Energy–Biosensor Synergy: Intrinsic Catalytic Reactions as Label-Free Signal Pathways
Seyyed Mehdi Khoshfetrat, Samaneh Mirsian, Amirreza Khodadadian, Wolfgang Hilber, Clemens Heitzinger

TL;DR
This review explores how energy-driven reactions can replace traditional labels in biosensors, simplifying detection and improving performance for various applications.
Contribution
The paper introduces energy-based electrochemical reactions as a novel, label-free alternative to conventional electroactive labels in biosensing.
Findings
Energy-based reactions like HER and ORR provide intrinsic electrochemical signals without synthetic redox mediators.
These reactions simplify assay design and improve compatibility with diverse target molecules.
Energy-driven approaches offer faster operation and lower costs compared to traditional methods.
Abstract
The selection of appropriate signal labels is a central consideration in electrochemical biosensing as it directly determines the achievable detection limits, dynamic range, and overall analytical performance. Conventional electroactive labels require low operating potentials, fast electron-transfer kinetics, and reliable attachment to electrode surfaces or recognition elements. Despite their extensive use, these labels present notable challenges for point-of-care applications, particularly in the detection of small molecules where target binding does not inherently generate a measurable electrochemical output. As a result, most sensing architectures depend on externally added redox reporters, introduced either freely into solution or covalently linked to recognition structures, which increases assay complexity and limits scalability. These limitations have motivated the transition…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
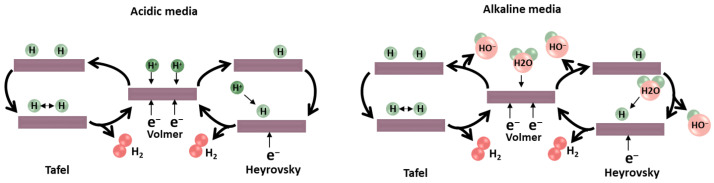 Figure 1
Figure 1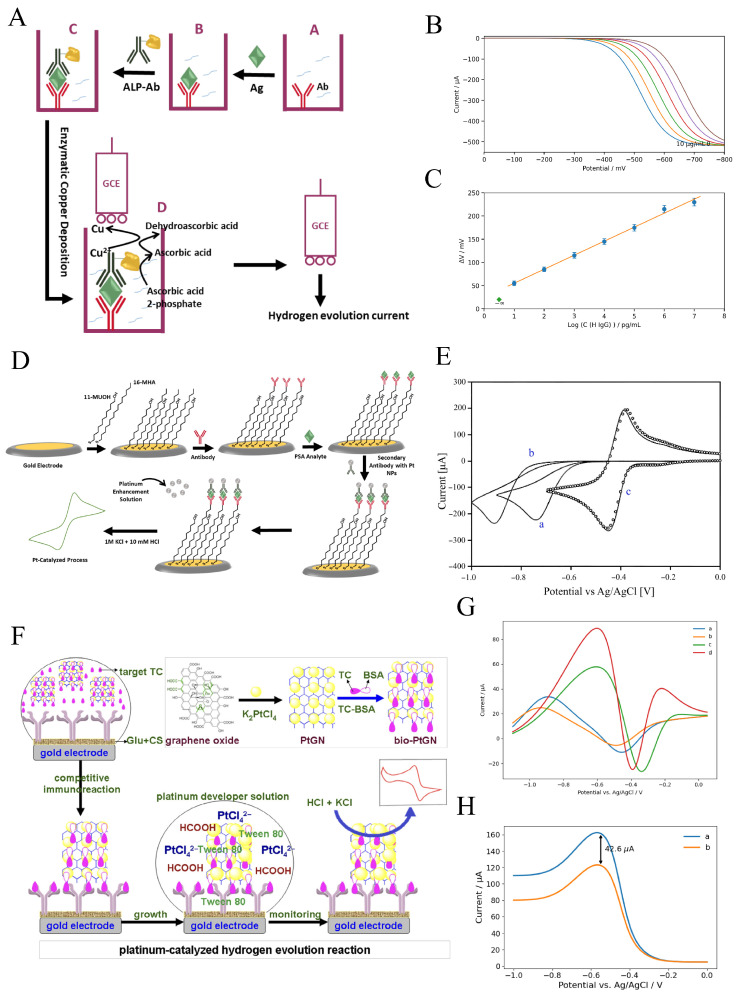 Figure 4
Figure 4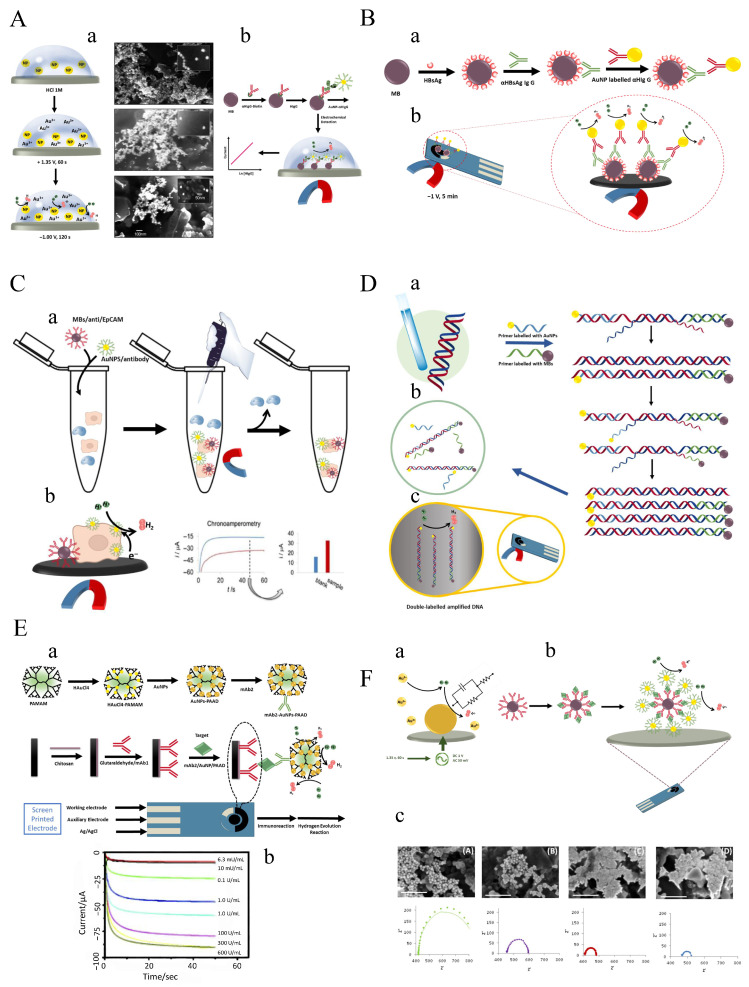 Figure 5
Figure 5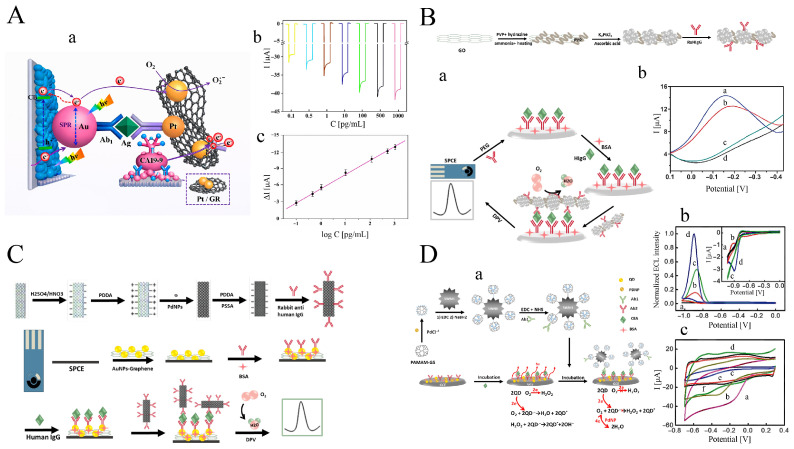 Figure 6
Figure 6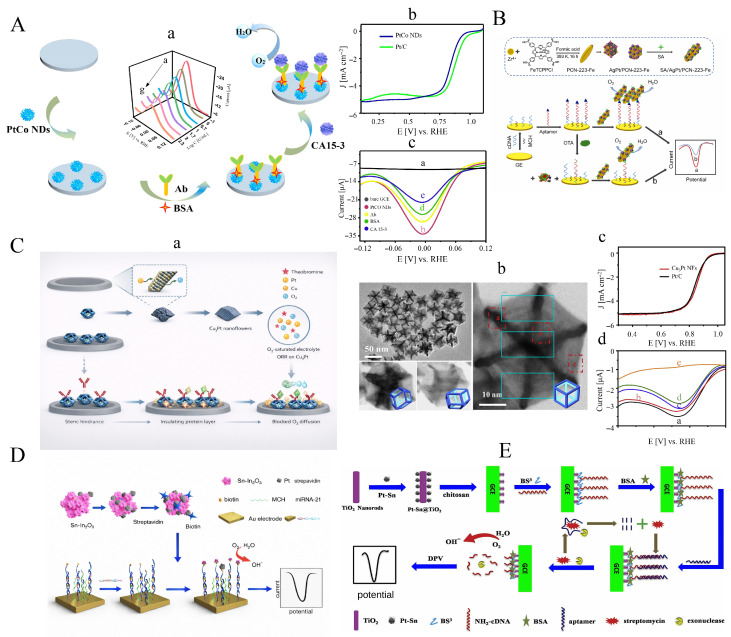 Figure 7
Figure 7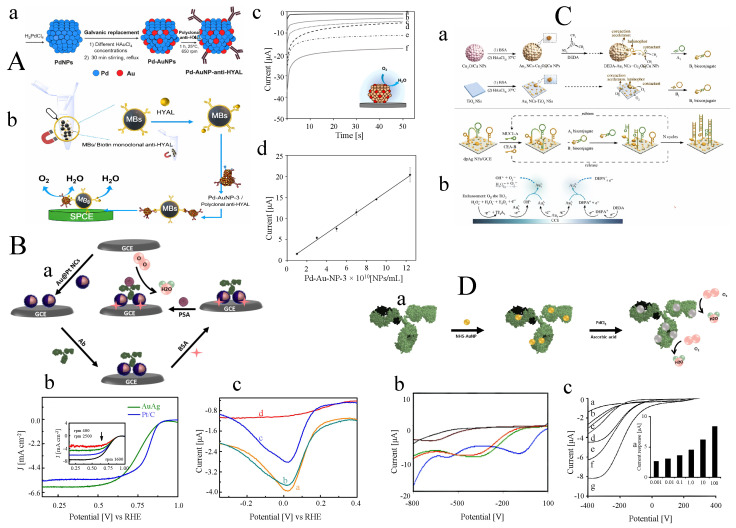 Figure 8
Figure 8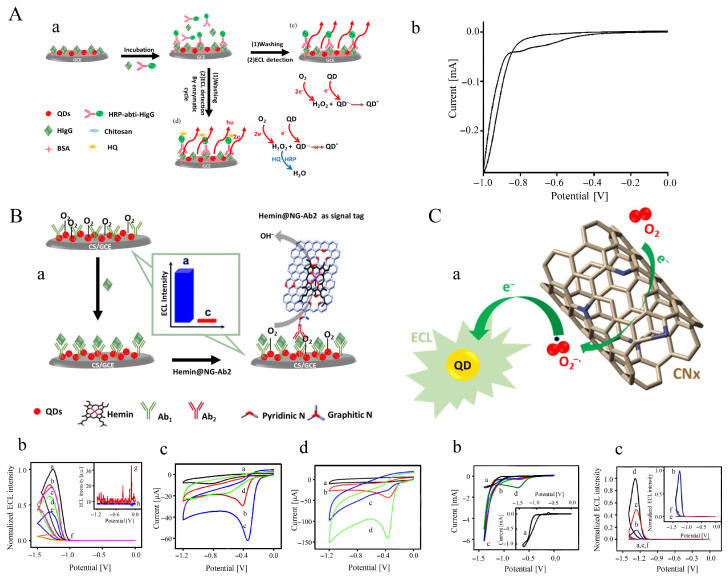 Figure 9
Figure 9 Figure 10
Figure 10- —Austrian Science Fund (FWF)
- —Iran National Science Foundation (INSF)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced biosensing and bioanalysis techniques · Electrochemical Analysis and Applications · Electrochemical sensors and biosensors
1. Introduction
Biosensors are analytical devices used to detect and measure specific biological substances or processes. They typically consist of two fundamental components: a biological recognition element that interacts with the target analyte and a transducer that converts this interaction into a measurable signal such as an electrical, optical, or electrochemical output. A significant challenge in biosensor development is that most biomolecules do not inherently produce a readily detectable signal upon binding. For instance, although antibody–antigen interactions do not generate photons or electrons, they produce only subtle conformational changes, making them difficult to detect directly. As a result, translating biomolecular binding events into clear, selective output signals, especially in complex biological samples containing potentially interfering species, remains difficult.
Upon recognition of biomolecule binding to target molecules, common methods for measuring refractive index, mass, steric bulk, and charge include surface plasmon resonance [1,2,3,4,5,6,7], quartz crystal microbalance [8,9], static microcantilever [10,11,12], impedance spectroscopy [13,14,15], and field-effect transistor (FET) sensors [16,17,18,19]. Among label-free transduction strategies, FET biosensors enable direct electrical readout of biomolecular binding without redox mediators or enzymatic labels. Early silicon nanowire (SiNW) sensors established highly sensitive detection via surface-charge-induced conductance modulation, later extended to sequence-specific DNA sensing and multiplexed biomarker detection [20,21,22]. CMOS-compatible implementations further demonstrated the scalability and practical viability of SiNW-FET immunodetection platforms. Related advances in field-effect biosensing have combined physics-based models, Bayesian inversion, and machine learning to enable quantitative interpretation and rational design of graphene and semiconductor FET sensors [23,24,25,26].
Each of these methods has specific limitations. For instance, surface plasmon resonance, a commercialized adsorption-based approach developed by Biacore [27,28,29], is effective in high-quality buffer samples that are free of interference from other components, leading to false positives in complex real samples. Because all of these proteins have mass, charge, etc., it is difficult, if not impossible, to distinguish between their nonspecific adsorption and binding of the true target [29]. The rapid results, low cost, and ease of use of electrochemical bioassays make them a vital diagnostic tool, especially for point-of-care frameworks, among the detection techniques employed in biosensor designs [30,31,32]. Improvements in detection methods have been the driving force for the development of electrochemical biosensors.
Label-free and label-based electrochemical methods are widely used in various applications and can be differentiated by their underlying operating principles and capabilities. Label-free approaches are generally simple and cost-effective because they detect and quantify analytes without requiring additional chemical tags or markers.
