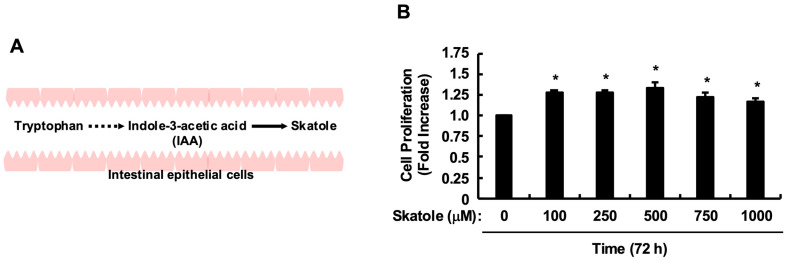 Figure 1
Figure 1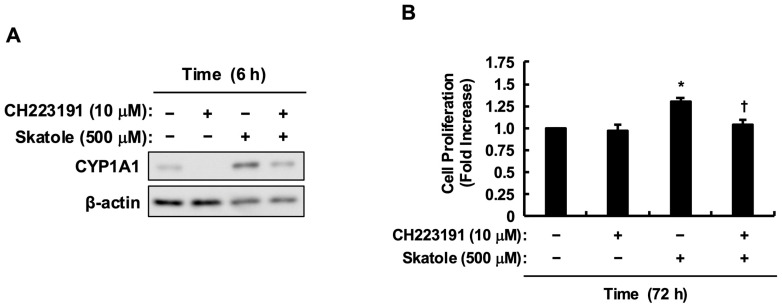 Figure 2
Figure 2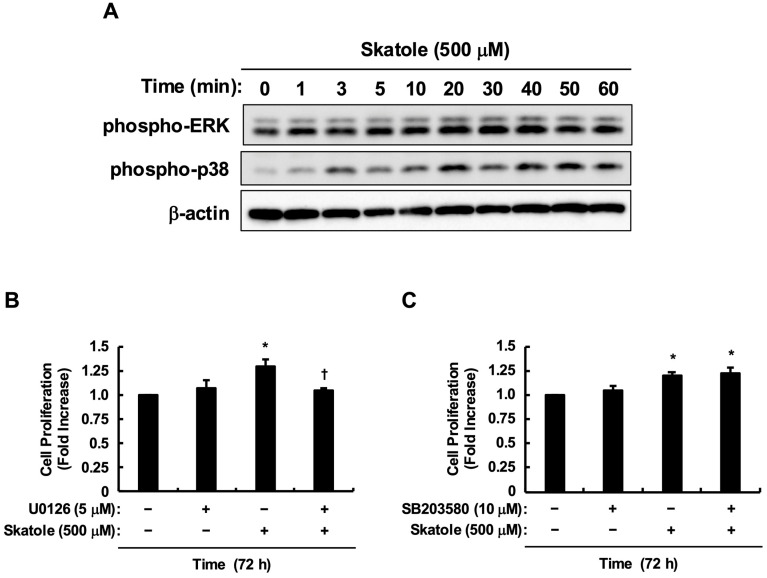 Figure 3
Figure 3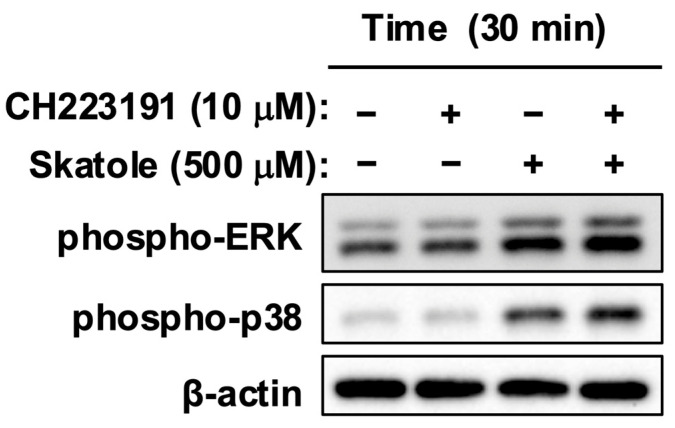 Figure 4
Figure 4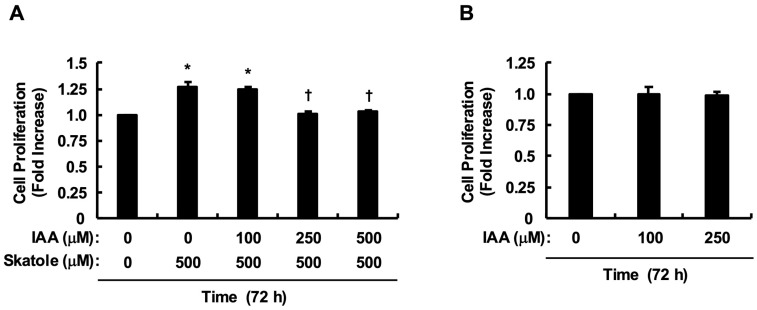 Figure 5
Figure 5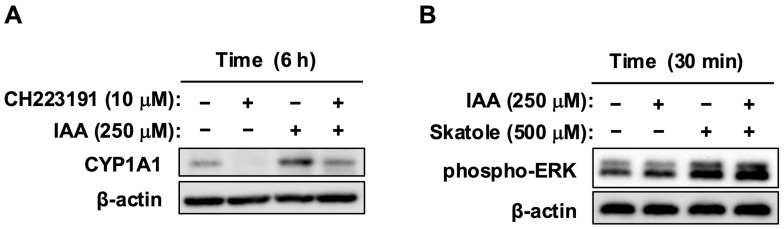 Figure 6
Figure 6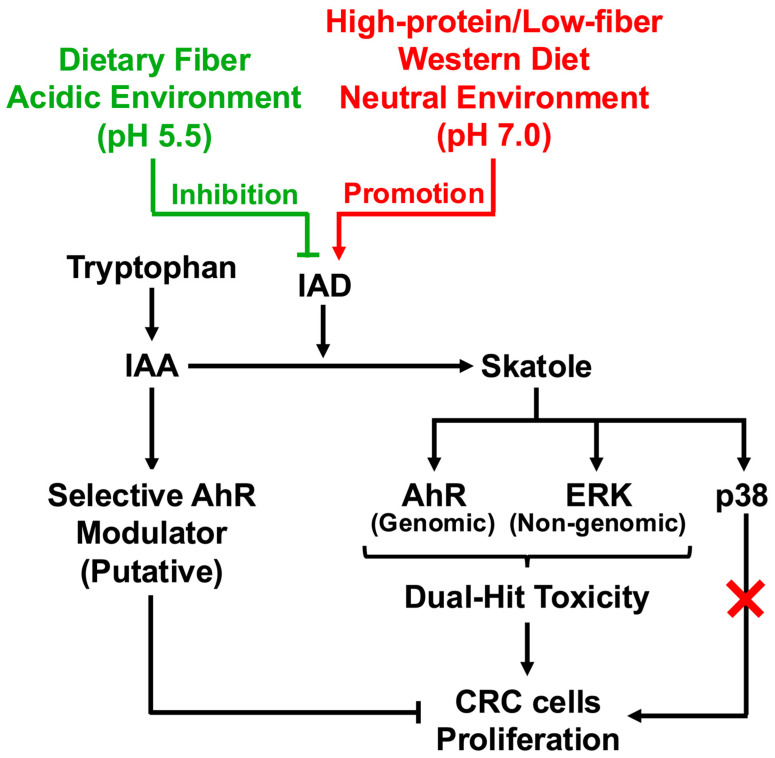 Figure 7
Figure 7- —Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsToxic Organic Pollutants Impact · Cancer, Hypoxia, and Metabolism · Epigenetics and DNA Methylation
1. Introduction
Colorectal cancer (CRC) stands as one of the leading causes of death in modern society [1,2,3]. The rapid rise in incidence, particularly in Asian countries including Japan, has escalated from a medical issue to a serious public health crisis [3]. Historically, Japan maintained a remarkably low global incidence of CRC, which was likely associated with a traditional diet rich in fermentable dietary fibers derived from brown rice, soybeans, and seaweeds. However, following the rapid economic growth after World War II, Japan’s dietary structure underwent a dramatic nutritional transition. The popularization of bread and dairy products, initiated by the School Lunch Act of 1954, and the dietary shift observed around 1985—where animal protein intake surpassed that of plant protein—fundamentally altered the Japanese intestinal environment [4]. Currently, the intake of red and processed meats among the Japanese population is on an upward trend [5], and it is widely acknowledged that this Westernization of diet correlates with the increase in inflammatory bowel diseases (IBD) and colorectal cancer risk in Japan [6,7,8,9].
