The Fires of Isengard Have Spread: Serratia sarumanii Is the Dominant Species in Clinical Isolates of the “Serratia marcescens Complex”
Levin Joe Klages, Julia Hassa, Tobias Busche, Olaf Kaup, Christiane Scherer, Claudia Christine Freytag, Thorsten Kaiser, Jörn Kalinowski, Christian Rückert-Reed

TL;DR
This study shows that Serratia sarumanii, not Serratia marcescens, is the most common species in clinical isolates of the Serratia marcescens complex.
Contribution
The paper identifies S. sarumanii as the dominant clinical species in the S. marcescens complex using genome sequencing and global genomic data.
Findings
Out of 21 isolates from hospitals in OWL, 10 were identified as S. sarumanii.
Approximately one-third of Serratia genomes in GenBank were reclassified as S. sarumanii.
S. sarumanii is the most dominant Serratia species in clinical settings globally.
Abstract
Recently, a new species, Serratia sarumanii, was described, belonging to a group of strains previously identified as Serratia marcescens in routine clinical analyses. It was shown that the identification of S. marcescens isolates by biochemical testing, mass spectrometry, or 16S rRNA gene sequencing was insufficient to resolve the ‘S. marcescens complex’, while sampling point analysis revealed that many genomes assigned to the S. sarumanii cluster were associated with a clinical context. Thus, here the clinical relevance and local as well as global distribution of S. sarumanii is analyzed. In total, 21 strains from three hospitals in Eastern Westphalia-Lippe (OWL), previously identified as S. marcescens and potential causative agents from severe bacterial infections, were analyzed by genome sequencing and species identification. It could be shown that only one isolate was confirmed as…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
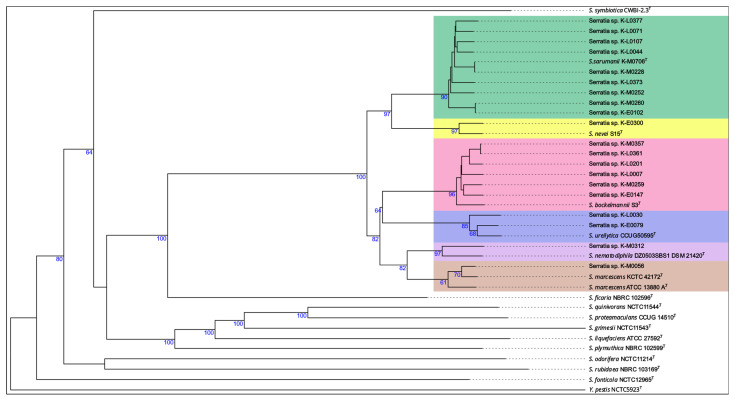 Figure 1
Figure 1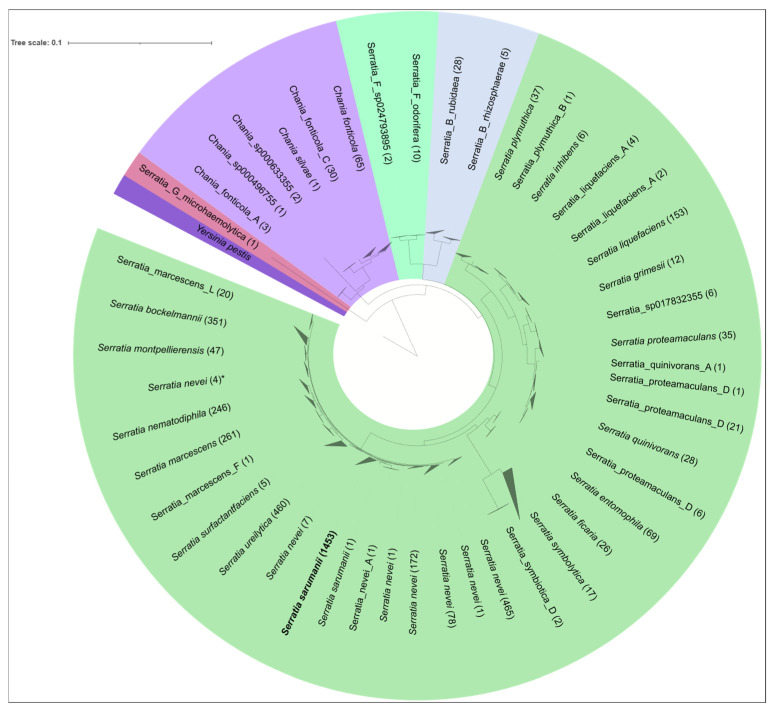 Figure 2
Figure 2- —German Federal Ministry of Health
- —German Research Foundation (DFG)
- —Open Access Publication Fund of Bielefeld University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Resistance in Staphylococcus · Antibiotic Resistance in Bacteria · Antibiotics Pharmacokinetics and Efficacy
1. Introduction
The genus Serratia, belonging to the order Enterobacterales, to date includes 24 species of Gram-negative, facultative anaerobic, rod-shaped bacteria [1,2,3]. The type strain of the genus is Serratia marcescens, which can cause infections in humans, animals, and insects [4]. S. marcescens is also well known for its red colony phenotype, caused by the secondary metabolite and pigment prodigiosin [1,5]. Interestingly, S. marcescens was used from the 1940s to the mid-1960s by the U.S. government as a tracer in medical experiments and biological warfare test agents before its pathogenicity was known [4]. Serratia spp. cause a wide range of asymptomatic and symptomatic infections in humans, such as respiratory, urinary, surgical wound, and bloodstream infections, as well as meningitis and keratitis [3,6].