Label-free techniques include certain voltammetric methods and electrochemical impedance spectroscopy (EIS), both of which can detect analytes directly. In contrast, label-based methods rely on the use of specific labels or tags that interact with the target analyte to enhance sensitivity. The use of redox-active molecules as labels is commonly referred to as electrochemical labeling. These label-based strategies are often integrated with other bioanalytical platforms, such as DNA biosensors and immunoassays. Incorporating a label typically improves detection sensitivity, allowing for the measurement of very low analyte concentrations, while the biological recognition element primarily governs selectivity.
In catalytic electrochemical biosensors, signal sensitivity and noise are strongly governed by electron-transfer kinetics and the underlying reaction pathways at the electrode–electrolyte interface [33,34]. Fast and well-defined electron-transfer processes enhance signal-to-noise ratios by producing sharp, reproducible electrochemical responses, whereas sluggish kinetics, multistep pathways, or competing side reactions can introduce background currents, temporal delays, and signal instabilities [23,34]. In particular, catalytic reactions involving multiple electron and proton transfer steps are highly sensitive to surface chemistry, mass transport, and local interfacial environments, all of which influence both amplification efficiency and noise characteristics [35]. Understanding and controlling electron-transfer rates and reaction mechanisms is therefore essential for optimizing sensitivity, minimizing false responses, and achieving reliable signal transduction in label-free and reagent-free catalytic biosensors.
1.1. Challenges of Label-Free, Label- and Reagent-Based Electrochemical Detection
1.1.1. Label-Free Electrochemical Techniques
Label-free biosensors can detect the target directly, allowing for rapid analyte detection with little or no sample preparation and enabling real-time evaluations. These types of sensors either rely on the intrinsic electroactivity of nucleic acid molecules in DNA biosensors or are associated with electrochemical changes caused by antibody–antigen interactions or hybridization processes [36]. Since label-free biosensors depend on direct biorecognition events, they require receptors with high specificity and affinity for the target molecule, as well as a sensitive transducer.
To achieve high sensitivity, signal amplification strategies such as the modification of electrodes with high-performance functional materials [37], enzyme-based isothermal amplification [38,39], and single-, dual-, or triple-signal amplification frameworks have been exploited [40,41]. These strategies enhance the detection capabilities of biosensors, making them more effective for identifying low-abundance targets in complex samples. However, to record the electrochemical signal, the use of reagent molecules in the solution or on the electrode surface is often required. For example, [Fe(CN)6]^3−/4−^ is a commonly employed negatively charged redox probe in label-free electrochemical biosensors, particularly in EIS, one of the most widely used label-free electrochemical techniques.
EIS-based biosensors monitor the impedance at the electrode/solution interface by imposing a small AC excitation over a constant DC voltage across a frequency range. Label-free electronic signal transduction using impedance biosensors detects target binding through changes in the biorecognition layer at the surface. A solid electrode’s surface develops an electrical double layer when exposed to an electrolyte solution, giving rise to a capacitor-like interfacial behavior. However, achieving signal efficiency in complex biological samples is a challenge for many impedance-based biosensors [34].
In complex biological systems, due to the shielding effect of the electrical double layer at the electrode–electrolyte interface, the effectiveness of signal transduction can be notably reduced. Consequently, the clinical usefulness of the sensors may be limited by reduced sensitivity. Furthermore, with these methodologies, the presence of any electroactive component in the solution has the potential to produce false-positive responses in electrochemical biosensors. These limitations hinder the full potential of label-free electrochemical sensors for personalized healthcare applications.
1.1.2. Label-Based Electrochemical Techniques
Label-based approaches detect a specific analyte in a matrix by using tags or labels [42]. In these assays, reliable and stable electrochemical labels are essential, particularly when the analyte is not electrochemically active. The selection of electrochemically active labels is generally based on two criteria: (i) redox potential and (ii) electron transfer kinetics. High redox potentials or slow electron transfer kinetics can lead to overlap with signals from interfering species in the sample, reducing selectivity and specificity [43,44,45]. Electrochemical labels are commonly divided into three functional categories:
- Enzyme-type labels responsible for catalyzing specific substrates. Examples include horseradish peroxidase (HRP) and alkaline phosphatase (ALP), which catalyze reactions that produce electroactive products detectable by the sensor. This approach increases sensitivity by generating higher concentrations of detectable species.
- Labels used as carriers for a high volume of electroactive substrates, increasing sensitivity by releasing or generating more detectable species [46,47].
- Labels that are intrinsically electrochemically active materials. Examples include metal nanoparticles, quantum dots, and other nanomaterials with strong and stable redox properties.
Enzymes produce redox-active molecules, which are commonly applied as bioassay labels. By providing a high number of detectable redox substances per label, enzymes contribute to lowering the limit of detection (LOD) in biosensing applications. However, enzymes can be delicate and susceptible to environmental factors such as pH and temperature. The detection time in enzyme-based assays is determined by the enzymatic turnover rate, which must be carefully controlled to ensure accurate and timely detection [48].
The utilization of compounds as carriers for electroactive materials is important in electrochemical methods. These carriers facilitate the efficient transport and interaction of electroactive components within the sensing system. In electrochemical sensors and supercapacitors, carrier compounds enable effective transport and high loading of redox labels, thereby enhancing overall device performance. The appropriate selection of carrier compounds is essential, as it can influence conductivity, stability, and label-loading capacity. Ongoing research continues to focus on developing improved carrier materials with enhanced properties to advance biosensor technologies and related fields. Based on the information presented thus far, the loading capacity of labels is regarded as the primary determinant of a carrier’s functionality. This is why nanoparticles play a particularly important role within this category. Their high surface-area-to-volume ratio allows efficient loading of electroactive materials, leading to enhanced sensitivity and performance in electrochemical devices.
Taking advantage of nanoparticles’ high surface area as carriers, increasing the loading number of redox labels on these nanoparticles can help overcome the low sensitivity of electroactive compounds. However, weak – interactions and the need for covalent attachment of redox labels on carrier nanoparticles lead to a limited loading capacity [49].
Alternatively, the high porosity and tunable pore sizes of metal–organic frameworks (MOFs) provide a suitable supporting matrix for incorporating high quantities of redox species inside the MOF structure [50,51,52,53,54,55,56,57]. However, this amplification alone is not sufficient for trace bioanalysis [58]. In addition, low conductivity and MOF instability in aqueous solutions impose further limitations on their use in electrochemical biosensors [59,60,61].
In another approach, electroactive labels such as nanoparticles and organic compounds are directly employed for detection by exploiting their inherent redox properties. The signal is substantially enhanced because each nanoparticle label contains large numbers of oxidizable or reducible electroactive centers. Increasing numbers of electrochemical immuno/biosensor designs have used biomolecules modified with redox-active reporters. Despite improved detection limits and the elimination of additional tags or redox indicators, challenges remain when using high-potential redox species or aggressive reagents during detection. For example, strong acids (such as HCl [62] or [47,63]) or powerful oxidizers (such as [64], [65,66], and [67]) may be required, increasing protocol complexity and reducing specificity and selectivity. Balancing enhanced sensitivity with safety, operational simplicity, and specificity is therefore crucial in biosensor design.
When considering organic materials, it is important to note that the redox center of many organic components is typically present in a 1:1 ratio with the analyte. This lack of intrinsic amplification can limit their utility in practical diagnostic devices, although exceptions exist. Furthermore, the performance of electrochemically active organic labels in complex environments is another challenge [68]. For example, while a sensor using the reporter methylene blue (MB) shows good stability in serum-containing media, other modified electrodes under similar conditions may exhibit a significant drop in current and lose around 50% of their original signal. This variability highlights the difficulty of achieving consistent and reliable performance for organic-based electrochemical labels in diverse biological environments. Efforts to address these issues include optimizing organic label design and improving the stability and sensitivity of the detection system.
To characterize the performance of electroactive compounds in biosensing applications, two key factors must be considered. First, it is important to select molecules with redox activities that fall within the potential window in which immuno/biosensors remain reasonably stable. In addition, these compounds should demonstrate a significant ability to conjugate with biomolecules or be efficiently converted into conjugatable forms. A wide variety of redox-active molecules can be used as labels in biosensors, including organic small molecules such as thionine [69,70,71], ferrocene [72,73,74,75,76,77], nile blue [78,79,80], anthraquinone [81,82], MB [83,84,85], gallocyanine [86], natural red [87], indophenol [88], carboxy-X-rhodamine (ROX) [89,90], diamino benzoic acid [91], and organometallic complexes such as pentamethylferrocene [86], [Ru(NH_3_)6]^3+^, and [Os(bpy)2]^3+^ [92]. Each of these compounds offers distinct redox properties and conjugation capabilities, making them suitable for a variety of biosensing applications. By selecting an appropriate electroactive label and optimizing the sensor architecture, researchers can develop highly sensitive and specific biosensors for a wide range of analytes.
Numerous research investigations have examined the development of labels in biological sensors, with particular emphasis on redox reporters linked to DNA in electrochemical biosensors. Further details on this topic will be discussed in the subsequent sections. In a comprehensive study, Plaxco’s group investigated various types of redox reporters conjugated to DNA in self-assembled electrochemical DNA sensors [86]. According to these studies, the redox reporters thionine, dabcyl, and ROX show no characteristic oxidation–reduction currents. Under basic conditions, free indophenol carboxylic acid is stable and electrochemically active; however, attaching indophenol to DNA proved challenging as it decomposed during conjugation to an amine-terminated DNA in a dimethyl sulfoxide (DMSO) medium. Furthermore, the redox potential of neutral red is approximately −0.7 V, which leads to instability of alkane thiols assembled on gold electrodes. In addition, the manner in which electroactive redox reporters are attached to DNA can influence their redox peak behavior. For example, only a small peak current was observed when the ferrocene reporter was conjugated to the 3’ end of DNA. The stability of these redox-conjugated DNA biosensors is also an essential consideration. The conjugation chemistry, the choice of redox reporter, and the operating conditions of the sensors all play crucial roles in determining the overall effectiveness and reliability of the biosensors.
The findings of Plaxco’s study demonstrate that after 100 cycles of square-wave voltammetric scans in buffer, the methylene-blue-modified DNA biosensor loses only about 20% of its current. In contrast, anthraquinone-, nile-blue-, or 5’-inked ferrocene-based sensors show moderate durability under the same conditions, exhibiting approximately 50% signal loss. Likewise, DNA self-assemblies modified with 3’-linked ferrocene, ferrocene-C5, or pentamethylferrocene display markedly reduced stability after 50 cycles, with a current decrease of roughly 50% of their initial signal. However, the signaling current of anthraquinone-based sensors rapidly decreases during the initial scans before reaching a plateau, whereas the signaling current of gallocyanine-based sensors increases substantially with repeated scanning.
Undoubtedly, degradation of the electroactive redox labels contributes to some of these instabilities. For example, the oxygen-containing environment of aqueous solutions renders the ferrocenium ion unstable, and the scan-induced increase in gallocyanine current is likely caused by redox-driven modification of the reporter. Additionally, part of the degradation can be attributed to the instability of the self-assembled monolayer (SAM) when repeatedly exposed to high or low electrostatic potentials. To address these issues—particularly the tendency of electroactive molecular labels to degrade or bind nonspecifically—researchers have exploited the electrical properties of double-helical DNA. Intercalated redox probe molecules have been employed to monitor and characterize disruptions in DNA base stacking. This strategy leverages the inherent stability and specificity of the DNA double helix, thereby improving the robustness and reliability of DNA-based electrochemical biosensors. Intercalators bind to DNA by inserting a planar aromatic chromophore between adjacent base pairs [93]. Redox-active intercalator-based electrochemical DNA biosensors, in terms of electron transfer through self-assembled DNA monolayers to reporter molecules, can be divided into three models [94], namely:
- Charge transfer to reporter molecules positioned far from the electrode surface and located above mismatch sites. These molecules are typically situated above regions where mismatches in the DNA sequence occur, enabling the detection of specific mismatches by monitoring changes in the redox activity of the intercalated reporters.
- Charge transfer to intercalated molecules located before the mismatch and positioned closer to the electrode surface. Electron transfer occurs to these intercalators, and their proximity to the mismatch influences the redox signal. This model helps identify the presence and approximate location of mismatches within the DNA sequence by observing the redox behavior of the intercalated molecules.
- Direct electron transfer to the electrode surface through pinholes in the monolayer. These pinholes allow direct contact between the electrode and the intercalated redox probes, enabling efficient electron transfer. This model benefits from enhanced electron-transfer rates and is suitable for detecting even minor disruptions in the DNA structure.
Clearly, to distinguish between fully complementary and mismatched targets, the intercalated reporter molecules must be located after the mismatch sites. In contrast, the two other models cannot discriminate between complementary and mismatched targets. This issue becomes more pronounced when attempting to differentiate thermodynamically stable mismatches such as G–A and G–T from complementary strands, as these mismatches remain well-stacked within the duplex [44,95].
Using MB, Barton and colleagues reported that it lacks the sensitivity required to detect the dynamic behavior of a G–A mismatch. To address this issue, the same group performed an electrocatalytic reduction of ferricyanide by MB [96]. In this electrocatalytic process, MB is electrochemically reduced through DNA -stack-mediated charge transport. The reduced form of MB (leucomethylene blue) is subsequently oxidized back to MB by the ferricyanide oxidizing agent. This cycle generates additional electrons that can flow through MB via DNA-mediated charge transport, thereby enhancing the detection of thermodynamically stable mismatches. However, the penetration of MB below the mismatch may hinder the electrocatalytic effect of ferricyanide due to repulsive interactions between the negatively charged DNA and ferricyanide.
Employing negatively charged intercalators can effectively address this issue. Due to charge repulsion, it is believed that these intercalators penetrate only the very top of the DNA duplex [94,97]. The use of such intercalators has shown that these assays are sufficiently sensitive to detect perturbations in the -stack, enabling the identification of all possible single-base mismatches, particularly the thermodynamically stable ones, without the need for a catalytic approach. However, despite their advanced detection capabilities and improved detection limits, methods based on redox-active intercalators often suffer from high background signals due to non-specific interactions of redox compounds with the electrode surface or ssDNA. Consequently, any factor that prevents electroactive species in the solution from reaching the electrode surface can hinder accurate identification of the target species, thereby limiting the performance of these methodologies.
As previously mentioned, several factors—such as non-specific adsorption of interfering molecules, steric hindrance, and diffusion-rate limitations—have been identified as contributors to delays in electrochemical response. These delays can lead to false-negative results or inaccurate analytical interpretations. Despite the wide applicability of electrochemical methods, such constraints have hindered the full advancement of personalized healthcare models that rely on these techniques for monitoring and diagnosis. Thus, progress in personalized medicine depends on the development of reliable and efficient electrochemical sensing devices capable of overcoming these obstacles. Due to the aforementioned limitations, it is necessary to employ alternative signal-recording strategies that do not rely on electroactive materials in the measuring medium. Reagent-free and label-free electrochemical approaches are promising candidates for developing versatile biosensors with improved practicality. Using energy-based readout mechanisms as signal-recording agents may be particularly advantageous in this context.