This dramatic dietary shift has fundamentally disrupted the metabolic balance of the essential amino acid tryptophan (Trp) by gut microbiota [10]. Unabsorbed Trp reaching the colon is converted into bioactive indole derivatives by various bacterial groups, but the metabolic pathway is heavily influenced by dietary content [10,11,12] (Figure 1A). Recent clinical studies highlight a critical finding: in healthy individuals, indole-3-acetic acid (IAA), which possesses anti-colorectal cancer activity and anti-inflammatory properties (e.g., via IL-35 induction [13]) that prevent tumorigenesis, is abundantly maintained. Specifically, IAA has been shown to inhibit tumorigenesis through IL-35 induction [13], ameliorate colitis via ERK signaling pathways [14], and maintain intestinal epithelial homeostasis through mucin sulfation [15]. This trend of IAA depletion is not limited to the local intestinal environment but has also been confirmed in the serum [16] and urine [17] of colorectal cancer patients, suggesting a systemic alteration in metabolic homeostasis. Conversely, in the feces of patients with a deteriorated intestinal environment, a metabolic divergence is observed where IAA levels significantly decrease while the concentration of the toxic metabolite skatole (3-methylindole) markedly increases [12]. Skatole is often discussed in the context of protein-bound uremic toxins that accumulate in chronic kidney disease (CKD) [18,19,20]; however, its local toxicity at the site of production—the gut—remains less understood. Skatole has been detected in the feces of patients with digestive disorders and colorectal cancer, reportedly reaching high concentrations of approximately 100 µg/g, which corresponds to roughly 1000 µM in molar concentration [11,12,21]. We hypothesize that this clinically observed profile... serves as a chemical driver of colorectal carcinogenesis, potentially through sustained perturbation of host AhR-dependent signaling pathways.
Furthermore, recent findings have revealed that CRC differs significantly in embryological origin and biological characteristics depending on the site of occurrence (proximal right side vs. distal left side). Right-sided colorectal cancer (RCC), in particular, has a poor prognosis and it is strongly suggested to involve the microbiota [6,22]. A pivotal role in the progression of RCC is played by the formation of bacterial biofilms specific to the right colon [22,23]. Within this biofilm microenvironment, it is plausible that specific IAD-expressing anaerobes, such as Olsenella scatoligenes and Clostridium species [24,25], catalyze the conversion of beneficial IAA into toxic skatole. Inside biofilms, diffusion of metabolites is restricted, suggesting that locally elevated concentrations of skatole may occur, continuously exposing host cell signal transduction systems. We propose that a Westernized diet promotes a synergistic pathological milieu where protective IAA is depleted while toxic skatole production is markedly increased. This metabolic shift from an ‘IAA-conserving’ to a ‘skatole-producing’ mode represents a critical turning point that may subvert the host’s Aryl Hydrocarbon Receptor (AhR) signaling from a protective mechanism into a driver of carcinogenesis. Elucidating the molecular consequences of this dysregulation is crucial for CRC prevention.
We have consistently investigated the impact of gut microbiota-derived tryptophan metabolites on colonic homeostasis and pathophysiology. Specifically, we have investigated complex inflammatory regulation mechanisms wherein skatole regulates intestinal epithelial cell functions via AhR and p38 pathways [26] and detailed the molecular mechanisms of skatole-induced inflammation [27,28]. We also reported that indoxyl sulfate acts as a toxic ligand promoting colorectal cancer cell proliferation via AhR/Akt and AhR/c-Myc pathways [29,30]. Conversely, regarding indole-3-acetic acid (IAA), we have demonstrated its anti-inflammatory and anti-proliferative effects via non-AhR pathways [31,32,33] in a cell-context dependent manner. Additionally, recent findings report that IAA reprograms systemic homeostasis and significantly ameliorates cachexia associated with IBD [34]. While our previous work established that toxic indoles function as potent AhR activators driving malignancy, the potential for IAA to qualitatively modulate AhR signaling itself remains a critical open question. Specifically, elucidating how a low-affinity ligand like IAA can functionally override the potent signaling driven by high concentrations of skatole constitutes the central mechanistic gap addressed in the present study. In this study, using HCT-116 cells which possess typical characteristics of RCC (MSI-H, KRAS mutation) [35], we aimed to elucidate the dual-hit proliferation mechanism induced by clinically relevant high concentrations of skatole. Furthermore, we aimed to evaluate whether IAA can act as a selective AhR modulator (SAhRM) by qualitatively modulating, rather than merely competing with, skatole-induced AhR signaling, thereby shifting cell fate from proliferation toward a tumor-suppressive response.