The clinical identification of such pathogens usually relies on phenotypical identification, biochemical assays, or mass spectrometry (MS) [7,8,9]. Therefore, mass spectrometry platforms like MALDI Biotyper (Bruker Daltonics) and biochemical analysis platforms like VITEK-MS (bioMérieux) are registered for clinical uses [7]. However, the clinical identification via VITEK2 and even MALDI-TOF-MS is not always sufficient for the correct species identification of certain clinically relevant pathogens, e.g., Serratia sarumanii [10]. As a result, S. marcescens was grouped in the ‘S. marcescens complex’ (SMC) together with closely related Serratia species, such as S. nematophilia and S. ureilytica [11] as well as S. bockelmannii and S. nevei [12]. The SMC can, currently, only be reliably subdivided with whole genome-based methods.
In our previous publication, we described the discovery of the new species Serratia sarumanii [10]. The species was previously classified as ‘S. marcescens’ and is therefore also part of the SMC. Furthermore, it has been shown that most strains in the Genome Taxonomy Database (GTDB) that were assigned to the new species originate from a clinical setting, raising the questions of Serratia sarumanii’s role in the SMC, both regionally and globally. To address these questions, Serratia isolates collected in three hospitals in Eastern Westphalia-Lippe (OWL) were analyzed using overall genome-relatedness indices as well as marker gene-based phylogenies. The approach was then expanded to reclassify all genomes submitted as Serratia species in the NCBI nucleotide database.
2. Materials and Methods
2.1. Clinical Sample Collection, DNA Isolation, and Initial Species Identification
A total of 21 samples were collected from patients in the context of diagnosed sepsis cases at three University Medical Center OWL hospitals: Klinikum Bielefeld (KBM), Evangelisches Klinikum Bethel (EvKB), and Klinikum Lippe (KL). The sampling was performed as standard clinical routine directly from blood, wound swabs, peritoneal wash fluent, or urine culture.
The cultivation and sample preparation of the eight samples originating from the hospital KBM were performed as described in a recent publication by Klages et al. [10].
All other samples were cultivated in a liquid blood culture medium at 37 °C. After the cultivation in blood culture, the samples were further grown on different solid media, and single colonies were selected for further analysis. Species identification of the cultivated microorganisms was performed in the hospital KBM biochemically on the VITEK2 system (bioMérieux, Marcy-l’Étoile, France) with the Gram-negative identification card (GN) and at the two other hospitals additionally via mass spectrometry on the MALDI Biotyper (Bruker Daltonics, Bremen, Germany).