Recording signals without relying on electroactive or enzymatic reactions is possible through the use of the HER, the oxygen reduction reaction (ORR), and water splitting as energy-based signal recorders [35]. These reactions offer high sensitivity, selectivity, and rapid response times, playing crucial roles in various electrochemical processes and attracting significant interest in biosensing applications. HER and ORR, along with water splitting, are fundamental to energy storage and conversion devices such as fuel cells and water electrolyzers, where they convert oxygen and hydrogen ions into water and hydrogen gas, respectively. In the context of biosensing, these reactions provide a distinct signal for detecting specific analytes, including biomolecules and chemical species, by altering the catalytic activity at the electrode–electrolyte interface. This analysis examines the roles of HER, ORR, and water splitting in electrochemical biosensing, exploring the underlying mechanisms and principles that govern these reactions and their interactions with relevant analytes. Integrating HER, ORR, and water splitting into biosensing strategies can enable the development of highly sensitive, fast-response, and cost-effective analytical tools with significant potential for healthcare, environmental monitoring, and other applications [98,99].
Understanding and using the unique characteristics of these reactions will continue to drive advancements in modern analytical research and biosensing technologies, opening new avenues for innovation and application.
Finally, unless otherwise stated, electrode potentials discussed in this review are reported versus the reference electrode employed in the original studies. Where comparison across different systems is required, potentials were converted to the reversible hydrogen electrode scale using standard conversion relationships. Throughout the manuscript, figure captions and data descriptions explicitly specify the reference electrode, electrolyte, and pH.
2. Hydrogen Evolution-Based Electrochemical Detection
2.1. Principles and Mechanism of HER
In general, the reduction of hydrogen and its conversion to hydrogen gas ( ) on the electrode surface, in both acidic and alkaline media, is understood through three mechanisms. Initially, the Volmer reaction ((1a) and (1b)) involves the formation of an adsorbed hydrogen atom ( ) through the capture of an electron by proton species at the electrode surface. Protons originate from hydronium ions ( ) in acidic environments and from water molecules in alkaline electrolytes (as illustrated in Figure 1). Subsequently, formation occurs either through the Heyrovsky reaction ((1c) and (1d)), the Tafel reaction (1e), or a combination of the two. In the Heyrovsky mechanism, additional protons diffuse from the solution to the electrode, where they react with an electron and to produce . In contrast, the Tafel process involves the recombination of two adjacent species to generate [100].
Mechanism HER in acidic and alkaline electrolytes [101].
Volmer reaction (electrochemical hydrogen adsorption):
Heyrovsky reaction (electrochemical desorption):
Tafel reaction (chemical desorption):
The response mechanism is determined by the slope of the Tafel plot, denoted as b. This slope represents the change in potential required for a tenfold increase or decrease in current density. Essentially, b serves as an intrinsic characteristic of the catalyst, directly associated with the rate of the HER, as defined by Equation (2). This relationship is established by plotting against j, where j denotes the current density and represents the exchange current density. A smaller value of b indicates faster electron-transfer kinetics, as a smaller increase in potential is needed to achieve a tenfold increase in current density.
The constant b can be defined as follows:
- If the Volmer (discharge) step is rapid and the Tafel (chemical desorption) step is rate-determining, .
- If the Volmer step is rapid and the Heyrovsky step is rate-determining, .
- If the Volmer step itself is rate-determining, .
It should be noted that HER-based sensing strategies often operate at potentials close to the hydrogen adsorption/desorption region, where the formation and consumption of adsorbed hydrogen intermediates ( ) coincide with significant capacitive contributions at the electrode–electrolyte interface. Under such conditions, faradaic currents associated with the HER may partially overlap with non-faradaic capacitive processes, increasing the risk of signal misinterpretation. Consequently, careful selection of the operating potential, together with appropriate control and blank measurements, is essential to ensure reliable signal attribution in HER-based biosensing. Under standard conditions, the Nernst potential for HER on the normal hydrogen electrode is zero. Nevertheless, to overcome kinetic limitations arising from factors such as high activation energy and low energy efficiency, practical HER requires additional potential. These factors cause an overpotential, defined as , where E (or ) is the applied potential. In practical systems, internal resistances—such as the resistance of the electrode material, electrolyte resistance, and contact resistances (e.g., wires, equipment)—introduce an additional potential drop. Therefore, an unavoidable series resistance ( ) generates an ohmic drop that must be corrected in potential–current density (E–j) curves. Accordingly, the potential required to drive HER is
where is the ohmic drop potential [33]. Typically, two specific values of are reported:
- , corresponding to a current density of 1 mA , reflecting the onset overpotential.
- , corresponding to 10 mA , commonly used to compare electrocatalysts.
Smaller and values indicate a lower onset potential and lower overpotential required to reach 10 mA , implying stronger electrocatalytic activity for HER.
2.2. Application of HER for Electrochemical Signal Tracing
Transition-Metal Dichalcogenides (TMDs)
Small labels with sizes comparable to immune molecules, catalytic activity, and long shelf life are ideal labels for probing biomolecular identification. Exfoliated nano TMDs, with the general chemical formula , where M is a transition metal (e.g., W, Mo, Hf) and X is a chalcogen (generally S, Se, or Te), have been used as strong candidates for biomolecule detection due to their size compatibility with biomolecules [102,103,104,105]. In their bulk form, however, TMDs exhibit negligible catalytic activity and therefore must be exfoliated to increase surface area and create catalytically active edge and defect sites. Several liquid-assisted mechanical and chemical exfoliation methods have been reported for reducing the size of TMDs [106,107,108,109,110]. Among these, a solution-dependent bipolar electrochemical exfoliation method based on powder precursors dispersed in aqueous solution has been developed [111,112]. Since the starting material is a powder and no electrode modification is required, this process offers a simple and low-cost exfoliation route.
Bipolar electrochemistry (BE), an electrochemical technique based on the polarization of electrically conducting materials (bipolar electrodes) exposed to an external electric field between two driving electrodes, is used in this process to break down powder precursors into nanosized material. Pumera’s research group has applied highly active TMDs such as [111], [112], [113], and their nanoparticles (NPs) as labels for magneto-immunoassays due to their high catalytic activity toward HER.
The synthesis of these NPs involved Li intercalation of corresponding TMD sheets using tert-butyl lithium (yielding t-BuLi). By applying a high potential between two platinum driving electrodes and initiating water oxidation at the electrode poles, NPs are fragmented. Linear sweep voltammetry (LSV) measurements of t-BuLi and NPs in at a scan rate of revealed that the HER onset potentials for t-BuLi and NPs were and versus the reversible hydrogen electrode (RHE), respectively (Figure 2A). This shift to more negative overpotential is associated with oxidation of NPs during synthesis.
A rabbit IgG/anti-rabbit IgG–MB complex was formed by binding rabbit IgG to anti-rabbit IgG-modified magnetic beads (MBs). After conjugation of the secondary anti-rabbit IgG with NPs, it recognized rabbit IgG, forming the magneto-sandwich complex anti-rabbit IgG–WS_2_ NPs/rabbit IgG/anti-rabbit IgG–MB, with the final amount of NPs proportional to the rabbit IgG concentration. The magneto-immunosandwich assay for rabbit IgG detection was then performed using nanolabels via HER catalysis and electrochemical impedance spectroscopy (EIS) as the transduction method (Figure 2B). To obtain the EIS polarization potential, Nyquist plots were recorded (Figure 2C) at , , and versus RHE. The semicircle diameter decreased with increasing potential.
Considering the mechanism of HER, its Nyquist equivalent circuit consists of two RC parallel elements in series: one associated with the adsorption step ( and ) and the other corresponding to hydrogen evolution to form ( and ). The calibration curve indicates that the net decreases at a polarization potential of as the protein concentration increases over a broad range between and , due to the concentration dependence of the impedimetric signal on the amount of rabbit IgG, with an LOD of . A relative standard deviation (RSD) of approximately 10% confirms the acceptable reproducibility of the protocol. Subsequently, based on a competitive approach, the same research group employed nanolabels for rabbit IgG detection using chronoamperometry [112]. In an acidic electrolyte, the HER catalytic efficiency of (t-BuLi) sheets and NPs, evaluated via LSV on a GC electrode, revealed overpotentials of and for (t-BuLi) sheets and NPs, respectively (Figure 2D).
Although the electrocatalytic activity of (t-BuLi) is higher than that of NPs, the NPs were used as nanolabels for measuring rabbit IgG. This choice is attributed to the size of the NPs, which is comparable to that of proteins and smaller than MBs, leading to improved assay reproducibility. To optimize experimental conditions for HER catalysis, the resulting NPs were exposed to three different potentials ( , , and versus RHE). Figure 2E shows that the current output at is highest when a potential of is applied. Furthermore, at this potential, the signal recorded using a bare GCE is negligible.
As shown in Figure 2F, the IgG–MoSe_2_ NPs/anti-IgG–MB complex was prepared by capturing IgG–MoSe_2_ NPs using anti-rabbit IgG conjugated with magnetic beads (MBs), and the concentration of rabbit IgG was determined based on the amount of NPs present. Although (t-BuLi) sheets show better HER catalytic efficiency, NPs were selected as labels because their size is comparable to proteins and smaller than MBs, which improves assay reproducibility. Good selectivity of this magneto-immunoassay was demonstrated by the acceptable linear response from 2 to , an LOD of , and a RSD of 9.7% across five protein concentrations when a potential of was applied in chronoamperometry.
Tafel analysis confirmed that the HER mechanism on NPs is based on the Volmer adsorption step. Although this biosensing system is simple and low-cost, the applied potential for HER is still relatively high compared with other reports. As shown in Figure 2G, exfoliated produced by BE was used to develop a competitive magneto-immunoassay based on HER on an SPE [113]. The electrocatalytic activity of NPs toward HER in showed better performance and a lower onset potential compared with bare GCE and (t-BuLi) (Figure 2H). This enhanced electrocatalytic activity results from the harsh synthesis conditions during BE ( ), which create new catalytic sites, edges, and defects. Since the slope of the Tafel curve for all three electrodes exceeds , the rate-limiting step in each case is the Volmer reaction. For IgG detection using chronoamperometry at a constant potential of , different concentrations of IgG protein were mixed with MBs/anti-rabbit IgG conjugate and and subsequently drop-cast onto the SPE. This potential was chosen because the highest absolute current was observed for the NP-modified electrode (Figure 2I). Preliminary studies show that a high current is obtained in chronoamperometry ( , ) even in the presence of only IgG protein, indicating high selectivity of the magneto-immunoassay. The amperometric calibration curve, obtained by measuring the current response after , showed a wide linear range from 5 to for label-free IgG with a low LOD of .
2.3. Black Phosphorus Nanoparticles (BP NPs)
Competitive magneto-immunoassay was used for the detection of free rabbit IgG based on the electrocatalysis of impacting black phosphorus nanoparticles (BP NPs) via HER (proton reduction) [114]. A single-step BE process was performed to synthesize BP NPs by exfoliating and downsizing layered black phosphorus microparticles in a solution containing BP crystals and , using two platinum electrodes spaced two centimeters apart with a potential difference of . The higher electrocatalytic activity of BP NPs toward HER was confirmed using LSV and EIS measurements in . The lower overpotential ( ) and the dramatic decrease in charge-transfer resistance ( ) observed for BP NPs ( , = 1.39 k ), compared with BP macroparticles ( , = 37.6 k ) and bare glassy carbon ( ), confirm that exfoliation produces more catalytically active edge sites and enhances HER performance.
As illustrated in Figure 3A, to detect IgG using BP NPs as labels, anti-rabbit IgG was first coupled to tosyl-activated paramagnetic beads (a). A competitive magneto-immunoassay was then designed by mixing rabbit IgG-modified BP NPs (a’) with anti-rabbit IgG-conjugated MBs in the presence of a desired concentration of free rabbit IgG (b). The resulting immune complex was subsequently drop-cast onto a screen-printed electrode in a solution (c). The acid induces denaturation of the immune complex, leading to the release of BP NPs, which migrate to and interact with the electrode surface (d). Finally, detection of the IgG concentration was performed via the nanoimpact [115,116,117,118,119] of the HER electrocatalytic behavior of the released BP NPs (e). The acidic medium serves not only as a supporting electrolyte but also as a denaturing agent that facilitates the liberation of BP NPs.
The acidic solution serves not only as a supporting electrolyte but also as a denaturing agent that facilitates the release of BP NPs. The number of spikes (i.e., the impacts generated by BP NPs on the electrode surface) is proportional to the number of nanoparticles attached to the protein, which in turn corresponds to the amount of rabbit IgG. As the concentration of free rabbit IgG increases, the number of spikes decreases due to competition between free antigens and IgG–BP NP conjugates for binding to the anti-rabbit IgG-conjugated MBs, based on the high affinity of the antigen–antibody interaction. At all tested concentrations of free rabbit IgG, the number of spikes generated by the magneto-immunoassay complex is greater than that of the blank control (Figure 3B, black line). The calibration curve obtained using chronoamperometry at an applied potential of , based on the spike count within the first 40 s (Figure 3C,D), shows a linear range of 2 to with an LOD of .
2.3.1. Ruthenium Nanoparticles (Ru NPs)
One effective strategy for achieving high sensitivity in bipolar electrochemistry (BE) is to reduce the potential difference between the anodic and cathodic poles. In this regard, various electrocatalysts in the cathodic compartment have been employed to catalyze the electroreduction of oxygen [122,123], thionine [124], and protons [120,121]. Among these systems, proton reduction using noble-metal electrocatalysts occurs at more positive potentials than the others, which significantly reduces and enhances sensitivity. To this end, our group developed a sensitive visual smartphone-based electrochemiluminescence (ECL) method for PSA detection [120] and the simultaneous detection of RASSF1A and SLC5A8 tumor suppressor gene methylation in plasma from thyroid cancer patients [121]. In this approach, ultrasensitive PSA detection was achieved using ECL derived from a high loading amount of luminol encapsulated in MIL-53(Fe) (L@MIL-53(Fe) ) acting as an accelerator at the anodic pole through a sandwich immunoassay between /L@MIL-53(Fe) and [120]. Alternatively, ruthenium nanoparticles electrodeposited on nitrogen-doped graphene-coated Cu foam (fCu/N-GN/Ru NPs) were used to decrease the HER overpotential at the cathodic pole (Figure 3E).