2. Results
2.1. Skatole Promotes Colorectal Cancer Cell Proliferation in an AhR-Dependent Manner
First, we validated the physiological relevance of the skatole concentration used in this study. It has been reported that fecal skatole concentrations in patients with severe dysbiosis reach approximately 100 µg/g [11,12,21]. Assuming a fecal water content of 75%, 100 µg/g of feces corresponds to approximately 133 µg per mL of liquid phase. Given the molecular weight of skatole (131.17 g/mol), this equates to approximately 1015 µM. Importantly, recent studies highlight that right-sided colorectal tumors are often covered by dense bacterial biofilms [22,23], creating a local microenvironment where metabolite diffusion is restricted. Consequently, local concentrations can substantially exceed those found in the bulk fecal stream. Therefore, we established 1000 µM as a physiologically plausible maximum. Under these conditions, proliferation assays revealed that skatole significantly promoted the proliferation of HCT-116 cells across this clinically relevant range (100–1000 µM) (Figure 1B). Since the growth-promoting effect was maximal at 500 µM, we selected this concentration as a physiologically relevant estimate of localized epithelial exposure for subsequent mechanistic analyses. Next, to confirm whether skatole functions as a ligand for AhR, we examined changes in the expression level of the target gene CYP1A1. Pretreatment with an AhR antagonist (CH223191) markedly suppressed the upregulation of CYP1A1 expression induced by skatole stimulation (Figure 2A). Furthermore, CH223191 significantly inhibited skatole-induced proliferation of HCT-116 cells, reducing the cell viability to levels comparable to the vehicle control (approximately 60–70% of the skatole-treated group) (Figure 2B). These results demonstrate that skatole at physiologically plausible concentrations drives the abnormal proliferation of colorectal cancer cells using AhR as a key mediator.
2.2. Activation of ERK Contributes to Skatole-Induced HCT-116 Cell Proliferation
Subsequently, we analyzed the intracellular signal transduction pathways induced by skatole. Given that ERK1/2 is a canonical driver of cell proliferation, and our previous findings identified p38 as a key downstream effector of skatole in Caco-2 cells [26], we focused our investigation on these two pathways. Addition of skatole (500 µM) resulted in rapid phosphorylation (activation) of both ERK1/2 and p38 (Figure 3A). To determine which MAPK pathway drives proliferation, we conducted experiments using specific inhibitors. The results showed that the ERK pathway inhibitor (U0126) significantly suppressed skatole-induced cell proliferation (Figure 3B). In contrast, the p38 inhibitor (SB203580) had no effect on proliferation, despite the observed phosphorylation of p38 (Figure 3C). Thus, these results indicate that while skatole activates multiple MAPK pathways, the activation of ERK specifically contributes to the induced proliferation of HCT-116 cells.
2.3. Skatole-Induced MAPK Activation Is Mediated via an AhR-Independent Pathway
Since skatole activated AhR and MAPKs (ERK and p38), we verified the possibility that AhR controls these kinase activities. As shown in Figure 4, pretreatment with the AhR antagonist (CH223191) did not alter the phosphorylation levels of either ERK or p38 induced by skatole. This indicates that AhR is not involved in skatole-mediated MAPK activation, suggesting the engagement of a parallel signal transduction pathway distinct from AhR. This finding implies that skatole drives proliferation signals by engaging an independent nongenomic pathway, separate from AhR-mediated genomic transcriptional activity. These results suggest that skatole toxicity involves a dual-hit mechanism that simultaneously mobilizes these two independent pathways.
2.4. IAA Abrogates Skatole-Induced Proliferation of HCT-116 Cells
We examined the effect of IAA, which is clinically depleted in colorectal cancer patients, on skatole-induced proliferation. Since skatole showed maximal growth promotion at 500 µM, we evaluated the antagonistic effect of IAA within the same concentration range. Notably, co-treatment with IAA dose-dependently suppressed skatole-induced proliferation. Notably, IAA at 250 µM was sufficient to effectively counteract the proliferation promotion, reducing cell numbers to control levels (p < 0.01 vs. skatole alone) (Figure 5A). Next, to rule out the possibility that this cancelation was due to nonspecific cytotoxicity, we examined the effect of IAA alone at the effective concentration range (0, 100, and 250 µM). As shown in Figure 5B, IAA alone did not affect cell proliferation, showing no significant difference compared to the control. These results support the notion that the rescue effect observed at 250 µM is due to the specific blockade of proliferation signals rather than cell death. Based on these findings, we selected 250 µM as the minimum effective and safe concentration for the subsequent mechanistic analyses.
2.5. IAA Modulates AhR Signaling and Blocks Proliferation While Maintaining ERK Activity
We confirmed that IAA is also a ligand for AhR in HCT-116 cells, similar to skatole, through CYP1A1 upregulation and its suppression by CH223191 (Figure 6A). Next, we examined whether IAA (250 µM) affects skatole-induced (500 µM) ERK activation. Interestingly, even while the proliferation-suppressive effect of IAA was being exerted, the activation of ERK—the proliferation accelerator induced by skatole—was maintained (Figure 6B). This paradoxical state suggests that IAA uncouples upstream MAPK survival signaling from the downstream cell cycle machinery. Typically, ERK activation drives proliferation; however, in the presence of IAA, this signal is uncoupled from cell division. These findings indicate that IAA does not merely turn off the signal source but rather induces a functional blockade at the receptor level, likely possessing properties of a SAhRM-like agent that induces a functional shift in AhR activity.