For MALDI-TOF analysis, the α-cyano-4-hydroxycinnamic acid matrix was dissolved in 50% acetonitrile and 2.5% trifluoroacetic acid (TFA). For the measurement, 1 μL each of the sample extract, formic acid, and the prepared matrix solution was consecutively pipetted onto a steel target plate (Bruker Daltonics, Bremen, Germany) and allowed to dry before applying the next solution. The target plate was then placed into the MALDI Microflex LT. The results were processed with the MBT compass IVD (Bruker Daltonics, Bremen, Germany).
Subsequently, at KBM, DNA extraction was performed using the GenoXtract device (HAIN Lifescience GmbH (now Bruker Daltonics, Bremen, Germany)), applying the ‘GXT NA Extraction as described in the recent publication by Klages et al. [10].
Genomic DNA isolation at KL and EvKB was performed using the Maxwell RSC Cultured Cells DNA Kit on the Maxwell RSC device (Promega, Madison, WI, USA), according to the manufacturer’s protocol.
2.2. Whole Genome Sequencing and Genome Assembly
Nanopore sequencing (Oxford Nanopore Technologies, ONT, Oxford, UK) was performed as described by Klages et al. [10]. Briefly, the DNA obtained from the cultivated organisms was used for sequencing library preparation, applying the ONT SQK-LSK112 kit with the native barcodes SQK-NBD112.24, according to the manufacturer’s protocol. The libraries were sequenced on an R10.4.1 flow cell using the GridION sequencing platform, and the resulting sequences were subsequently basecalled with either GUPPY v6.2.11, v6.2.8, or v6.3.7 in super high accuracy (SUP) mode. To obtain the whole genome sequences, the raw sequencing data were assembled using FLYE v2.9-b1768 [13] and manually curated using Bandage v. 0.8.1 [14] to obtain complete genome assemblies. The genomes were annotated using PGAP 2025-05-06.build7983 [15] and are available via BioProject PRJNA1274668 (National Center for Biotechnology Information, NCBI, Bethesda, MD, USA).
2.3. Overall Genome-Relatedness Indices
The genome sequences were uploaded to the TYpe strain Genome Server (TYGS, https://tygs.dsmz.de, 14 August 2023) for a whole genome dDDH-based taxonomic analysis [16,17]. Information on nomenclature, synonymy, and the associated taxonomic literature was provided by TYGS’s sister database, the List of Prokaryotic names with Standing in Nomenclature (LPSN, https://lpsn.dsmz.de, 14 August 2023) [17,18].
For the phylogenetic analysis, all genomes of Serratia species were downloaded from the National Center for Biotechnology Information (NCBI) repository [accessed at 29 January 2025] and used for a comprehensive genome-wide comparison with GTDB-tk v.2.4.0 [19] and GTDB [20] release 226, applying the workflows classify_wf and de_novo_wf with the parameters–taxa_filter g__Serratia–outgroup_taxon “s__Yersinia pestis”.
3. Results
3.1. Prevalence of Species Belonging to the Serratia marcescens Complex (SMC) Within the Regional Clinical Isolates
To determine the prevalent Serratia species of the SMC in the regional hospitals, samples were taken from patients in the three cooperating facilities (Klinikum Bielefeld, Ev. Klinikum Bethel, and Klinikum Lippe) over the course of one year (mid-2021 to mid-2022). From these samples, bacterial isolates were cultivated on blood agar as described above.
In total, 21 bacterial isolates from sepsis patients were identified as “Serratia marcescens” (Table 1) using routine clinical identification methods. The majority of these 21 isolates originated from blood samples, but some were also derived from wound swabs, urine, or peritoneal lavage in the three hospitals (Table 1).
In general, the subdivision of SMC strains identified via biochemical assays or mass spectrometry can partially be performed based on the phenotype of this pathogen during cultivation, as cultures of S. marcescens and S. nematodiphila show a blood-red color, while the other members are white. As only one of the 21 “S. marcescens” cultures showed a red colony phenotype, this was a first indication that the clinically relevant regional strains belong to the latter group.