The electrocatalytic activity of fCu/N-GN/Ru NPs toward HER at each modification step was assessed using LSV in at room temperature with a potential scan rate of . Compared with fCu ( ), fCu/N-GN exhibits higher electrocatalytic activity, with an overpotential of at , due to the reduced resistance and enhanced metallic character arising from the injection of p electrons from pyridinic nitrogen into the graphene system. Moreover, the nanostructure of Ru NPs provides additional catalytic benefits. The most impressive HER activity is observed for fCu/N-GN/Ru NPs, which display an overpotential of —corresponding to reductions of , , and relative to fCu, fCu/N-GN, and fCu/Ru NPs, respectively. In addition, a much smaller Tafel slope was obtained for fCu/N-GN/Ru NPs ( ) compared with fCu ( ), fCu/N-GN ( ), and fCu/Ru NPs ( ), indicating facilitated mass transport between the catalyst and electrolyte as well as faster electron-transfer kinetics at increasing overpotentials. To evaluate the electrochemical behavior of fCu/N-GN/Ru NPs for enhancing the ECL signal at the anodic pole by lowering the voltage required to drive the reactions (i.e., reducing ), the ECL intensity of the system was recorded using a PMT detector in the absence and presence of fCu/N-GN/Ru NPs. In the absence of the electrocatalyst, where a Cu sheet served as the cathodic pole of the BPE, luminol emission produced only a weak ECL signal. In contrast, a markedly enhanced ECL intensity—approximately 3.7-fold higher—was observed in the presence of fCu/N-GN/Ru NPs, confirming that the fCu/N-GN/Ru NPs electrode can effectively catalyze HER, facilitate electron transfer through the BPE, and enable sensitive ECL detection.
Due to the urgent demand for portable point-of-care testing, the luminol emission from the MNP/Ab_1_–PSA–Ab_2_/L@MIL-53(Fe)-modified anodic pole of the BPE was captured using a smartphone (Figure 3F). Figure 3G demonstrates a linear correlation between the ECL gray value and PSA concentration, with a detection limit of . Furthermore, our group introduced a novel method for the simultaneous detection of 5-methylcytosine (5mC) in the tumor suppressor genes RASSF1A and SLC5A8 in papillary thyroid cancer using an integrated BE–ECL platform, where the emitted light was recorded by a smartphone camera. In this system, @UiO-66-functionalized luminol was placed on the anodic pole of the BPE, and gold nanorods loaded on graphite-like carbon nitride nanosheets ( NS) were placed on the cathodic pole (Figure 3H) [121]. As a result of HER at the fCu/N-GN/Ru NPs cathode and electrooxidation of hydrazine at the PC-rGO/PGE anode, this approach not only maintains the same potential difference between poles but also enhances sensitivity by decreasing the potential difference.
2.3.2. Platinum Nanoparticles (Pt NPs)
Electrochemical immunosensors generally use either enzymatic or nanomaterial-based tags to record the signal generated by the analyte. Enzyme-linked electrochemical immunoassays (ELEIA) have shown substantial potential for detecting molecular targets in clinical diagnostics, food analysis, and other fields. In general, the response of ELEIA is measured either through the amperometric detection of electroactive species or through potentiometric monitoring of ionic products generated by enzymatic reactions. However, due to the inherent limitations of amperometric and potentiometric methods in achieving highly sensitive detection, the development of more sensitive strategies remains a major challenge. Signal amplification based on the catalytic capability of Pt to produce strong electrocatalytic currents for the HER offers a promising new approach for enhancing biosensor sensitivity. Since Pt NPs are well known as efficient electrocatalysts for HER, the presence of additional heavy metals can poison their catalytic activity. In this regard, in 2008, Huang and co-workers [125] reported a sensitive method for detecting human IgG (hIgG) based on HER inhibition through enzymatic Cu deposition on Pt NP-modified GCE in an alkaline medium containing ascorbic acid 2-phosphatase (AAP) and ions. In this system, after the target hIgG was sandwiched between the capture antibody immobilized on the microtiter plate wells and the alkaline phosphatase (ALP)-conjugated detection antibody, the enzymatic reaction of ALP generated the reducing agent ascorbic acid in glycine–NaOH buffer (pH = 10). Consequently, the reduction of ions in solution ((3a)–(3c)) resulted in the selective deposition of copper onto the Pt NP electrode (Figure 4A).
The reason for using GL–NaOH buffer is that ions do not precipitate in alkaline media and instead form a stable complex with GL. Owing to the poisoning of the Pt catalyst through hIgG-dependent enzymatic copper deposition, a clear potential shift was observed between the copper-deposited electrode and the undeveloped (copper-free) electrode (Figure 4B). The response of the immunosensor at a constant current of showed a linear relationship over the range of to with a low detection limit of (Figure 4C). In contrast to silver and organic precipitates [126,127,128], copper is chemically and selectively deposited onto Pt NPs, yielding significantly greater sensitivity. Sharma et al. [129] developed a similar strategy for detecting Staphylococcal Enterotoxin B (SEB) using Pt NP-modified GCE based on the HER inhibition approach. Anti-SEB polyclonal antibody-modified SPEs were exposed to various concentrations of SEB toxin to enable selective antigen–antibody binding. After the addition of secondary mouse anti-SEB monoclonal antibodies, the resulting immune complex was incubated with ALP-conjugated anti-mouse antibodies. Finally, the indirect sandwich enzyme-linked immunoassay (ELISA) assembly, together with the Pt NP-modified GCE, was immersed in GL–NaOH buffer (pH = 10) containing the copper deposition solution and AAP.
(A) Schematic inhibition of HER using the enzymatic Cu deposition on Pt NP-modified GCE. (B) LSV response of the immunosensor vs. concentration of hIgG and (C) related calibration curve [125]. (D) Amplified detection of the PSA based on Pt-catalyzed HER. (E) CV of the proton redox; bare Au electrode (a), thiol-modified Au electrode (b), bare Pt electrode (c), and immunosensor in the presence of 10nM PSA [130]. (F) Pt-catalyzed HER with Pt-mediated seed growth for detection of TC. (G) CV of (a) TC/Glu/CS-modified Au, modified Au in the presence of (b) 50ng/mL TC, and with (c) excess bioPtGN, (d) electrode ‘c’ after incubation with Pt developer solution. (H) LSV responses of the electrochemical immunosensors toward (a) zero analyte and (b) 1.0ng/mL TC standards using various labeling probes bio PtGN [131]. The figures were adapted from the cited references.
Under optimized conditions, the enzymatic reaction led to the hydrolysis of ascorbic acid 2-phosphate, which consequently resulted in copper deposition on the Pt NP-modified GCE according to the above mechanism. A negative potential shift in HCl was observed for SEB concentrations ranging from to , with a detection limit of . Detection of SEB in buffer samples, without the addition of external tracing labels, provided a simple and reproducible signal. In addition, the ability of Pt NPs to reduce protons and produce high HER currents contributes to increased sensitivity in field analysis. Nanoparticles have also been used as seeds to catalyze further metal precipitation to enhance signal amplification by increasing the amount of deposited material. In most cases, catalytic deposition of Au [132] or Ag [133] on colloidal gold labels has been reported. After dissolution of the deposited metal in acid, highly sensitive biomolecule detection can be achieved using stripping voltammetry. Electrocatalytic reduction of using platinum is advantageous for several reasons:
- Hydrogen undergoes reversible electrochemical reduction on the surface of platinum.
- Background electroreduction of on non-Pt electrodes is negligible because the catalytic activity of platinum is substantially higher than that of other materials.
- The diffusion coefficient of hydrogen ( ) is at least one order of magnitude higher than that of many other redox species.
- Inorganic acids are highly soluble and stable in aqueous media.
Regarding these benefits, the gap between the electrode surface and the Pt NPs resulting from the presence of antibodies causes slow electron tunneling, which reduces the measurable current. Therefore, a Pt enhancement step based on seed-mediated Pt deposition is necessary. To this end, Zhang et al. [130] proposed a prostate-specific antigen (PSA) immunosensor using Pt seed-mediated growth in a platinum developer solution containing formate, , and Tween 80 (pH 6.5, ) (Figure 4D).
As shown in Figure 4E, the platinum electrode exhibits a lower overpotential for hydrogen reduction, with a formal potential of vs. Ag/AgCl, compared with both the bare Au electrode and the thiol-modified Au electrode. The similarity between the voltammogram obtained in the presence of PSA and that of the platinum electrode indicates that the Pt NPs generated by the developer solution were active for the hydrogen evolution reaction. Pt enhancement time is one of the critical factors influencing the electrochemical response of the immunosensor. At short enhancement times (2–5 min), radial diffusion controls mass transport, and a sigmoidal steady-state voltammogram is observed because the spacing between small Pt particles is approximately ten times their diameter, causing each Pt NP to behave as an individual microelectrode. However, when enhancement time exceeds one hour, the Pt NPs completely cover the electrode surface and planar diffusion becomes dominant. The calibration curves show that increasing the enhancement time improves the sensitivity of the PSA immunosensor. The response range improved from – to – when Pt enhancement time increased from 10 to 30 min. For further signal amplification, nanoparticles can be coupled with two-dimensional materials. Recent studies show that NP-decorated 2D materials provide promising strategies for biosensing applications. Graphene, as a two-dimensional material, has attracted significant interest owing to its high electron-transfer rate, large surface-to-volume ratio, and excellent electrical conductivity. Que et al. [131] reported the synthesis of platinum/graphene nanosheets (PtGN) via a redox reaction between GO and and employed them to label tetracycline–bovine serum albumin conjugates (TC–BSA) for the electrochemical determination of tetracycline (TC) as a signal amplification strategy (Figure 4F).
To construct the competitive immunoassay, the anti-TC antibody was first covalently conjugated onto the chitosan-modified Au electrode through glutaraldehyde cross-linking. Subsequently, to determine TC residues on the anti-TC/Glu/CS/Au electrode, the immunosensor was immersed in the platinum developer solution described above. Compared with the anti-TC/Glu/CS/Au electrode in the absence and presence of TC, the TC/anti-TC/Glu/CS/Au in the presence of TC–PtGN exhibited a pair of redox peaks at and in KCl and HCl, originating from the Pt NP-based HER. Interestingly, further increases in the redox peak currents were observed after Pt-mediated growth in the platinum developer solution.
Moreover, the advantages of PtGN over the previous immunoassay were demonstrated by employing two labeling strategies: PtGN-labeled TC–BSA and Pt-labeled TC–BSA. The higher peak current of TC–PtGN compared with TC–PtNP can be attributed to the higher loading of Pt NPs and TC–BSA conjugates on the high-surface-area graphene nanosheets (Figure 4G).
When one TC–BSA conjugate binds to anti-TC, additional Pt NPs located on the same graphene nanosheet participate in the HER, resulting in enhanced peak currents (Figure 4H). The sensitivity and linear range of the immunosensor in HCl + KCl, based on the electrocatalytic properties of TC–PtGN toward platinum-catalyzed HER, showed a useful analytical range from to with a low detection limit of . Although this method allows determination of various antibiotics by adjusting the target antibodies without relying on enzyme substrates, performing measurements in aqueous environments remains challenging because Pt NPs are highly active in water and are readily influenced by external conditions.
A sensitive label-free electrochemical detection strategy for PSA using electrocatalytic Pt NPs conjugated to a recombinant scFv antibody was reported by Spain et al. [134]. While the bare gold electrode exhibited oxidation and reduction peaks at and in , the L-cysteine-modified electrode showed an increased peak current for gold oxide reduction. The increase in microscopic electrode area exposed after L-cysteine modification ( ), relative to the pristine surface ( ), was attributed to the loss of surface gold atoms induced by cysteine binding, leading to surface roughening. The presence of PSA resulted in a decrease in the gold oxide reduction peak current. Formation of the sandwich complex with scFv-labeled electrocatalytic Pt NPs generated metallic platinum at approximately through reduction of platinum oxide, and produced hydrogen adsorption/desorption features at about . In addition to the hydrogen adsorption/desorption peaks, the peak associated with platinum oxide at increased with increasing PSA concentration.
2.3.3. Gold Nanoparticles (Au NPs)
The gold electrode is generally inert toward redox reactions because of the repulsion between hydrogen and oxygen molecular orbitals and the fully occupied d-states of gold. In contrast, Au NPs behave differently owing to the large number of unsaturated atoms at their edge sites. In addition, quantum-size effects arising from the electronic structure of Au NPs, including the presence of d-orbital electrons, promote enhanced electrostatic interactions. Altogether, these features give Au NPs significant electrocatalytic properties. Their good biocompatibility and high conductivity also make them attractive for biosensor applications. To investigate the electrocatalytic properties of Au NPs toward HER, different concentrations of 20 nm Au NPs were drop-cast onto screen-printed carbon electrodes (SPCEs) and tested in HCl, where no evidence of Au NP aggregation was observed [135].
The results showed that with increasing nanoparticle concentration, the HER overpotential shifted to more positive values, and the cathodic current at increased. Studies also demonstrated that pre-oxidation of Au NPs at enhances their electrocatalytic activity toward HER. This enhancement is attributed to oxidation of surface gold atoms to Au(III) species. The coexistence of these Au(III) ions with Au NPs promotes an increase in HER electrocatalysis. SEM images collected before and after the pre-oxidation step confirmed the presence of Au NPs (Figure 5A(a)). As shown in Figure 5A(b), the electrocatalytic properties of Au NPs were used for the determination of human IgG (HIgG) based on the HER.
Streptavidin-modified MBs were used to immobilize biotinylated anti-human IgG (HIgG-B). After capture of HIgG from the sample, secondary antibodies conjugated with Au NPs were added to form the sandwich complex (Au NP–HIgG). The relationship between the HER signal recorded by chronoamperometry at and HIgG concentration exhibited a dynamic range of to with a detection limit of . In a similar sandwich-type design, anti-hepatitis B surface antigen (HBsAg) was sandwiched between tosyl-activated magnetic bead platforms and electroactive Au NP labels. Electrochemical detection was then achieved by exploiting the catalytic hydrogen evolution properties of Au NPs in acidic media without prior dissolution of the nanoparticles (Figure 5B(a)) [136].
The HBsAg-immobilized MBs (MB/HBsAg) were exposed to HBsAg IgG antibodies present in serum samples, resulting in the formation of the MB/HBsAg–HBsAg IgG complex. Subsequently, HBsAg IgG antibodies were preferentially captured by Au NP-conjugated goat polyclonal anti-human IgG (HIgG) antibodies, where the amount of HBsAg IgG was directly correlated with the quantity of Au NPs. A catalytic current was then generated by proton reduction, dependent on the amount of -HBsAg IgG, by maintaining the electrode at followed by applying (Figure 5B(b)). Muiz et al. [137] investigated and optimized a chronoamperometric Au NP measurement approach. At an applied potential of , proton reduction occurs preferentially only in the presence of Au NPs; therefore, the cathodic current recorded at a fixed time is proportional to the number of Au NPs. This strategy was successfully applied for detecting cancer cells cultured on SPEs using specific antibodies tagged with Au NPs, enabling detection of 4000 tumor cells in 700 ^−^L of suspension. A similar approach was later used to identify circulating tumor cells (CTCs) [138].
CTCs are circulating blood cells originating from primary tumors or metastatic tissues. CTC quantification is crucial for monitoring cancer progression, prognosis, and therapeutic response [139,140]. Specific membrane proteins overexpressed on cancer cells are widely exploited as recognition targets in CTC biosensing [141]. The CellSearch^®^ system is the only FDA-approved platform for CTC detection, using ferrofluidic nanoparticles conjugated with epithelial cell adhesion molecule (EpCAM) to enrich tumor cells, followed by fluorescence staining for enumeration [142,143]. However, fluorescence detection is time-consuming, costly, and equipment-dependent. To address these limitations, Costa and colleagues [138] developed an electrochemical detection strategy based on the HER activity of Au NPs, combined with anti-EpCAM-modified MBs as a cell-capture platform. EpCAM is highly expressed in colon adenocarcinoma cells (nearly 100%), making it an ideal biomarker for the Caco-2 cell line. Sandwiching Caco-2 cells between Au NP-labeled primary anti-EpCAM antibodies and secondary anti-EpCAM-modified MBs enabled electrocatalytic hydrogen reduction at in HCl by chronoamperometry, allowing quantification of labeled cancer cells (Figure 5C).