3. Discussion
3.1. Summary of Key Findings
The present study elucidates the molecular mechanisms by which modern dietary changes drive bacterial metabolic switching toward skatole overproduction, and how resulting metabolite fluctuations shape host cell fate. We demonstrate that skatole, markedly elevated in CRC patient guts, potently drives HCT-116 cell proliferation through a dual-hit mechanism simultaneously engaging AhR-dependent genomic transcription and nongenomic MAPK (ERK1/2) activation. Critically, indole-3-acetic acid (IAA)—clinically depleted in CRC—appears to act as a functional selective antagonist. IAA qualitatively modulates AhR signaling at the receptor level while preserving skatole-induced MAPK activation, functioning as a putative selective AhR modulator (SAhRM) that potently counteracts skatole-driven proliferation. These findings suggest that bacterial IAD inhibition and IAA preservation via dietary intervention may offer a mechanistically rational strategy to disrupt CRC progression at the receptor level, as summarized in our proposed graphical model (Figure 7).
3.2. Mechanisms of Metabolic Dysregulation
The observation that skatole levels are pathologically elevated in CRC patients while IAA is depleted suggests a marked dysregulation of tryptophan metabolism homeostasis. We propose that the Westernized diet drives this “metabolic switch” through three converging mechanisms: enzymatic, genetic, and physical control. First, enzymatic regulation via pH plays a critical role. High dietary protein intake increases colonic ammonia production, elevating luminal pH [36]. Biochemical characterization of the skatole-producing enzyme, indoleacetate decarboxylase (IAD), reveals a strict pH optimum of approximately 7.5 (neutral to slightly alkaline), with sharply reduced activity in acidic conditions [24,37]. Consequently, the elevation of colonic pH typical of dysbiosis creates a highly favorable biochemical environment for IAD activity, accelerating the irreversible decarboxylation of the precursor IAA into toxic skatole. Second, genetic and ecological regulation exacerbates this process. In the absence of fermentable carbohydrates (dietary fiber), gut bacteria are released from carbon catabolite repression (CCR), triggering the expression of the tryptophanase gene (tnaA) to utilize amino acids as alternative energy sources [38]. Crucially, IAD expression is distinct from tnaA and is primarily governed by substrate induction; the accumulation of the precursor IAA paradoxically promotes the upregulation of IAD in specific anaerobes, such as Olsenella scatoligenes and Clostridium species [24,25,39]. This creates a pathological feed-forward loop where initial IAA accumulation fuels its own conversion to skatole. Third, physical retention amplifies metabolite accumulation. Low-fiber diets are associated with prolonged intestinal transit time (stasis) [40,41,42,43,44]. This stasis provides an extended fermentation window, favoring the proliferation of slow-growing anaerobes responsible for secondary metabolism and allowing skatole to accumulate to toxic concentrations within the lumen [45,46,47]. Collectively, these mechanisms provide a mechanistic basis for why the “High Skatole/Low IAA” metabolic profile is a hallmark of the Westernized gut environment.
3.3. Skatole-Induced Dual-Hit Proliferation Mechanism
AhR antagonism by CH223191 potently suppressed skatole-induced HCT-116 proliferation (Figure 2B), consistent with Karasová et al. [48], who demonstrated that AhR is an essential survival factor in these cells, regulating fatty acid synthase (FASN) to supply lipids for rapid membrane biogenesis. However, skatole-induced MAPK (ERK and p38) activation persisted despite complete AhR blockade (Figure 4). This indicates the existence of a parallel, AhR-independent MAPK activation pathway. While direct ROS quantification was not performed in this study, the metabolism of skatole by cytochrome P450 enzymes (e.g., CYP1A1, CYP2F1)—which are potently induced by skatole itself [49]—is well-established to generate reactive intermediates (3-methyleneindolenine) and ROS [12,49,50,51]. Critically, Schroyer et al. demonstrated that in HCT-116 cells, oxidative stress explicitly triggers ERK1/2 phosphorylation, which is requisite for driving a malignant phenotype [52]. This aligns with our finding that ERK inhibition significantly abolished skatole-induced proliferation (Figure 3B). Thus, our data suggest that skatole exerts a coordinated “dual-hit” control—simultaneously driving AhR-dependent transcriptional programs and ROS-mediated AhR-independent ERK hyperactivity [52]—which cooperatively fuels sustained CRC cell proliferation. Furthermore, we recently reported that indoxyl sulfate, another tryptophan-derived uremic toxin, exacerbates CRC progression by stabilizing the proto-oncogene c-Myc via an AhR/Akt-dependent pathway [30]. Given that ERK phosphorylation is a known stabilizer of c-Myc, it is highly plausible that skatole similarly exploits this “dual-hit” (AhR/ERK) axis to sustain high c-Myc levels, thereby fueling the rapid cell cycle progression characteristic of CRC. Verifying this downstream axis will be a critical objective of our future studies.