3.2. Genome-Based Classification of the Regional SMC Strains
To resolve the taxonomy of these SMC strains, their genomes were sequenced, assembled, and used for taxonomic classification. As we recently demonstrated [10], 16S-based comparisons are insufficient to resolve species in the SMC. This is validated here, as 16S rRNA comparisons for the 21 isolates based on the Type Strain Genome Server (TYGS) were also inconclusive (Supplementary Figure SA).
The first overall genome-relatedness index used to identify the species was to calculate the digital DNA-DNA hybridization (dDDH) values (Figure 1). These correlate to the classic DNA-DNA hybridization method and were also performed via the TYGS in an all-versus-all manner (Supplementary Table SA). Due to the fact that the genome of the S. nevei type strain was missing in the TYGS, we manually included it (GCA_037948395.1; available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_037948395.1/ (accessed on 30 August 2022)). The analysis identified one genome as belonging to S. marcescens. Furthermore, six genomes were identified as S. bockelmannii, one as S. nematodiphila, two genomes as S. ureilytica, one as S. nevei, and ten genomes as belonging to the recently published species S. sarumanii (Table 2).
Remarkably, the TYGS marks S. nematodiphila as a potential subspecies of S. marcescens, which aligns with the phenotypic similarity of the two species based on the presence of the prodigiosin gene cluster. Another noteworthy observation is the extremely high degree of similarity between the strains K-M0260 and K-E0102, as well as between strains K-M0706 and K-M0228, each being reported with 100.0% dDDH, indicating they might be identical. A closer analysis of both pairs using blastn revealed that K-M0706 and K-M0228, identified in the same hospital, are indeed basically identical, with just 114 mismatches. In contrast, despite their similarity, K-M0260 and K-E0102 were collected from different hospitals and differ by about 370 single nucleotide polymorphisms (SNPs) and deletion/insertion polymorphisms (DIPs), as well as in the presence of 5 larger regions in each genome (between 6 kb and 66 kb for K-M0260 and between 15 kb and 72 kb for K-E0102) that are absent in the other genome. This is noteworthy because a 100.0% dDDH should indicate basically identical genomes, yet they contain approximately 181 and 202 kbp of unique sequences, respectively. Additionally, in contrast to K-E0102, two plasmids with a size of 15.7 and 76.5 kbp were found in K-M0260.
For visualization, the genomes of the 21 samples, the genomes of all Serratia type strains, as well as the Yersinia pestis type strain as an outgroup, were used for analysis using the TYGS with distance formula d_5_ [21]. The species belonging to the SMC form a distinct group within the tree that is clearly separated from the other Serratia (Figure 1). Within the cluster, the S. marcescens and S. nematodiphila type strains, as well as K-M0312 and K-M0056, cluster even closer together, indicating that S. nematodiphila might be a subspecies of S. marcescens, as their dDDH value of 73.6% is above the cutoff of 70%.
To verify the dDDH distance-based tree, the genomes used for the TYGS-based analyses were also used for a taxonomic analysis applying GTDB-tk v2.4.0 and GTDB and release 226. The results of the classify_wf workflow as well as the de_novo_wf workflow closely support the results reported by the TYGS (Supplementary Table SB, Supplementary Figure SB). As indicated by the TYGS, the ANI and alignment fraction (AF) of nematodiphila and S. marcescens indicate that the former might be considered to be a subspecies of the latter (96.8% ANI, 91.0% AF).
Besides the chromosomes, we were able to identify seven distinct plasmids in the 21 samples (Supplementary Table SC). While five of them were found in a single sample and the fifth in two, one plasmid of 83.6 kbp was observed to be present in a total of ten samples, nine of which carried an almost identical version (while in the tenth, it is about 7 kbp larger due to two additional regions of about 11 kbp total and the loss of a 4 kbp region). Interestingly, those ten samples consist of seven S. sarumanii, two S. bockelmannii, and one S. ureilytica, indicating that this plasmid has the capability to be transferred between the different species. Indeed, based on the annotation, almost half of the plasmid encodes for genes known to be involved in mobilization and plasmid transfer (see annotated GenBank entries, IDs listed in Supplementary Table SC).