Electrochemical analysis of Caco-2 cells showed a linear range of to cells with an LOD of cells. Using MBs to preconcentrate CTCs on SPEs also enabled detection of approximately cells [144]. Due to the strong catalytic HER activity of Au NP labels, electrochemical detection of isothermally amplified Leishmania DNA was demonstrated using primers simultaneously tagged with Au NPs and MBs (Figure 5D) [145]. After amplification, the double-labeled MB/amplified DNA/Au NP complexes were magnetically separated and drop-cast onto SPCEs. Following electrochemical pretreatment at , HER currents were recorded at , enabling highly sensitive and reproducible detection down to parasites per amplification reaction and successful discrimination between infected and healthy dog blood samples. A simple and specific immune-sandwich assay based on Au NP-catalyzed HER and superparamagnetic MB platforms was also developed for rapid detection of Escherichia coli O157:H7 in meat and water [146].
(A) (a) Schematic illustration of Au NP-catalyzed HER (left) and corresponding SEM images (right); (b) magneto-sandwich immunoassay for detection of α-HIgG-B [135]. (B) (a) Scheme of the sandwich-type immunosensing of human IgG antibodies; (b) electrochemical detection based on HER [136]. (C) (a) Capture of Caco-2 cells by MBs–anti-EpCAM and antibody-conjugated Au NPs in the presence of control cells; (b) detection of labeled Caco-2 cells through HER electrocatalysis by the Au NP labels [138]. (D) Scheme of the experimental procedure for detection of isothermally amplified DNA using primers labeled with Au NPs and MBs: (a) DNA extraction from dog blood; (b) isothermal amplification of a kinetoplast-specific region via RPA using dual-labeled primers; (c) magnetic capture of the double-labeled amplified product (MB/amplified DNA/Au NP complex) on the reverse side of the SPCE working electrode [145]. (E) (a) Schematic illustration of CA 19-9 detection via Au NP-stimulated HER; (b) chronoamperograms of the developed electrochemical immuno-HER assay at different CA 19-9 concentrations [147]. (F) (a) Principle of EIS sensing via Au NP-induced HER; (b) Au NP-induced HER signal tracer for HIgG detection; (c) SEM images and Nyquist plots of Au NPs after potentiostatic cathodic polarization at (A) −0.6V, (B) −0.8V, (C) −1.0V, and (D) −1.2V [148]. The figures were adapted from the cited references.
Chronoamperometric measurements in HCl, after pre-oxidation for at and subsequent application of a negative potential of for , revealed a broad detection range of with LODs of 148, 457, and 309 CFU/mL in buffer solution, minced beef, and tap water samples, respectively. This was achieved using capture MBs conjugated with anti-E. coli O157 antibodies (MBs–pECAb) and sandwiching the E. coli O157 cells with Au NPs modified with secondary antibodies (Au NPs–sECAb). The same strategy was employed for the detection of Alzheimer’s disease (AD) biomarkers, ApoE and human cerebrospinal fluid (CSF), by using Au NP-based HER as the tracer signal and porous magnetic microspheres (PMM) as the preconcentration platform [149]. The high porosity of PMMs increases the surface area available for antibody immobilization, enhancing the catalytic activity of the captured Au NP electrocatalytic labels and enabling sensitive detection. To achieve high sensitivity in electrochemical immuno-HER assays for low-abundance targets, signal amplification remains a critical requirement.
Due to the limited number of catalytic active sites on individual Au NPs, higher loading of Au NPs in immunosensor construction can significantly enhance electrocatalytic performance by increasing the density of active sites. To this end, Ai-Li Sun [147] employed poly(amidoamine) dendrimer (PAAD) to achieve high-density decoration of Au NPs, leveraging the hundreds of potential conjugation sites available on each dendrimer. Herein, Au NP-catalyzed HER was used to quantify carbohydrate antigen 19-9 (CA 19-9) based on the sandwich immunocomplex formed between -modified SPE and –AuNP–PAAD (Figure 5E(a)). Under optimized conditions—pre-oxidation at for followed by holding at for in chronoamperometry—the immuno-HER assay exhibited a linear range from to with a detection limit of in medium. The 3D Au NP–PAAD nanostructures functioned both as nanocatalysts for HER and as platforms for assembly of the detection antibody (Figure 5E(b)). Importantly, the proposed immuno-HER assay was simple, low-cost, enzyme-free, and required no complex instrumentation, making it a versatile detection methodology. Sensitive detection of IgG based on EIS through HER induced by Au NPs was demonstrated by the Merkoçi group [148] (Figure 5E(a,b)). This EIS–HER approach was developed as an alternative to traditional EIS systems relying on redox couples such as [Fe(CN)6]^3−/4−^ or [Ru(NH_3_)6]^3+/2+^.
Since the impedance behavior of Au NPs depends on both their concentration and size, initial experiments were performed using of Au NPs in HCl. The electrodes were held at a DC potential of for (electro-oxidizing Au to ), followed by application of various DC potentials while recording impedance spectra from to . A conventional Randles circuit—including solution resistance ( ), double-layer capacitance ( ), and charge-transfer resistance ( )—was used to evaluate HER, since hydrogen adsorption on Au NPs is not apparent.
SEM images of Au NP-modified SPEs after applying different DC potentials showed that Nyquist diagrams obtained at potentials more negative than exhibited increased irregularity and reduced stability (Figure 5E(c)). Conversely, at low AC amplitudes ( and ), the impedance response was unstable. Because an AC amplitude of was too large, an AC perturbation of was selected as optimal. Under these optimized conditions, Nyquist plots recorded at varying Au NP concentrations demonstrated that decreased with increasing Au NP concentration. After establishing impedimetric detection of Au NP electrocatalysis and confirming its utility as a transduction method, the system was applied to a conventional immunoassay for protein detection. The Au NP signal was proportional to the number of formed immunoconjugates. The inverse of the measured yielded a linear biosensing response over a wide dynamic range of IgG, with an LOD of . Finally, Table 1 provides a compact comparison of HER-based electrochemical biosensors, including assay formats, electrolytes, operating conditions, readout modes, and analytical figures of merit, highlighting the versatility of HER as a signal transduction and amplification mechanism.
3. Oxygen Reduction-Based Electrochemical Detection
3.1. Principles and Mechanism of ORR
The most common element in the earth’s crust is oxygen ( ). The oxygen reduction reaction (ORR) is a key process in energy conversion devices, such as fuel cells, and in biological respiration. ORR proceeds via two main pathways: a two-electron mechanism, which converts oxygen to hydrogen peroxide, and a four-electron mechanism, which reduces oxygen directly to water. The corresponding ORR processes are summarized in Table 2 together with their thermodynamic potentials (TP) under standard conditions. The electrochemical reduction of oxygen is a complex multistep process involving several intermediates, and its behavior depends on various factors including the catalyst type, electrode material, and electrolyte composition [52,150].
The use of nanoparticles as labels in HER catalysis typically requires highly acidic conditions, introducing additional steps that increase the overall analysis time. Consequently, employing nanoparticles capable of electrocatalyzing reactions under neutral conditions can overcome this limitation. A new generation of nanoparticles has therefore been developed to catalyze naturally occurring neutral-pH processes such as the water oxidation reaction (WOR) and the ORR.
3.2. Application of ORR for Electrochemical Signal Tracing
One of the key goals in developing biosensors is to achieve high sensitivity for detecting low-abundance species. Consequently, signal amplification approaches based on nanoparticles have been widely reported in recent years. Owing to their high surface-to-volume ratio, nanoparticles can load large quantities of enzymes such as horseradish peroxidase, thereby increasing signal intensity and overall sensitivity. However, because oxygen is electrochemically generated during the electrode process, horseradish peroxidase-based electrochemical immunosensors require deoxygenation, which limits their clinical applicability, particularly in point-of-care settings [151,152]. In addition, these systems often require extra reactants to generate a measurable signal, making them less suitable for routine analysis. To address these limitations, gold or silver nanoparticles have been employed as labels, and enzymes such as glucose oxidase have also been proposed. Nonetheless, enzyme-based approaches can be adversely affected by harsh environmental conditions such as extreme pH or elevated temperatures, which may compromise enzyme bioactivity and reduce catalytic performance. The use of Au NPs typically requires chemical oxidation in [47] or electrochemical oxidation in HCl [153], followed by reduction to produce a detectable signal. Moreover, the high potentials needed for Au NP oxidation (around ) may limit specificity and selectivity. As a result, these methods are difficult to implement for routine or urgent clinical analyses. One strategy to overcome these constraints is to use enzyme-free and label-free systems in electrolytes by employing Pt NPs.
3.2.1. Pt NPs
Due to their inherent resistance to interference from reductive species, photocathodic bioassays have shown significant potential for use in real biosample detection. To develop a high-performance photocathodic immunoassay, an enhanced signal amplifier based on a platinum-based nanocatalyst with robust oxygen electroreduction capability was investigated. Typically, a nanofilm was potentiostatically deposited on an ITO electrode at a constant potential of vs. in an electrolyte solution containing Cu(NO_3_)2 and Bi(NO_3_)3 (Figure 6A) [154].
After modification of the nanofilm with Au NPs, the resulting electrode served as the anchoring matrix for capturing the CA19-9 antibody ( ). The signal antibody ( ) was conjugated with Pt NP-decorated graphene oxide nanosheets as the signal amplifier. The Pt/GR nanocatalyst efficiently promoted the electroreduction of oxygen acting as the electron acceptor in the electrolyte when the sandwich immunoreaction occurred, leading to a marked increase in the cathodic photocurrent signal. Using this enhanced signal-amplification strategy, CA19-9 was detected with high sensitivity and specificity using the anti-interference photocathodic immunoassay. As shown in Figure 6B, high loading of Pt NPs onto highly conductive graphene nanosheets (PtNDs@GS) enabled amplified ORR signals for sensitive detection of HIgG in a sandwich-type immunoassay format [155].
The electrocatalytic performance of SPCEs modified with PVP–GS (Figure 6B(b), curve a), PtNDs@GS (curve b), and PtNDs@GS-labeled AbHIgG (curve c) was evaluated in air-saturated PBS (pH 7.4) using DPV. The PtNDs@GS electrode displayed the highest current enhancement. Due to the non-conductive nature of the antibody, a slight current decrease was observed for the PtNDs@GS-labeled AbHIgG electrode. In -saturated PBS, the PtNDs@GS-labeled AbHIgG-modified SPCE (Figure 6B(b), curve d) exhibited negligible electrocatalytic current. These results indicate that the catalytic reduction of dissolved to O via a four-electron pathway on PtNDs is responsible for most of the amperometric response. Using the oxygen-reduction-based signal tracer, HIgG could be quantified without deoxygenation over the range of to .
3.2.2. PdNPs
Using Pd NP-decorated CNTs as labels for the secondary antibody, a highly sensitive sandwich immunosensor was constructed by immobilizing the capture antibody on Au NP-decorated graphene nanosheet-modified SPEs for HIgG detection, based on an efficient ORR signaling tracer (Figure 6C) [156]. The current generated from the electrochemical reduction of oxygen increased with the number of Pd NPs bound to the immunocomplex, enabling HIgG quantification over the range of to with an LOD of . The most significant advantage of this detection approach is that it neither requires a dedicated signal reporter nor demands deoxygenation of the solution. Because of their exceptional benefits—such as high detection sensitivity, spatial separation of excitation and emission, ease of controllability, low background intensity, and the absence of a need for an external excitation light source—ECL sensors have attracted significant interest among electrochemical sensing platforms.
Co-reactant systems have been extensively investigated due to their high ECL efficiency and ability to operate without potential cycling. However, these systems still have drawbacks, including the need for co-reactant transport to the emitter, short radical lifetimes, and possible interference with analyte stability. A promising strategy to reduce the distance between radicals generated from the co-reactant and the luminophore is to decrease the amount of dissolved oxygen or to embed the co-reactant within the luminophore matrix. Quantum dot-based ECL approaches using cathodic oxygen reduction have been employed to enhance ECL intensity. During cathodic potential scanning, the reduction of dissolved oxygen produces , which reacts with electron-injected quantum dots (QDs) to form excited QDs, thereby generating ECL emission. Denoting the formed radical anions as , the quantum dot-based ECL pathway in the presence of as the co-reactant is described in Equation (4).
Meanwhile, any factor that reduces oxygen to water via a four-electron process will suppress the QD ECL emission (“turn-off” effect). This principle was exploited to develop a signal amplification strategy for CEA detection by reducing the co-reactant using highly loaded poly(amidoamine) (PAMAM) dendrimer-encapsulated Pd NPs anchored on single-walled carbon nanohorns (SWNHs or SWCNHs), as shown in Figure 6D(a) [157]. The CdTe QD-modified GCE exhibited strong ECL intensity in air-saturated PBS due to the electroreduction of to . As shown in Figure 6D(b), sequential modification of the electrode with , BSA, and CEA caused a decrease in ECL intensity because the inert protein layers acted as an electron barrier and hindered mass transport of the co-reactant. The greatest decrease in ECL intensity was observed when the sandwich-type immunoassay was completed in the presence of /Pd NPs@PAMAM5/SWNH. The significantly enhanced quenching efficiency resulted from the electrocatalytic activity of the Pd NPs, which lowered the concentration of the co-reactant by promoting its four-electron reduction to water during ORR, thereby suppressing QD ECL emission.
(A) (a) Photoelectrochemical (PEC) detection of CA19-9 based on Pt/GR-catalyzed ORR as a signal amplifier, (b) photocurrent response, and (c) corresponding calibration curve [154]. (B) (a) Signal amplification using PtNDs@GS for sensitive electrochemical immunoassay, (b) DPV responses of SPCEs modified with (a) as-prepared PtNDs@GS and (b) PVP-GS in air-saturated PBS, and PtNDs@GS-labeled RaHIgG-bound SPCEs in (c) air- and (d) N2-saturated PBS [155]. (C) Schematic representation of Pd NPs-decorated CNTs as an efficient ORR signaling tracer for sensitive detection of HIgG. (D) (a) Electrocatalytic reduction of O2 at Pd NPs@PAMAM5/SWNH nanohybrids for CEA detection via an ECL annihilation strategy, (b) ECL–potential curves of QD-modified GCE in N2-saturated (a), air-saturated (b), 320mMH2O2 + N2-saturated (c), and O2-saturated (d) pH 9.0 PBS. Inset: corresponding CVs; (c) CVs of Pd NPs@PAMAM5/SWNHs (a), Pd NPs@PAMAM4/SWNHs (b), Pd NPs@PAMAM5 (c), Pd NPs/SWNHs (d), PAMAM5/SWNHs (e), and SWNHs (f) modified GCE in air-saturated pH 9.0 PBS [157]. The figures were adapted from the cited references.