3.4. Cell-Specific Context of Skatole Toxicity
Remarkably, skatole at physiologically relevant concentrations (up to 1000 µM) drove robust proliferation in HCT-116 cells (Figure 1B), whereas we previously reported that it induces apoptosis in Caco-2 cells [26]. This dichotomous response likely reflects cell-specific metabolic vulnerabilities dictated by genetic background. Caco-2 cells (KRAS wild-type) express highly inducible CYP1A1, rapidly converting skatole to toxic 3-methyleneindolenine and generating overwhelming ROS [12,49], which exceeds their survival threshold. Conversely, HCT-116 cells (KRAS G13D mutant, MSI-H) [35], which represent a model of right-sided CRC, possess constitutively active survival signaling. We postulate that this oncogenic drive allows HCT-116 cells to tolerate skatole-derived oxidative stress and repurpose the concurrent AhR/ERK activation to fuel proliferation rather than cell death. This cell context-dependent fate switching provides mechanistic insight into the clinical paradox where skatole accumulation selectively promotes malignancy in specific CRC subsets, particularly within the biofilm-rich environment of the right colon [22,23].
3.5. IAA Functions as a Putative Selective AhR Modulator
Our data indicate that IAA does not merely compete with skatole for receptor binding but functions as a putative selective AhR modulator (SAhRM) that qualitatively reprograms downstream signaling [53,54,55]. The structural distinction between the flexible hydrophilic sidechain of IAA (-CH_2_COOH) and the hydrophobic methyl group of skatole (-CH_3_) likely induces thermodynamically divergent AhR conformations within the ligand-binding pocket [56,57]. Key evidence for this functional selectivity is the uncoupling of upstream signaling from downstream proliferation. As shown in Figure 6B, IAA completely abolished skatole-induced proliferation without inhibiting skatole-induced ERK phosphorylation. We postulate that IAA-bound AhR functionally overrides the proliferative ERK “accelerator”, potentially by acting as a dominant “molecular brake.” Mechanistically, ligand-activated AhR is known to directly induce the expression of the cyclin-dependent kinase inhibitor p27Kip1, leading to G1 cell cycle arrest [58]. Additionally, nongenomic modulation of other pathways [59] or interaction with the Hsp90 chaperone complex [60,61], and activation of the AhR E3 ubiquitin ligase function [62] may further contribute to this regulatory shift.
3.6. Metabolic Counter-Attack Hypothesis: Skatole-Driven Bioactivation of IAA
Beyond receptor competition, skatole metabolism by CYP1A1 generates reactive intermediates (e.g., 3-methyleneindolenine) and ROS [12,49], establishing a pro-oxidant intracellular milieu. Crucially, the metabolic fates of skatole and IAA are diametrically opposed. Dong et al. recently demonstrated that unlike classical high-affinity ligands, IAA is not a substrate for CYP1A1 despite inducing its expression [63]. Structural simulations reveal that IAA binds to an allosteric pocket distant from the catalytic heme center, thereby evading enzyme-dependent clearance [63]. Consequently, while skatole is rapidly metabolized, IAA persists intracellularly. This stability may allow IAA to undergo non-enzymatic ROS-mediated oxidation, yielding quinone-imine or dihydroxyindole derivatives with potentially enhanced bioactivity. Tintelnot et al. recently demonstrated that peroxidase-oxidized IAA intermediates suppress pancreatic malignancy [64], providing a direct precedent for this mechanism. It is tempting to speculate that the pathological ROS output driven by skatole might bioactivate its own metabolic antagonist IAA, turning the hostile microenvironment against the tumor cell.
3.7. Physiological Relevance of Metabolite Concentrations
A critical consideration for in vitro studies is the physiological relevance of the concentrations used. As summarized in Supplementary Table S1, reported fecal skatole concentrations in humans vary but can reach pathological levels in disease states. Notably, Karlin et al. reported skatole levels of approximately 100 µg/g feces in patients with digestive disorders [21]. Assuming a fecal water content of 75%, this corresponds to approximately 1000 µM in the aqueous phase. Furthermore, considering the restricted diffusion within the dense bacterial biofilms characteristic of right-sided CRC [22,23], epithelial cells are likely exposed to stable microgradients of skatole exceeding 1000 µM. Thus, the 500 µM concentration selected for our mechanistic experiments represents a conservative and highly relevant exposure level. Similarly, considering the stoichiometry of the reaction catalyzed by IAD (direct decarboxylation of IAA to skatole), the accumulation of skatole inherently implies an equimolar availability of its precursor, IAA. Therefore, suppressing IAD activity would theoretically allow the local IAA pool to recover to concentrations well exceeding the 250 µM used in our rescue experiments.