3.3. Analysis of the Abundance of Serratia sarumanii Within All Publicly Available Serratia Genome Sequences
The frequent occurrence of S. sarumanii (10 out of 21 samples) and S. bockelmannii (6 out of 21 samples) is intriguing due to their recent description as valid species, resulting in the obvious question of their distribution on a global scale.
To address this question, all Serratia genomes in the NCBI genomes database (accessed on 29 January 2025), 4172 in total, were downloaded and, together with the genomes of the 21 samples in this study, classified using GTDB-tk. Based on the classify_wf workflow, most genomes in the database belong to the SMC (3574 = 85.7%), while other Serratia and those assigned to other genera are a small minority (433 Serratia sp., 115 Chania sp, 33 Serratia_B, 13 Serratia_F). Surprisingly, more than a third of all genomes belong to S. sarumanii (1453 = 34.8%), which equals 40.7% of the SMC genomes. The following most abundant species, S. nevei, contributes just half that number (729 = 17.5% total/20.4% SMC), followed by S. ureilytica (460 = 11.0%/12.9% SMC) and S. bockelmannii (351 = 8.4%/9.8% SMC), each contributing about half of that, respectively. Genomes of the name-giving S. marcescens only make up 261 entries (6.3%/7.3% SMC), barely more than those from S. nematodiphila (246 = 5.9%/6.9% SMC).
To obtain a clearer understanding of the data, a phylogenetic tree was calculated using the de_novo_wf workflow of GTDB-TK, which was then visualized with iTOL (https://itol.embl.de/, 6 May 2025) (Figure 2). Except for a fair number of S. nevei genomes (and a single S. sarumanii genome) that form multiple clades besides those belonging to the respective type species within the SMC clade, all genomes fall into distinct clades with that of the corresponding type species (Figure 2). The split within S. nevei was already observed by Klages et al. [10], who described three distinct groups based on average nucleotide identity (ANI) and alignment fraction (AF) for this species.
Also of interest is the observation that 9 of the 10 S. sarumanii are distinct isolates, with only one pair (K-M0706 and K-M0228, both isolated at the same hospital) showing so little difference that they are likely part of an infection chain, either directly from patient to patient or from a common, unidentified source. Given that the two patients were admitted three months apart, the latter explanation is more likely. Even more interesting is a second pair of strains, K-M0260 and K-E0102, which were isolated from two different hospitals eight months apart. They differ by just about 370 SNPs/DIPs, but each harbors five larger regions (6 to 72 kbp in length), containing a total of 181 and 202 kbp of unique sequences, respectively. In addition, K-M0260 harbors two plasmids of 15.7 and 76.5 kbp. This indicates that even if they are derived from a common source, which is likely, they each either gained a significant amount of information (3.5 to 5.5% of their total genome size) via HTG events or lost it while diverging from the common ancestor. Given its prevalence in the databases, another important piece of information regarding the clinical significance of the various species within the SMC is their actual “origin” (i.e., the source material the respective strain was isolated from). Unfortunately, this information is often not available; e.g., in [10], the isolation source was present for 87.0% of the genomes of S. sarumanii strains. While this restricts a comprehensive analysis, we focused on several studies addressing the species distribution within the SMC [11,12,22,23,24] that included in-depth information on the source of the respective samples. In all of them, the authors observed a large group of genomes that was clearly separate from the type species validly described at the respective time. We used either the assemblies presented in these studies or, if only raw data were available from SRA, we created draft assemblies with SPAdes v3.13.0 [25] using standard parameters. Running the classify_wf and the de_novo_wf workflow, we could show in all these studies that the “unknown” group consisted of strains belonging to S. sarumanii (Supplementary Table SD, Supplementary Figure SC). For example, 166 of 455 isolates (36.5%) in the outbreak investigation by Taxt et al. [24] belong to S. sarumanii. Likewise, in the studies of [11,12], the largest cluster, consisting of 47 of 225 samples (20.9%), respectively, 41 of 165 samples (24.8%), is S. sarumanii.
