Evaluation of Multi-Target Genotyping (ITS-hsp70-cpb) for Detecting Population Heterogeneity Within Mediterranean Leishmania infantum, with a Focus on Zymodeme MON-24
Trentina Di Muccio, Daniele Tonanzi, Gert Van der Auwera, Eleonora Fiorentino, Luigi Gradoni, Marina Gramiccia, Giuseppe La Rosa

TL;DR
This study evaluates a multi-target genotyping method to better detect population differences in Mediterranean Leishmania infantum, focusing on the ZMON-24 zymodeme.
Contribution
The study introduces a multi-target genotyping approach combining ITS, hsp70, and cpb for improved discrimination of L. infantum populations.
Findings
L. infantum hsp70 showed unexpected variability with 8 sequence variants.
Combining ITS-hsp70-cpb sequence variants identified distinct genotypes for L. infantum strains.
Four genotypes were associated with ZMON-24, including clusters from Mediterranean and North African regions.
Abstract
L. infantum and L. donovani, distinct species in the L. donovani complex, show high phenotypic and genotypic polymorphism and share molecular traits. Therefore, genotyping by a single molecular target can give uncertain results. This study focuses on genotyping a set of L. donovani complex strains, including 18 zymodemes classified according to Montpellier nomenclature (ZMONs) and different clinical forms, by internal transcribed spacer (ITS), heat shock protein 70 (hsp70), and cysteine proteinase b (cpb) sequencing to evaluate their ability in species discrimination. We found an unexpected L. infantum hsp70 variability, with 8 sequence variants. Cpb-PCR could not distinguish L. donovani complex species, due to a L. infantum intraspecific allelic (cpbEF) polymorphism. By combining ITS-hsp70-cpb sequence variants, we obtained different genotypes. ITS(A)-hsp70inf(2)-cpbE identified 69.9%…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
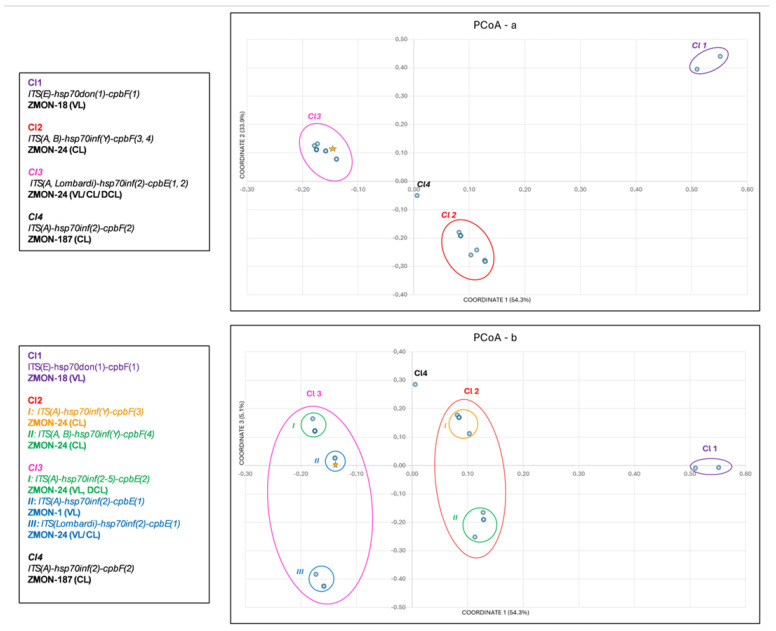 Figure 1
Figure 1- —EU funding within the NextGeneration EU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsResearch on Leishmaniasis Studies · Trypanosoma species research and implications · Parasites and Host Interactions
1. Introduction
Leishmaniasis is considered a neglected infectious disease that affects, kills or disables millions of people worldwide [1,2]. The disease affects both humans and animals and is caused by protozoan parasites of phagocytic cells belonging to Leishmania genus (Kinetoplastida, Trypanosomatidae) transmitted by bite of several sandfly Phlebotomus species. In the WHO European Region, the estimated number of visceral (VL) and cutaneous (CL) leishmaniasis cases is 1100–1900 and 10,000–17,000 cases per 100,000 population, respectively [3]. In this region, leishmaniasis is considered emergent and re-emergent due to different epidemiological factors such as human immunodepression, human migration, transport of infected animals, climate and environmental changes which affect the vectorial capacity and geographic distribution of sandfly populations [4,5].
Leishmania infantum (L. donovani complex) is the main agent of zoonotic VL in Mediterranean areas where dogs represent the main reservoir hosts for human infection. It has long been considered a viscerotropic species, but many cutaneous cases have been recorded throughout the Mediterranean basin which were mostly attributed to dermotropic variants of L. infantum, according to isoenzyme based-classification of parasite populations known as Montpellier zymodemes (ZMON) [6,7,8,9,10].