3.8. Limitations of the Present Study
While this study presents evidence for the “dual-hit” toxicity of skatole and the protective role of IAA, several limitations must be acknowledged. First, our functional assays primarily relied on the HCT-116 cell line. While this cell line models the specific context of right-sided, MSI-high, KRAS-mutated CRC, verifying these findings in patient-derived organoids would strengthen clinical translatability. However, it is noteworthy that human AhR has evolutionarily adapted to bind dietary indoles with higher specificity than murine AhR [65], suggesting that human cell models may provide more relevant mechanistic insights than rodent models. Second, although pharmacological inhibition using CH223191 effectively phenocopied AhR knockout effects [48], genetic ablation (CRISPR/Cas9) of AhR would provide definitive confirmation. Third, the designation of IAA as a “SAhRM” is currently based on functional outcomes (proliferation vs. signaling uncoupling); structural biology studies (e.g., X-ray crystallography) are required to elucidate the precise conformational changes induced by IAA binding. Finally, given the physicochemical distinction between lipophilic skatole and anionic IAA, their cellular uptake mechanisms likely differ. Therefore, elucidating the specific transporters responsible for IAA influx and examining how their expression levels influence its anti-proliferative efficacy remain important subjects for future investigation.
3.9. Clinical Translation: Precision Nutrition Strategy
Our findings suggest that the bacterial metabolic switch is a potential driver in the progression of CRC. High-fat, high-protein Western diets induce dysbiosis and disrupt protective tryptophan metabolic pathways [6,66]. A potential intervention strategy involves the restoration of physiological colonic pH. Daily intake of resistant starch promotes the production of short-chain fatty acids (SCFAs) and lowers colonic pH (<6.0) [39,67]. Since IAD belongs to the glycyl radical enzyme family and loses catalytic activity in acidic environments [24,37], luminal acidification could serve as a “metabolic brake,” inhibiting skatole production and conserving IAA [45]. Furthermore, microbial tryptophan metabolites have been shown to modulate intestinal immunity [68,69,70], engaging AhR to balance mucosal reactivity [69,71]. Thus, targeting bacterial IAD via precision nutrition may shift colonic metabolism from skatole-generation to IAA-conservation, potentially restoring the protective AhR axis.
4. Conclusions
The present study indicates that skatole drives HCT-116 cell proliferation via a dual-hit mechanism involving AhR-dependent genomic signaling and AhR-independent ERK activation. IAA counteracts this toxicity, functioning as a putative SAhRM that qualitatively reprograms receptor output without inhibiting upstream MAPK signaling. These findings provide a molecular basis for understanding how diet-induced dysbiosis contributes to CRC progression and suggest that restoring the IAA/skatole balance may offer a potential preventative strategy.
5. Materials and Methods
5.1. Materials
Various reagents and antibodies were obtained from suppliers for the present study. 3-Methylindole (Skatole; >98.0% GC) and 3-Indoleacetic Acid (>98.0%(T)) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Cayman Chemical (Ann Arbor, MI, USA) provided the aryl hydrocarbon receptor (AhR) antagonist CH223191. The MEK1/2 (ERK pathway) inhibitor U0126, the p38 inhibitor SB203580, Dulbecco’s modified Eagle’s medium (DMEM), penicillin-streptomycin solution, and dimethyl sulfoxide (DMSO) were obtained from Wako Pure Chemical Industries (Osaka, Japan). Anti-β-actin antibody (C4) was supplied by Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) and anti-phospho-p38 (Thr180/Tyr182) antibodies were supplied by Cell Signaling Technology, Inc. (Danvers, MA, USA). Anti-CYP1A1 antibody was obtained from Proteintech Group Inc. (Chicago, IL, USA). Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) and Anti-Mouse IgG (H + L) antibodies were acquired from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). Nacalai Tesque (Kyoto, Japan) supplied Protease Inhibitor Cocktail (EDTA-free), Phosphatase Inhibitor Cocktail, and the chemiluminescent reagent Chemi-Lumi One L/Super. Fetal Bovine Serum (FBS) was procured from Biowest (Nuaillé, France).
5.2. Cell Culture
As this study utilized established cell lines in vitro, clinical inclusion/exclusion criteria are not applicable. The human colorectal cancer cell line HCT-116 was provided by the RIKEN BioResource Center (Tsukuba, Japan). Cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin at 37 °C in a 5% CO2 atmosphere. Twenty-four hours prior to the start of experiments, cells were switched to serum-free DMEM to induce starvation and stabilize the signal baseline. DMSO was used as the solvent for all compounds (skatole, IAA, CH223191, inhibitors), and the final DMSO concentration in the medium was unified at 0.1% (v/v). The control group received the same concentration of DMSO (vehicle control).