We also used BLAST v2.15.0+ to search for the plasmid we identified to be present in several species and were able to detect it (i.e., >75% coverage of the query sequence) in 48 assemblies of Serratia spp. in the NCBI database (Supplementary Table SE). Again, it was found in a variety of species, consisting of 23 S. sarumanii, 16 S. bockelmannii, 4 S. nevei, 3 S. ureilytica, and 2 S. nematodiphila, based on our classification above.
4. Discussion
The genome-based analysis of 21 “Serratia marcescens” isolates from clinical samples associated with bacterial infections in three local hospitals in OWL revealed that they belong predominantly to Serratia species other than S. marcescens. While this is hardly a novel observation, having been reported, e.g., by Ono et al. [11] and Aracil-Gisbert et al. [12], we are the first to identify the dominant species as S. sarumanii. While the number of occurrences in the regional hospitals is low, with just 21 isolates of the SMC in one year, the number of S. sarumanii isolates among them is still significant (10 out of 21 isolates = 47.6%). Of the remaining 11, only one really belongs to the species S. marcescens, while the rest comprises S. bockelmannii (6), S. ureilytica (2), S. nevei (1), and S. nematodiphila (1), although dDDH, ANI, and AF values support that S. nematodiphila should be reclassified as a subspecies of S. marcescens.
Analysis of all assembled Serratia genomes in GenBank (NCBI) confirms the dominance of S. sarumanii among the strains of scientific interest, as approximately 40% of the genomes from species currently classified as belonging to the SMC originate from S. sarumanii. This “dominance” can be attributed to a high clinical significance, both in terms of prevalence in clinical samples overall and in its dissemination in patient-derived samples [11,12,24]. Besides this prevalence in samples overall, S. sarumanii is noteworthy since, in contrast to most other members of the SMC in larger studies, the samples are usually derived from patients [22], predominantly from blood samples [10,11]. This coincides with S. sarumanii being often found in intensive care units [12,22], indicating that this species either is especially problematic for patients in already critical condition or is responsible for said critical condition. Another marker that indicates the increased pathogenicity of S. sarumanii compared to the second major Serratia species in a clinical context, Serratia nevei, is the smaller average genome size of the former: 5.1 ± 0.1 Mbp for S. sarumanii and 5.7 ± 0.2 Mbp for S. nevei. While the actual cause(s) for this correlation are still unclear, it can be observed regardless of the taxonomic level [26].
Surprisingly, though the strains we identified to be S. sarumanii harbor fewer resistance markers on average than S. neveii, the latter often occurs in hospital environments and has been shown to persist for extended periods in hospital sinks, acquiring highly conserved plasmids that carry multiple resistance-conferring genes [12]. This reservoir function poses a significant risk in hospital hygiene and patient safety, as recent findings by Zhang et al. [27] and Khalifa et al. [23] indicate that the relative absence of resistance markers is counteracted by the apparent ability of “S. marcescens”, identified by us as S. sarumanii, to quickly acquire and transfer resistance genes, both intra- and interspecies.
In combination with the findings of [11], who identified additional Serratia species in hospital settings harboring multiple resistance genes, and those of Aracil-Gisbert et al. [12], who report S. neveii and a group of strains we identified as S. sarumanii as the most frequently detected Serratia species in patient samples, a deeply concerning picture emerges. S. sarumanii appears particularly dangerous, not only due to its clinical prevalence but also because it might serve as a center for resistance gene acquisition and dissemination [27]. Furthermore, Khalifa et al. [23] confirmed that S. nevei and the group of strains we showed to be S. sarumanii are the most common Serratia in hospitals harboring several resistance genes, showing that all carbapenem-resistant clinical strains from the Serratia marcescens complex can be assigned to the species S. nevei and S. sarumanii. As a possible route of transfer, plasmids that replicate in different Serratia species are a strong possibility. For example, the 83.6 kbp plasmid, which was found in 10 out of 21 samples in our study, was also found in about 1.4% of SMC strains in the NCBI database (48 out of 3574). While this plasmid lacks resistance markers (at least in our samples), those could be easily acquired via horizontal gene transfer (HTG). The occurrence of several HTG events in S. sarumanii, possibly in a very short time (as measured by SNP occurrence), is evidenced by the strains K-M0260 and K-E0102, each of which gained about 5% additional genetic information in 5 islands each.