In order to investigate the effect of different electroreduction pathways of , various nanomaterial-modified GCEs were examined using CV in air-saturated pH 9.0 PBS. Pd NPs@PAMAM5 (Figure 6D(c), curve d) exhibits a greater ORR current than Pd NPs/SWNHs (Figure 6D(c), curve c), indicating that electron transport is enhanced in the presence of SWNHs due to their high conductivity. In contrast, the absence of ORR current for SWNHs and PAMAM5/SWNHs (Figure 6D(c), curves e and f) suggests that SWNHs alone are unable to reduce oxygen. The highest ORR current is observed for the Pd NPs@PAMAM5/SWNHs-modified GCE (Figure 6D(c)), confirming the synergistic effect between Pd NPs, the dendrimer, and the conductive nanohorn scaffold. Pd NPs@PAMAM5/SWNHs efficiently consume the coreactant via ORR in the QD-based ECL system, suppressing the formation of the excited state and thereby quenching the ECL emission. Thus, Pd NPs@PAMAM5/SWNHs can serve as an effective signal-tracing tag in QD-based ECL immunoassays. This immunosensor not only provides a wide linear range (from to ) and high sensitivity (LOD: ) but, importantly, does not require deoxygenation during the assay.
3.3. Pt-Based Bimetallic
Although noble metal nanoparticles provide low detection limits with high sensitivity, their high cost and potential activity degradation limit their practical applications. To address these issues, alloying noble metals with 3d-transition metals offers an effective strategy to reduce cost while enhancing catalytic activity and electrical conductivity due to the synergistic effects between the bimetals [158,159,160]. Using the synergistic conductivity and catalytic properties of alloyed Pt–Ag atoms, an ultrasensitive, label-free electrochemical immunosensor based on bimetallic PtAg nanowire-catalyzed oxygen reduction was developed for the detection of alpha-fetoprotein (AFP) [161].
Exposure of chitosan-modified GCE to one-dimensional Pt–Ag nanowires (PtAgNW/CSE) in air-saturated PBS produced enhanced oxygen-reduction peak currents. The electrode behavior showed a decrease in ORR current after conjugation of anti-AFP via physico-electrostatic adsorption onto PtAgNW/CSE, followed by interaction between AFP and anti-AFP. Furthermore, no ORR peak was observed for the AFP/anti-AFP/PtAgNW/CSE electrode in -saturated PBS, confirming that these nanoparticles can be used as electrochemical tags for AFP detection. The higher current and more positive onset potential of the PtAgNW/CSE-modified GCE, compared to commercial Pt/C in -saturated at a defined rotation rate, can be attributed to the increased number of active Pt sites arising from Ag incorporation and the synergistic Pt–Ag interactions. Based on the Koutecký–Levich equation and corresponding plots at different rotation rates, the number of electrons transferred (n) was calculated as 4.01 (approximately 4), indicating that oxygen is reduced to water via a four-electron pathway. A continuous decrease in ORR current with increasing AFP concentration, within the linear range of to and with a detection limit of , highlights the promise of PtAg nanowires as highly sensitive biomarker detection platforms.
Due to the enhanced ORR signals generated by Pt–Co nanodendrites (NDs), a highly sensitive label-free electrochemical CA15-3 assay was developed (Figure 7A(a)) [162]. The ORR performance of PtCo NDs was evaluated using LSV in an -saturated electrolyte (rotation rate: 1600 rpm; scan rate: ). As illustrated in Figure 7A(b), the PtCo NDs exhibited a substantially higher onset potential than Pt/C ( versus vs. RHE). These results demonstrate the exceptional catalytic activity of PtCo NDs toward the ORR, attributed primarily to their uniform dendritic nanostructures, high specific surface area, and abundance of accessible active sites.
The analytical performance of PtCo NDs for electrocatalytic detection of CA15-3 was assessed through stepwise modification of the GCE (Figure 7A(c)). After immobilization of PtCo NDs (curve b), the peak current at increased markedly. Subsequent incubation with the antibody (curve c) and BSA (curve d) caused significant current decreases. A further decline was observed upon binding of CA15-3 (curve e), attributable to the immunocomplex creating substantial hindrance to electron transport. The electrochemical responses obtained with increasing CA15-3 concentration clearly display a “signal-off” behavior (Figure 7A), showing a logarithmic dependence on CA15-3 concentration over the range , with a low detection limit of . Using AgPt bimetallic nanoparticles decorated on an iron-porphyrinic MOF (PCN-223-Fe), an electrochemical aptasensor for ochratoxin A (OTA) has been developed (Figure 7B) [163]. The electrocatalytic activity of differently modified electrodes toward oxygen reduction was investigated by DPV in pH 6.0 oxygen-saturated PBS. The results demonstrated a pronounced synergistic effect between PCN-223-Fe and AgPt, leading to a substantial enhancement in the catalytic activity of PtAg/PCN-223-Fe/GCE toward the ORR. The biotinylated OTA aptamer-modified GCE captured streptavidin (SA)-modified AgPt/PCN-223-Fe, producing a significant ORR current. Owing to the strong affinity between the aptamer and OTA, formation of the aptamer–OTA complex caused detachment of the AgPt/PCN-223-Fe composite, resulting in a decrease in catalytic current. The proposed method exhibited a wide linear range from to and a low detection limit of .
Beyond compositional tuning, morphological control represents another critical aspect for improving nanocatalysts due to the structure–activity relationship. Metal nanoframes (NFs) have recently gained considerable attention owing to their large surface area, three-dimensional (3D) architecture, and chemical stability [164,165,166,167]. Leveraging the enhanced catalytic currents of the ORR on rhombic dodecahedral Pt NFs, a novel label-free immunosensor was established for the quantification of AFP (Figure 7C(a)) [168]. Here, solvothermally synthesized Pt NFs (Figure 7C(b)) were obtained using theobromine as a structure-directing agent and reductant, together with cetyltrimethylammonium chloride (CTAC) as a co-structure-director. The synthesis involves two main steps:
- Selective formation of Cu solid nanocrystals with a dodecahedron-like morphology directed by theobromine.
- Galvanic displacement of surface Cu atoms with platinum salt under high temperature and pressure.
ORR in -saturated KOH solution using linear sweep voltammetry (LSV) shows behavior similar to commercial Pt/C, with two distinct regions: a diffusion-limiting current region below and a mixed kinetic–diffusion-controlled region between and , indicating that Pt NFs exhibit significantly enhanced activity toward the ORR (Figure 7C(c)). The linearity of the Koutecký–Levich plots of Pt NFs at various rotation rates indicates that the total electron transfer number (n) is 4, demonstrating that most oxygen molecules are directly reduced to water via a four-electron pathway. Incorporation of Cu into Pt typically increases lattice strain, decreasing the surface affinity for and thereby improving ORR efficiency. ORR, a crucial reaction in electrochemical energy systems and an important process in human physiology, provides a promising platform for signal amplification in immunosensing.
As shown in Figure 7C, the Pt NFs/GCE exhibits the highest peak current in air-saturated PBS (pH 7.4) at (Figure 7C, curve a). In contrast, the current disappears in -saturated electrolyte (Figure 7C, curve e), confirming that the peak originates from electroreduction. Sequential electrode modification with Ab (Figure 7C, curve b), BSA (Figure 7C, curve c), and AFP (Figure 7C, curve d) results in a progressive decrease in the ORR signal due to steric hindrance and insulating effects from protein layers. The current decreases linearly over the AFP concentration range of , with a LOD of , demonstrating that the proposed immunosensor enables ultrasensitive detection of AFP. Because surfactants such as CTAB are difficult to remove completely from nanoparticle surfaces, their use in fabricating architected nanomaterials for enhanced electrocatalytic performance can negatively affect catalytic activity.
(A) (a) Detection of CA15-3 based on PtCo NDs-catalyzed O2 reduction, (b) LSV curves of PtCo NDs and Pt/C in O2-saturated 0.1mol/L KOH electrolyte, (c) Step-by-step modification of the GCE in air-saturated PBS [162]. (B) Electrochemical aptasensing of OTA using the catalytic activity of PtAg/PCN-223-Fe/GCE toward ORR [163]. (C) (a) Label-free Cu3Pt NFs-based immunosensor for AFP quantification, (b) EM images of Cu3Pt nanoflowers adapted from Ref. [168], showing (i) a low-magnification overview and (ii) a high-resolution TEM image with selected surface regions (Regions I–III) highlighted. Corresponding magnified views illustrate local structural features of the nanoflowers, (c) ORR polarization curves of Cu3Pt NFs and Pt/C catalysts in O2-saturated electrolyte, (d) DPV curves of Cu3Pt NFs (curve a), Ab/Cu3Pt NFs (curve b), BSA/Ab/Cu3Pt NFs (curve c), and AFP/BSA/Ab/Cu3Pt NFs (curve d) modified electrodes in air-saturated PBS, and Cu3Pt NFs-modified electrode (curve e) in N2-saturated PBS [168]. (D) Design of an efficient Pt/Sn–In2O3 electrochemical tracer to promote the ORR for miRNA detection [169]. (E) Streptomycin aptasensor based on amplified electrocatalytic activity of Pt–Sn@TiO2 for ORR [170]. The figures were adapted from the cited references.
Zhang et al. [169] designed an efficient Pt/Sn–In_2_O_3_ electrochemical tracer to promote the ORR for miRNA detection (Figure 7D). Due to strong steric hindrance, the immobilized hairpin structure of the biotinylated capture probe does not interact with streptavidin (SA)-functionalized Pt/Sn–In_2_O_3_. Upon hybridization of the capture probe with the target miRNA, the hairpin structure opens and the biotin group moves away from the electrode surface. Consequently, the modified electrode captures SA-functionalized Pt/Sn–In_2_O_3_, producing a significant ORR signal in -saturated solution. Using SA/Pt/Sn–In_2_O_3_ as an electrochemical tracer, a miRNA biosensor was developed with a detection limit of and a linear range from to .
Due to their large surface area, excellent stability, and ability to preserve biomolecular activity, nanoparticles are increasingly used as scaffolds in biosensors [171,172]. As reported in [171,172], the Pt–Sn@TiO_2_ nanocomposite exhibits enhanced electrocatalytic activity toward the ORR owing to the synergistic interaction between Pt and Sn (Figure 7). The composite was synthesized by decorating nanorods with Pt and Sn nanoparticles. A detection limit of 20 pM for streptomycin was achieved through exonuclease-assisted target recycling combined with Pt–Sn@TiO_2_-based signal amplification. In this approach, ORR catalysis is suppressed by dsDNA on the Pt–Sn@TiO_2_-modified electrode. In the presence of streptomycin, exonuclease digests the aptamer and the streptomycin–aptamer complex, exposing the Pt–Sn surface and restoring ORR. The use of a bimetallic Pt–Sn alloy resistant to oxidation, together with a support that provides excellent stability, is a key feature of this sensing platform.
In [173], sodium pyrrolidone-5-carboxylate (PCA–Na), an environmentally benign growth-directing agent, was used to synthesize core–shell Au@Pt nanocrystals (NCs) to enhance electrocatalytic performance by controlling nanoparticle morphology. The electrocatalytic activity of Au@Pt NCs toward the ORR, comparable to that of commercial Pt/C, makes them strong candidates for signal amplification in electrochemical biosensors. A prostate-specific antigen (PSA) immunosensor was fabricated by immobilizing PSA antibodies on Au@Pt NC-modified GCE. The ORR current decreased markedly following the specific antigen–antibody interaction. The immunocomplex hindered access of the redox probe in -saturated buffer (0.1 M PBS containing 0.15 M NaCl, pH 7.4) to the electrode surface, enabling ultrasensitive PSA detection.
3.4. Au-Based Bimetallic
Toyos-Rodríguez et al. [174] reported that bimetallic Pd–Au NPs with an optimal Pd–Au ratio exhibit outstanding electroactivity toward the ORR. Pd NPs were first synthesized in the presence of polyvinylpyrrolidone using an alcohol reducing agent, followed by selective incorporation of Au atoms through a galvanic replacement reaction (Figure 8A(a)). The effect of Au incorporation into Pd nanoclusters on ORR performance was initially evaluated using linear sweep voltammetry (LSV) on SPCEs in 10 mM PBS (pH 7.4). The results showed that the highest oxygen-reduction activity occurs when the Au content reaches approximately 30%. Notably, the onset potential of the Pd–Au NP-3 (30%) modified SPCE shifts to , accompanied by a substantially increased current. Using magnetic beads as platforms, these optimized Pd–Au NPs were subsequently employed as electrochemical labels for hyaluronidase wound infection detection (Figure 8A(b)). At an applied potential of 0.45 V, the amperometric response recorded at 5 s increases with Pd–Au NP-3 concentration due to their catalytic activity toward the ORR (Figure 8A(c,d)). The electrochemical signal increased linearly with hyaluronidase concentration from to , yielding a good linear correlation and a detection limit of 50 ng/mL.
In 2017, Wang et al. [175] introduced hollow-architecture AuAg nanocrystals (AuAg HNCs) prepared using ascorbic acid and hydrazine as reducing agents, together with polycytidylic acid (PCA) as a green growth-directing agent (Figure 8B(a)). Owing to its amine and phosphate functional groups, PCA provides strong electrostatic and chelating interactions with metallic precursors, enabling effective control over nanostructure size and morphology. Compared with Pt/C, the ORR polarization curve of AuAg HNCs exhibits markedly enhanced electrocatalytic activity—showing higher current density and more positive onset potential—in -saturated 0.1 mol/L KOH (Figure 8B(b)). This enhanced activity is attributed to the unique hollow morphology, which provides abundant active sites, as well as synergistic effects between Au and Ag.
To investigate the number of electrons involved, the ORR was recorded at different rotation rates. Based on the Koutecky–Levich (K–L) equation and the corresponding plots of current density versus the square root of the rotation rate, the number of transferred electrons was calculated to be approximately 4, demonstrating that is directly reduced to O via a four-electron pathway. Owing to the high electrocatalytic activity of AuAg NCs toward ORR, a signal-off, label-free electrochemical CA19-9 immunosensor was proposed. To this end, the performance of the AuAg NCs-modified electrode in PBS (pH 7.4) was evaluated. A sharp reduction peak in the air-saturated electrolyte (Figure 8B, curve a), in contrast to the negligible peak in -saturated PBS (Figure 8B, curve d), together with the decreased peak current after immobilization (Figure 8B, curve b) and subsequent CA19-9 binding, clearly indicates that the proposed approach enables CA19-9 detection using -reduction as the signal tracer. The improved analytical performance—reflected in a low LOD of and a wide linear range of —is comparable to or even superior to previous reports.