5.3. Quantitation of Cell Proliferation
The proliferative capacity of HCT-116 cells was evaluated using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Cells were seeded in 96-well plates and starved, then treated with various concentrations of skatole (0–1000 µM) or IAA (0, 100, 250, 500 µM) alone or in combination for 72 h. When using the antagonist CH223191 (10 μM) or each signaling inhibitor, pretreatment was performed 30–60 min before ligand stimulation. After treatment, CCK-8 solution was added, and absorbance at 450 nm was measured using a microplate reader. Results were calculated as relative absorbance (% of control) or cell proliferation rate (fold increase) relative to the vehicle control.
5.4. Immunoblotting
Immunoblotting was performed to analyze signal transduction and target proteins. Stimulated HCT-116 cells were lysed on ice using a buffer as described in previous studies [29,30]. Proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto Immobilon-P polyvinylidene fluoride membranes (Millipore Inc., Bedford, MA, USA). After blocking, membranes were incubated with primary antibodies (anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204), anti-CYP1A1, and anti-β-actin at 1:5000 dilution; anti-phospho-p38 (Thr180/Tyr182) at 1:1000 dilution). After washing, membranes were incubated with the corresponding HRP-conjugated secondary antibodies (Goat Anti-Rabbit IgG or Anti-Mouse IgG; 1:5000 dilution). Target proteins were visualized using the Chemi-Lumi One system. Images were captured using an ImageQuant LAS 4010 (GE Healthcare). Membranes were stripped and reprobed to detect multiple proteins (CYP1A1, phospho-ERK, phospho-p38, and β-actin) on the same blot.
5.5. Statistical Analysis
All data are presented as mean ± standard error (SE). Data represent the mean of quadruplicates (n = 4 wells) from at least three independent experiments (N = 3–6 separate passages, as indicated in each figure legend). Statistical significance was evaluated using one-way analysis of variance (ANOVA), followed by post hoc tests. Specifically, Dunnett’s test was used for Figure 1, while the Tukey–Kramer test was applied for Figure 2A, Figure 3B,C and Figure 5A,B. All statistical analyses were performed using Microsoft Excel and Statcel 4 software (OMS Publishing, Saitama, Japan), with p < 0.05 considered statistically significant.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Siegel R.L. Giaquinto A.N. Jemal A. Cancer statistics, 2024 CA Cancer J. Clin.202474124910.3322/caac.2182038230766 · doi ↗ · pubmed ↗
- 2Bray F. Laversanne M. Sung H. Ferlay J. Siegel R.L. Soerjomataram I. Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20247422926310.3322/caac.2183438572751 · doi ↗ · pubmed ↗
- 3Xi Y. Xu P. Global colorectal cancer burden in 2020 and projections to 2040 Transl. Oncol.20211410117410.1016/j.tranon.2021.10117434243011 PMC 8273208 · doi ↗ · pubmed ↗
- 4Otsuka R. Yatsuya H. Tamakoshi K. Imai T. Nishio N. Suzuki S. Chikaraishi Y. Kato Y. Ando M. Murata C. Descriptive epidemiological study of food intake among Japanese adults: Analyses by age, time and birth cohort model BMC Public Health 20141432810.1186/1471-2458-14-32824712924 PMC 3997235 · doi ↗ · pubmed ↗
- 5Sasaki K. Motoyama M. Watanabe G. Nakajima I. Meat consumption and consumer attitudes in Japan: An overview Meat Sci.202219210887910.1016/j.meatsci.2022.10887935687968 · doi ↗ · pubmed ↗
- 6Brennan C.A. Garrett W.S. Gut Microbiota, Inflammation, and Colorectal Cancer Annu. Rev. Microbiol.20167039541110.1146/annurev-micro-102215-09551327607555 PMC 5541233 · doi ↗ · pubmed ↗
- 7Zhang Y. Yu Y. Jiang Z. Yu J. Zhang Z. An Z. Du Y. Mao Y. Hu L. Tang X. The impact of red meat and processed meat consumption on the risk of development and relapse of ulcerative colitis: A systematic review and dose-response meta-analysis Front. Nutr.202512166830210.3389/fnut.2025.166830241019562 PMC 12463588 · doi ↗ · pubmed ↗
- 8Chen H. Fu T. Dan L. Chen X. Sun Y. Chen J. Wang X. Hesketh T. Meat consumption and all-cause mortality in 5763 patients with inflammatory bowel disease: A retrospective cohort study E Clinical Medicine 20224710140610.1016/j.eclinm.2022.10140635497068 PMC 9046121 · doi ↗ · pubmed ↗