5. Conclusions
This study demonstrates that genome-based species identification provides a more accurate picture of clinically relevant species within the ‘Serratia marcescens complex’ (SMC) than the conventional clinical classification. Our analysis of clinical isolates from regional hospitals, supported by comparative data from publicly available genomes, reveals that several Serratia species contribute to infections previously attributed to S. marcescens. In particular, S. sarumanii emerges as the predominant species, showing a strong association with patient-derived samples, intensive care units, and a high potential for acquiring and spreading resistance genes. These findings underscore the clinical importance of precise taxonomic resolution for epidemiological surveillance, infection control, and antimicrobial stewardship.
Given the clinical dominance of S. sarumanii, capacity for resistance gene transfer, strong association with hospital-acquired infections, and scarcity of S. marcescens in clinical isolates, we propose that the term Serratia marcescens complex (SMC) is no longer scientifically appropriate or optimal for use in clinical or genomic contexts. Based on the presented evidence, we propose to rename the SMC to Serratia sarumanii complex (SSC), to reflect its true composition and allow for more accurate identification, surveillance, and treatment strategies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1de Araújo H.W. Fukushima K. Takaki G.M. Prodigiosin Production by Serratia marcescens UCP 1549 Using Renewable-Resources as a Low Cost Substrate Molecules 2010156931694010.3390/molecules 1510693120938403 PMC 6259207 · doi ↗ · pubmed ↗
- 2Adeolu M. Alnajar S. Naushad S. Gupta R.S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov Int. J. Syst. Evol. Microbiol.2016665575559910.1099/ijsem.0.00148527620848 · doi ↗ · pubmed ↗
- 3Ioannou P. Alexakis K. Spentzouri D. Kofteridis D.P. Infective endocarditis by Serratia species: A systematic review J. Chemother.20223434735910.1080/1120009 X.2022.204351335209804 · doi ↗ · pubmed ↗
- 4Mahlen S.D. Serratia infections: From military experiments to current practice Clin. Microbiol. Rev.20112475579110.1128/CMR.00017-1121976608 PMC 3194826 · doi ↗ · pubmed ↗
- 5Paul T. Mondal A. Bandyopadhyay T.K. Bhunia B. Prodigiosin production and recovery from Serratia marcescens: Process development and cost–benefit analysis Biomass Convers. Biorefinery 2024144091411010.1007/s 13399-022-02639-2 · doi ↗
- 6Piccirilli A. Cherubini S. Brisdelli F. Fazii P. Stanziale A. Di Valerio S. Chiavaroli V. Principe L. Perilli M. Molecular Characterization by Whole-Genome Sequencing of Clinical and Environmental Serratia marcescens Strains Isolated during an Outbreak in a Neonatal Intensive Care Unit (NICU)Diagnostics 202212218010.3390/diagnostics 1209218036140580 PMC 9498040 · doi ↗ · pubmed ↗
- 7Li D. Yi J. Han G. Qiao L. MALDI-TOF Mass Spectrometry in Clinical Analysis and Research ACS Meas. Sci. Au 2022238540410.1021/acsmeasuresciau.2c 0001936785658 PMC 9885950 · doi ↗ · pubmed ↗
- 8Bizzini A. Greub G. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry, a revolution in clinical microbial identification Clin. Microbiol. Infect.2010161614161910.1111/j.1469-0691.2010.03311.x 20636422 · doi ↗ · pubmed ↗