(A) (a) Synthesis route of polyclonal anti-hyaluronidase (HYAL)-modified Pd–Au NPs, (b) Signal amplification for sensitive detection of HYAL using Pd–Au NP-3 tags, (c) Chronoamperograms recorded at −0.45V for 50 s at different Pd–Au NP-3 concentrations, (d) Recorded currents at 5s versus Pd–Au NP-3 concentration [174]. (B) (a) AuAg HNCs-catalyzed ORR for construction of the CA19-9 immunosensor, (b) Comparison of ORR polarization curves of AuAg HNCs and Pt/C catalysts in O2, (c) DPV curves of AuAg HNCs (curve a), Ab1/AuAg HNCs (curve b), and Ab1/CA19-9/AuAg HNCs (curve c) modified electrodes in air-saturated PBS, and AuAg HNCs-modified electrode (curve d) in N2-saturated PBS [175]. (C) (a,b) Schematic diagram showing fabrication of the ECL aptasensor [176]. (D) (a) Preparation of Au/Pd-modified antibodies, (b) LSV of GCE modified with NP–antibody at 0, 15, 30, and 60 min Pd deposition (brown, blue, green, and orange lines, respectively) and at 15 min deposition after degassing (black line), (c) LSV response with increasing concentrations of TNF-α [177]. The figures were adapted from the cited references.
Zhou et al. [176] employed serum–albumin-stabilized nanoclusters ( NCs) as bipolar ECL probes for the simultaneous detection of CEA and MUC1 (Figure 8C). In this strategy, nanosheets served as a cathodic coreactant accelerator by catalyzing the reduction of , thereby enhancing cathodic ECL emission. Conversely, O@Cu functioned as an anodic coreactant accelerator, boosting anodic ECL emission by promoting the oxidation of N,N-diethylethylenediamine (DEDA). This dual-accelerator design avoids cross-reactions between luminophores and enables simultaneous detection of both biomarkers. As a result, CEA and MUC1 were detected with LODs of and , respectively.
Seed-mediated growth was employed to prepare Au/Pd core–shell nanoparticles in the presence of antibody. To obtain Au NPs-modified anti-tumor necrosis factor (TNF- ) antibody, sulfo-NHS-activated Au NPs were reacted with the antibody (Figure 8D(a)) [177]. For this strategy, each NP must act as a single functional unit to avoid antibody cross-linking and aggregation. Subsequently, Au/Pd core–shell NPs were synthesized by depositing onto the Au NP-modified antibody using ascorbic acid as the reducing agent. As shown in Figure 8D(b), different Pd deposition times yield distinct electrocatalytic behaviors toward the ORR. As expected, the Au NP/Ab-modified GCE exhibits only a small oxygen reduction wave at approximately mV (Figure 8D(b), brown) in 0.1 M PBS (pH 7.4). After 15 min of Pd deposition, the Au NP diameter increases to roughly 2–2.5 nm due to reduction, resulting in two reduction peaks at mV and mV corresponding to the two-electron reduction of oxygen to and subsequent reduction to O, respectively (Figure 8D(b), blue). With longer deposition times, the nanoparticle diameter increases to approximately 2–8 nm, producing a broad oxygen reduction peak near mV (Figure 8D(b), green and orange). The lack of further changes at prolonged deposition (Figure 8D(b), orange, 30 min) indicates full coverage of the Au core and behavior characteristic of bulk Pd. The lower ORR potential of the electrocatalytic antibody makes it more suitable for sensing applications than a non-catalytic antibody (Figure 8D(b), brown).
In conventional sandwich immunosensors, efficient electron transfer requires that the electrocatalytic NP-modified detection antibodies remain sufficiently close to the electrode surface. However, as reported in [177], immobilization of primary antibodies via EDC/NHS coupling between carboxyl groups of electropolymerized aminobenzoic acid and the amine groups of capture antibodies showed no significant response change, even at a saturated TNF- concentration ( ). Alternatively, immobilizing diazonium-modified antibodies through electrodeposition produced a porous film with unique pinhole sites ideally suited for immunosensing. Examination of sandwich immunoassays using diazonium-based capture antibody immobilization revealed that increasing TNF- concentration (from to ) led to higher ORR peak currents due to the accumulation of Au/Pd core–shell NPs. Using electrocatalyzed ORR as the readout not only eliminates the need for external reagents but also simplifies sensor fabrication (Figure 8D(c)).
3.5. QDs-Based Detection
Competitive immunoassay was constructed by immobilizing HIgG antigen on a meso-2,3-dimercaptosuccinic acid (DMSA)-stabilized CdTe QDs-modified GCE, followed by incubation with different concentrations of HIgG and HRP–anti-HIgG (Figure 9A(a)) [178]. The voltammogram of the QDs/chitosan–HIgG/HRP–anti-HIgG-modified GCE in air-saturated Tris–HCl buffer (pH 9.0) shows two reduction peaks corresponding to dissolved oxygen and DMSA–CdTe QDs at and , respectively (Figure 9A(b)). Through the reaction of -generated ORR intermediates with in (5a), an intense ECL emission from the CdTe QDs is produced. In the presence of hydroquinone (HQ), consumption of via its enzymatic reduction to water by HRP leads to a quenching effect, as described in (5b).
Here, the cyclic enzymatic strategy that consumes the self-produced co-reactant, generated by the reduction of dissolved oxygen, results in highly sensitive detection of HIgG in the competitive format, with a low LOD of over a dynamic range of to . It is worth noting that, as the quantity of antigen increases, smaller amounts of antibody are deposited on the surface, leading to reduced catalytic activity of the enzyme and, consequently, an increased signal.
The ORR has been used to quantify biomarkers using high-resolution, ultrasensitive ECL. In these reactions, consumption of the ECL co-reactant (i.e., reduction) leads to quenching of the ECL emission. Hemin-functionalized nitrogen-doped graphene-catalyzed ORR has been introduced for ultrasensitive detection of CEA through ECL quenching of CdTe quantum dots (Figure 9B(a)) [179]. While the cathodic light emission of the CdTe QDs-modified GCE in air-saturated PBS (0.1 M, pH 8.0) is prominent due to oxygen electroreduction to the co-reactant (Figure 9B(b), curve a), a sequential decrease in emission is observed for QDs-modified GCEs subsequently modified with (curve b), NG (curve c), HRP (curve d), hemin@GO (curve e), and hemin@NG-labeled (curve f) in the presence of 10 ng/mL antigen. The decrease in ECL emission for the HRP-modified electrode is attributed to steric hindrance, whereas the reduced ECL intensities for the carbon-based nanomaterial electrodes arise from enhanced ECL sensitization in the presence of these nanomaterials. To clarify the mechanism, the electroreduction of dissolved oxygen was examined in air-saturated PBS using CV. As shown in Figure 9B(c), the highest electrocatalytic activity toward reduction of dissolved oxygen to O or is observed for the sandwich-type immunoreaction using the hemin@NG-modified electrode. This arises from the increased hemin loading on NG and from electron donation by nitrogen atoms in NG, which enhances hemin adsorption and facilitates reduction by electrocatalysis [180,181].
In comparison to the hemin-modified GCE, which exhibits negligible current in -saturated solution (Figure 9B(d), curve a), the reduced hemin can be efficiently oxidized by dissolved oxygen under basic conditions, converting into and producing an increased reduction peak (Figure 9B(d), curve b). As expected, a substantially larger reduction peak for hemin in -saturated electrolyte (Figure 9B(d), curve c), along with a strong electroreduction response toward in air-saturated solution, is observed on the hemin@NG-modified GCE (Figure 9B(d), curve d). Based on these results, the electrochemical processes can be described by the equations in (6).
Therefore, during the ORR and its conversion to (6a), the formation of as the ECL co-reactant is inhibited, leading to quenched ECL emission. As a result, the hemin@NG signal tag is highly effective for the ECL immunoassay. Under optimized conditions, the ECL intensity of the immunosensor decreased with increasing CEA concentration over the range of to . The detection limit of is approximately 20-fold lower than those reported for other amplified electrochemical/ECL approaches [157,182].
QDs-based ECL systems can also be enhanced using as the co-reactant generated from the ORR. To further increase QD-based ECL efficiency, functionalized nanomaterials can be used as catalytic substrates to accelerate emission through enzyme-mimicking activity. Delocalization of nitrogen lone-pair electrons in heteroatom-doped nanotubes can change the chemisorption mode of from end-on monoatomic adsorption to side-on diatomic adsorption, effectively weakening the O–O bond and facilitating its reduction to the superoxide intermediate in the ORR [180].
Polystyrene sulfonate (PSS)-functionalized nitrogen-doped carbon nanotubes (NCNTs) have been introduced as labels for secondary antibodies ( ) (Figure 9C(a)), serving as a new ECL signal tag to amplify CdS QD-based ECL in a sandwich immunocomplex formed on an -modified GCE. In this strategy, no strong oxidant is required as a co-reactant, since NCNT-catalyzed generation of from dissolved provides an efficient ECL pathway [183]. Unlike signal-off approaches, the signal-on ECL immunosensing configuration avoids false positives that typically arise from steric hindrance associated with immunocomplex formation. The high reduction current and corresponding strong ECL intensity of the CdS-modified GCE in -saturated solution (Figure 9C(b,d), curve d), compared with -saturated PBS + (curve c), demonstrate that dissolved is a more effective co-reactant for enhancing QD-based ECL emission. These results also confirm that is not produced from the reduction of via a two-electron pathway.
(A) (a) Enzymatic amplification through consumption of H2O2. (b) CV curve of GCE/QDs/chitosan–HIgG/HRP–anti-HIgG in air-saturated buffer [178]. (B) (a) QD-based ECL immunoassay using hemin@NG as a signal tracer. (b) ECL responses of the immunosensor in air-saturated 0.1M pH 8.0 PBS: (a) before and after incubation with 10ng/mL CEA, followed by modification with (b) Ab2, (c) NG-labeled Ab2, (d) HRP-labeled Ab2, (e) hemin@GO-labeled Ab2, and (f) hemin@NG-labeled Ab2. Inset: ECL behaviors of (g) GO- and (h) NG-modified GCEs. (c) CVs of the immunosensor in air-saturated 0.1M pH 8.0 PBS after incubation with 10ng/mL CEA and subsequent modification with (a) HRP-, (b) hemin@GO-, (c) hemin@NG-, and (d) NG-labeled Ab2. (d) CVs of (a,b) hemin- and (c,d) hemin@NG-modified GCEs in (a,c) N2-saturated and (b,d) air-saturated 0.1M pH 8.0 PBS [179]. (C) (a) NCNT-catalyzed reduction of O2 for enhanced ECL emission. (b) CVs and (c) ECL curves of CdS QDs-modified GCE in (a) N2-saturated, (b) air-saturated, (c) 320mMH2O2 + N2-saturated, (d) O2-saturated, (e) 1mM L-cysteine + air-saturated, (f) 0.2mg/mL SOD + air-saturated, and (g) 0.1M pH 8.0 PBS [183]. The figures were adapted from the cited references.
Introducing L-cysteine (Figure 9 curve e), a scavenger of both OH and superoxide radicals, and superoxide dismutase (SOD) (Figure 9 curve f) led to a decrease in the ECL responses. These results confirmed that the ECL intensity originated from species generated by dissolved . Additional electron paramagnetic resonance (EPR) experiments also verified the formation of on the CdS-modified GCE. Because of the nonconductive characteristics of BSA, , and CEA on the CdS/chitosan-modified electrode, the ECL intensity was considerably reduced. Interestingly, a significant increase in ECL intensity was observed in the presence of –PNCNTs. This enhancement arose from the high conversion efficiency of dissolved to by the PNCNTs. The signal-on ECL response increased with the CEA concentration over a wide range ( to ), with a LOD of , demonstrating that the immunoassay possesses excellent practical applicability in clinical diagnosis without the need for deoxygenation.
3.6. Ir Based Single Atom Catalysts (SACs)
Self-powered sensing systems (SPSSs) refer to devices or systems capable of generating and storing the necessary energy to perform sensing functions without relying on external power supplies. SPSSs have been extensively studied in biotechnology as innovative detection platforms. Nevertheless, creating a dependable and long-lasting power source for SPSSs—which must harvest energy from surrounding media such as sunlight, water waves, and air currents—remains a significant challenge. In particular, numerous bioelectrochemical fuel cells have been developed for SPSSs, but their low open-circuit potentials are not suitable for accurate detection [184,185].
Several Ir-based single-atom catalysts (Ir-SACs) have been identified as highly efficient catalysts for the ORR, enabling sensitive analyte detection in SPSSs with stable output signals. The Zhu group successfully developed a hierarchically porous Ir-SAC catalyst that exhibits exceptional ORR performance and remarkable long-term durability in neutral electrolytes. Density functional theory (DFT) calculations revealed that Ir-SAC possesses moderate adsorption energies for ORR intermediates, facilitating the four-electron pathway. Notably, this work demonstrated the feasibility of integrating SPSSs with glucose oxidase and Ir-SAC for the accurate detection of glucose [185].
They also synthesized a nanozymatic biofuel cell with SPSS functionality, composed of Ir-SAC as the cathode and Au nanozymes as the anode. This biofuel cell exhibited exceptional sensitivity and specificity toward prostate-specific antigen detection. Experimental studies confirmed that a single iridium atom catalyst shows outstanding catalytic activity for the ORR under neutral pH conditions. When combined with Au nanozymes at the anode, the Ir-SAC biofuel cell outperformed Pt/C-based biofuel cells. This highly active nanozymatic biofuel cell was therefore employed for the ultrasensitive detection of prostate-specific antigen in an SPSS configuration [184]. Finally, Table 3 summarizes ORR-based biosensors that exploit noble-metal nanoparticle electrocatalysis to generate current or ECL signals directly from dissolved under air-saturated conditions, eliminating the need for deoxygenation or external reporters. These platforms achieve low pg/mL sensitivity and broad dynamic ranges, demonstrating the effectiveness of ORR as a robust signal-tracing and amplification mechanism in electrochemical and ECL biosensing.
4. Water Splitting-Based Electrochemical Detection
4.1. Principles and Mechanism of Water Splitting
Water electrolysis, or electrochemical water splitting, is used as an alternative, clean, and sustainable strategy to replace fossil fuels for the production of hydrogen and oxygen. This process involves the combination of the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER), as shown in (7) [186,187].
The possibility of electrochemically detecting various biomarkers without the use of acidic solutions as a proton source for HER or relying on stripping voltammetric detection of completely dissolved nanoparticles is particularly appealing. In addition to the obvious safety concerns, the use of strong acidic media introduces an extra step that not only lengthens the analytical workflow but also poses a substantial obstacle for the development of integrated sensing systems, especially those based on lab-on-a-chip or lateral-flow platforms. A further drawback of HER-based biosensing devices is the formation of hydrogen bubbles during the reaction, which can destabilize the signal and impair assay reproducibility. These limitations highlight the need for novel nanoparticle tags that can be readily detected in immunoreaction media—typically saline buffers with neutral pH. In this context, nanoparticles capable of catalyzing the water oxidation reaction represent a promising solution.
4.2. Application of Water-Splitting for Electrochemical Signal Tracing
4.2.1. IrO2 NPs
In the investigation reported by Merkoçi’s group, chronoamperometry and cyclic voltammetry were employed to examine the electrocatalytic activity of NPs in neutral media toward the water oxidation reaction [188]. The voltammetric results demonstrate that as the concentration of NPs increases, the overpotential decreases and the corresponding currents increase. The amount of NPs on the SPE transducer correlates with the magnitude of the current recorded in chronoamperometric mode when an appropriate oxidative potential is applied. For ApoE detection—a recognized biomarker for Alzheimer’s disease—the researchers first coated magnetic beads with anti-ApoE antibodies and subsequently introduced NPs conjugated with secondary anti-ApoE antibodies. The same group later applied an immunoassay based on the same principles to identify a flame-retardant compound, BDE-47 (a polybrominated diphenyl ether, PBDE), achieving detection levels in the ppb range, which meet environmental monitoring requirements [189].
4.2.2. Au NPs
The detection of microRNA was demonstrated using a cascade amplification strategy that combined electrocatalytic water-splitting as a signal tracer with duplex-specific nuclease (DSN)-assisted target recycling. The DNA probe was self-assembled onto Au NPs through Au–S chemistry (Figure 10A(a,b)) [190]. In the presence of the microRNA target, the resulting DNA:RNA duplex is specifically hydrolyzed by DSN. During this enzymatic process, the DNA strand is cleaved while the RNA strand remains intact. The surviving RNA target then hybridizes with additional probe DNA strands on the nanoparticle surface, enabling continuous probe digestion and thereby exposing an increasing portion of the Au NP surface. The centrifuge-collected Au NPs were subsequently mixed with graphene oxide (GO) and spin-coated onto the ITO electrode, after which the cathodic amperometric water-splitting reaction was recorded at vs. Ag/AgCl in PBS (1 M, pH 7.4) for . The three-dimensional architecture formed with GO enhances the loading of Au NPs and thus amplifies the signal.
A larger exposed Au NP surface provides more active sites for hydrogen generation, markedly increasing the corresponding current. Water molecules are reduced to and adsorbed hydrogen ( ) intermediates on the Au NP surface, and subsequently recombines to produce on the Au NP/GO-modified electrode at the applied potential. The electrocatalytic activity of the Au NP–probe/GO–ITO electrode is lower than that of the Au /GO–ITO electrode (Figure 10A(c)), confirming successful immobilization of the non-conductive DNA probe layer. Upon microRNA hybridization and DSN-assisted recycling, increasingly larger Au NP surface areas become exposed, thereby initiating more efficient cathodic water-splitting catalysis as the probe layer is hydrolyzed. With target microRNA concentrations ranging from to , the catalytic current progressively increases. Owing to the water-splitting electrocatalytic signal and the recycling amplification mechanism, the proposed Au NP/GO-based biosensor achieves a low LOD of , outperforming other DSN-based microRNA detection approaches that rely on a single amplification step.
4.2.3. Cu(II)-Based Complexs
Caspase-3 is regarded as an essential biological biomarker and therapeutic target in the diagnosis and prognosis of apoptosis-related diseases. In this context, a specific caspase-3 peptide substrate (Ac-GDEVDSK(FFFF)H) is anchored onto a graphene sheet through hydrophobic and – interactions [191]. When caspase-3 cleaves the peptide between the D and S residues, the Ac-GDEVD fragment dissociates from the electrode surface, exposing an ATCUN motif (SKH). Consequently, the remaining peptide segment on the electrode is capable of forming a complex with Cu(II), which enhances electrocatalytic water splitting. Unlike existing electrochemical approaches for caspase-3 detection, this signal-on strategy offers a higher signal-to-background ratio and does not require peptide labeling with an electrochemical tracer. Moreover, the close proximity of the ATCUN motif to graphene facilitates electron transfer between Cu(II) and the electrode.
This method enables caspase-3 analysis over a linear range of to with a detection limit of . Proteases are enzymes that catalyze peptide bond cleavage, producing shorter peptide fragments. Numerous metal ions are known to bind specific peptide sequences, and protease-mediated cleavage can therefore modulate peptide–metal interactions. Liu’s group investigated the redox properties of various ATCUN–Cu(II) complexes and demonstrated that, under neutral pH conditions, these metallopeptides exhibit strong electrocatalytic activity toward water oxidation. Building on this finding, they developed an immunosensor in which an /protease-modified carbon nanotube (CNT) catalyzes the formation of ATCUN–Cu(II) complexes for water oxidation [192]. In the presence of PSA as a model analyte, the peptide substrate GARGGH is cleaved by trypsin, releasing an ATCUN-containing fragment (GGH) capable of forming a copper complex that catalyzes the water-splitting reaction. PSA concentrations as low as were detected based on the water electrooxidation activity of the generated ATCUN–Cu(II) complexes.
4.2.4. CoP-Nanowires (CoP-NWs)
The photochemical driving force has been employed as a cost-effective and environmentally sustainable energy source for the sequence-specific detection of DNA using water splitting as an electrochemical tracer [193]. Unlike fluorescence resonance energy transfer (FRET), which requires an external light source and the labeling of sequences with donor–acceptor pairs, photochemical water splitting eliminates these requirements. This feature simplifies and automates the measurement system. Based on the photocatalytic hydrogen generation ability of CoP nanowires (CoP-NWs), the non-covalent attachment of a fluorescein-based dye (FAM)-modified capture probe HIV (PHIV) onto negatively charged CoP-NWs was used for HIV detection.
Upon illumination, an anodic photocurrent was generated by fluorine-doped tin oxide (FTO) modified with CoP-NWs (Figure 10B(a), curve a), indicating that photogenerated electrons were transferred from the n-type CoP-NWs to the FTO electrode. Because the lowest unoccupied molecular orbital (LUMO) level of FAM is more negative than the conduction band (CB) edge of CoP, electron injection from photoexcited FAM into CoP occurs via photoinduced electron transfer (PET), resulting in an enhanced photocurrent on the PHIV/CoP-modified FTO (Figure 10B(a), curve b). The increased photochemical hydrogen evolution under visible light for PHIV/CoP hybrids, compared with CoP-NW- modified FTO in the presence of triethanolamine (TEOA) as a sacrificial agent, further confirms that high electron accumulation in the CB of CoP-NWs originates from PET from the excited dye’s LUMO (Figure 10B(b)).
With increasing concentrations of the complementary HIV target, the affinity of CoP-NWs toward the resulting double-stranded DNA (dsDNA) decreases due to the absence of mismatched nucleobases. Consequently, electron accumulation in CoP-NWs is reduced because electron transfer from FAM to the CB of CoP-NWs is hindered. This process leads to a decrease in photocatalytic generation from water as the target concentration increases (Figure 10B(c)). The good electrical conductivity of CoP-NWs, combined with the strong photocatalytic generation in the absence of any electrochemical signal tracer, makes this strategy highly suitable for sensitive point-of-care detection.
(A) (a) DSN-assisted target recycling in the presence of Au NPs, (b) Electrocatalytic detection using an Au-NP/GO-modified electrode, (c) Water-splitting reactions of differently modified electrodes [190]. (B) (a) H2 generation under visible light (λ>420nm) with (a) CoP and (b) PHIV/CoP hybrids, (b) Photocurrent of (a) CoP-NWs and (b) PHIV/CoP hybrids measured at a bias of 0.0V vs. Ag/AgCl, (c) Mechanism of H2 generation using the PHIV/CoP hybrid [193]. (C) (a) Sweatband glucose sensing, (b) Pd@ZIF-67-catalyzed water-splitting electrocatalysis (WSE) for glucose sensing, (c) Integration of the sweatband with Pd@ZIF-67, (d) Glucose sensing on the sweatband, (e) Smartphone-based perspiration analysis, (f) Monitoring of glucose concentration in perspiration and blood from a human subject over 10 days [194]. The figures were adapted from the cited references.
4.2.5. Pd-Based NPs
Wearable perspiration glucose sensing is being developed using an enzyme-free electrochemical sensor based on a water-splitting signal. Unlike previously reported nonenzymatic glucose sensors that detect glucose in alkaline buffers, Zhu et al. [194] proposed a glucose sensor operating at physiological pH without the need for additional reagents, enabling long-term, wearable, and maintenance-free glucose monitoring. In this work, a screen-printed Pd nanoparticle-encapsulated Co-based zeolitic imidazolate framework (Pd@ZIF-67) carbon conductive ink on a flexible PET substrate was employed for highly sensitive and reproducible detection of glucose in perspiration using a water-splitting-assisted electrocatalytic (WSE) reaction (Figure 10C). In this regard, a multistep potential protocol versus PVA/KCl–Ag/AgCl was applied to the electrode, including a high negative potential ( ) to generate localized alkalinity via electrochemical water splitting, a moderate potential ( ) to oxidize glucose through the conversion of [Co(III)(mim)2(OH)]n/[Co(IV)(mim)2(OH)2]n, and a highly positive potential ( ) to regenerate the electrode:
Application of a high negative potential generates hydrogen bubbles, which can introduce uncertainty into glucose measurement. In this study, this challenge was mitigated using Pd@ZIF-67: the “spillover” characteristics of Pd NPs embedded in ZIF-67 catalytically dissociate the generated into monatomic hydrogen, which subsequently combines with the Pd matrix [195,196,197]. The wearable sweatband incorporating Pd@ZIF-67 enabled nonenzymatic glucose detection in perspiration with an acceptable shelf life (2 months under ambient conditions), a linear response from to , and a limit of detection of .
Table 4 provides a comparative overview of water-splitting and -based electrochemical biosensors, summarizing key experimental conditions, signal readout modes, and reported analytical performance.
While the preceding sections discuss representative case studies for HER-, ORR-, and water-splitting-based biosensing strategies, cross-comparison of analytical performance across different signal transduction approaches remains challenging due to variations in assay formats and experimental conditions. To provide quantitative context, it is therefore instructive to benchmark energy-based electrocatalytic biosensing strategies against conventional redox-label and enzyme-based electrochemical techniques at the order-of-magnitude level.
Accordingly, Table 5 summarizes the overall ranges of LOD and linear/dynamic ranges reported for energy-based electrocatalytic biosensing strategies and compares them with traditional redox-label and enzyme-based electrochemical techniques. The reported values are extracted from representative recent studies and reviews and are intended to reflect typical order-of-magnitude performance observed across different analytes, matrices, and assay formats, rather than direct head-to-head experimental comparisons. Overall, this comparison indicates that energy-based electrocatalytic biosensing strategies can achieve sensitivities and dynamic ranges comparable, at the order-of-magnitude level, to conventional redox-label and enzyme-based electrochemical techniques, while offering additional advantages such as reagentless operation and simplified assay design.
5. Conclusions
This study’s main purpose is to provide a comprehensive, authoritative, and essential resource for researchers on the interdisciplinary between energy and biosensors. Research progress into the desire for new signal recording pathways using the versatility of the HER/ORR and water splitting during the last few decades is highlighted. Current challenges in the field of biosensors are presented together with strategies to overcome these obstacles; one strategy is using energy-to-biosensor conversion. Another goal of this study is to bridge the gap between biosensors and commercialization, which is currently one of the “hottest” challenges in the field of POC bioassays. As indicated in this review, the increase in the introduction of new materials for recording the signal in only supporting electrolytes without any electroactive labels has been particularly encouraging to date.
The field of HER/ORR/water splitting-based materials for signal recording in reagentless and electrochemical redox reporter-free biosensors is very promising. However, there are several challenges and unanswered questions that require further exploration in future research. It is essential to continue investigating and addressing these issues to fully realize the potential of these materials in biosensor applications.
In particular, the reproducibility of catalytic biosensors—especially those employing nanoparticle labels—remains a critical issue, as batch-to-batch variability and surface heterogeneity can significantly affect signal consistency and hinder reliable translation. Second, using both crystalline and amorphous structures to capture signals has proven to be an effective method for achieving exceptional material performance. The enhanced catalytic activity and strengthening effect stem from the distinctive atomic arrangements. Hence, it is crucial to design nanomaterials with unique structures for the use of HER/ORR/water splitting to comprehend the relationship between the structure and properties of nanomaterials. However, conducting a precise structural analysis is challenging because of the difficulty in accurately determining the atomic arrangement in both amorphous and crystalline structures, as well as at phase interfaces. A second factor influencing the durability of nanostructures is their thermal stability, which has hardly been investigated in engineering applications. In fact, the applications of these nanoparticles at human body temperature and above to fight diseases should be investigated. Also, creating flexible and stretchable biosensors may appear to be a viable solution, but it may not be feasible for devices that require a steady power source for ongoing monitoring of physiological functions. In this context, generating power from the dynamic micromotions of implanted tissues and the human body is highly desirable for sustaining device functionality. However, substantial progress is needed to obtain sufficient energy to power soft functional devices. Finally, seeking nanoparticles that can function properly in biological pH conditions is another challenge that should be considered.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mayer K.M. Hafner J.H. Localized surface plasmon resonance sensors Chem. Rev.20111113828385710.1021/cr 100313 v 21648956 · doi ↗ · pubmed ↗
- 2Hou W. Cronin S.B. A review of surface plasmon resonance-enhanced photocatalysis Adv. Funct. Mater.2013231612161910.1002/adfm.201202148 · doi ↗
- 3Homola J. Present and future of surface plasmon resonance biosensors Anal. Bioanal. Chem.200337752853910.1007/s 00216-003-2101-012879189 · doi ↗ · pubmed ↗
- 4Wang Q. Ren Z.-H. Zhao W.-M. Wang L. Yan X. Zhu A.-S. Qiu F.-M. Zhang K.-K. Research advances on surface plasmon resonance biosensors Nanoscale 2022145645913494076610.1039/d 1nr 05400 g · doi ↗ · pubmed ↗
- 5Azzouz A. Hejji L. Kim K.-H. Kukkar D. Souhail B. Bhardwaj N. Brown R.J.C. Zhang W. Advances in surface plasmon resonance–based biosensor technologies for cancer biomarker detection Biosens. Bioelectron.202219711376710.1016/j.bios.2021.11376734768064 · doi ↗ · pubmed ↗
- 6Chen T. Xin J. Chang S.J. Chen C.-J. Liu J.-T. Surface plasmon resonance (SPR) combined technology: A powerful tool for investigating interface phenomena Adv. Mater. Interfaces 202310220220210.1002/admi.202202202 · doi ↗
- 7Khoshfetrat S.M. Khodadadian A. Mirsian S. Hilber W. Heitzinger C. The marriage between energy and biosensor Chem Rxiv 2025 preprint 10.26434/chemrxiv-2025-2b 065 · doi ↗
- 8Lim H.J. Saha T. Tey B.T. Tan W.S. Ooi C.W. Quartz crystal microbalance-based biosensors as rapid diagnostic devices for infectious diseases Biosens. Bioelectron.202016811251310.1016/j.bios.2020.11251332889395 PMC 7443316 · doi ↗ · pubmed ↗