Clinical manifestations of the human leishmaniasis are highly polymorphic including asymptomatic infections, spontaneously healing localized skin nodules or cutaneous ulcers (cutaneous leishmaniasis, CL), multiple nodules (diffuse cutaneous leishmaniasis, DCL), infection of the mucous membranes (mucosal leishmaniasis, ML), and serious systemic visceral forms (visceral leishmaniasis, VL) which have a high mortality rate if not treated. Hosts’ immune response and cell-mediated immunity establish the outcome of the disease. Immunodeficiency syndromes from human immunodeficiency virus (HIV) infection, immunosuppressive treatments, and organ transplantation or malnutrition in childhood are major risk factors for VL by L. infantum [11,12,13]. Occasionally, VL may also exhibit epidemic characteristics among immunocompetent adults without natural acquired immunity to the parasite [3,8,14].
Leishmania identification has important implications both clinical and epidemiological in disease control and prevention, considering the emergence and re-emergence of Leishmania in different Mediterranean areas and the possible introduction of new species and variants [15,16,17,18,19,20,21,22]. Some genetic evidence can affect the molecular identification of Leishmania species. In fact, in this area, L. infantum shows a high polymorphism highlighted in the past by Multilocus Enzyme Electrophoresis (MLEE) typing [6,7,8,9,10] and more recently by molecular and genomic typing tools, such as Multi Locus Sequence Typing (MLST), Multilocus Microsatellite Typing (MLMT), and Next Generation Sequencing (NGS) [23,24,25,26,27,28,29,30,31]. Moreover, different hybrids such as L. infantum/L. major and L. infantum/L. donovani have been also demonstrated [32,33]. Therefore, the single marker-based approach can display uncertain results in close species, such as L. infantum and L. donovani, which are not clearly distinguished from a genetic point of view, given that they are known to form a continuum of populations in L. donovani species complex [34,35]. Consequently, the validation of different molecular targets and approaches should be considered as an ongoing process, and it should be evaluated in different epidemiological and geographical settings, considering phenotypic and genotypic polymorphisms, as well as the high genomic plasticity and adaptive capacity of Leishmania parasites [36,37].
For these reasons, the aim of this study was to genotype L. donovani complex strains by the most widely used diagnostic targets such as internal transcribed spacers ITS1 and ITS2 (ITS), heat-shock protein 70 (hsp70) and cathepsin L-like cysteine proteinase B (cpb) genes [38,39,40,41,42] also by combining ITS-hsp70-cpb sequence variants to gain more informative genetic data on L. infantum populations. For that, we selected a representative set of L. donovani complex strains, isolated over a 30-year period from CL and VL cases, occurred either as sporadic clinical episodes or during disease outbreaks, in order to account not only for the intraspecific variability, but also to rule out geographical bias due to the focal epidemiological aspect of this disease. We believe that our findings can contribute to the discussion on the variability and utility of these specific molecular targets, in single and multi-target-based approaches, in identifying Leishmania populations in the Mediterranean region.
2. Materials and Methods
2.1. Ethics Statement
Ethical review and approval were not required, as genome sequence analysis on Leishmania isolates is part of surveillance activities, conducted within the scope of the public health practice in Italy. Human clinical samples were collected and analyzed within the mandate of the Department of Infectious Diseases of Istituto Superiore di Sanità (ISS) as conferred by the Italian Ministry of Health (Protocol number 33122-14/10/2020-DGPRE-DGPRE-P ‘Prevention and control of Leishmaniasis in Italy’). Patient’s informed written consent was obtained from all subjects at the time of the clinical examination. Patient information was anonymized and de-identified prior to analysis, according to the European Union General Data Protection Regulation (Regulation (EU) 2016/679) (https://eur-lex.europa.eu/eli/reg/2016/679/oj, accessed on 30 November 2023) adopted by ISS (www.iss.it) and in agreement with the Data Protection Officer of ISS.
2.2. Samples
A total of 88 strains has been studied. Eighty-six strains were previously isolated, cultured and cryopreserved at Istituto Superiore di Sanità in Rome, during a period between 1982 and 2017. Leishmania strains originated from different geographical areas, from the main medical forms of Leishmaniases (VL, CL, and DCL), isolated from human cases of all clinical categories (infants, adults, immunocompetent and immune compromised patients); a few strains from animal hosts (dog, cat and Phlebotomus sand flies) were included. As shown in Table S1A, all the strains have been classified by ITS sequencing according to Kuhls et al., 2005 and Chicharro et al., 2013 [14,43]: 82 L. infantum strains from Mediterranean areas (n = 68 from Italy, n = 10 from Spain, n = 1 from Malta, n = 3 from North Africa) and 4 L. donovani strains from Africa (Table S1A). Furthermore, 2 reference strains were included in the analysis: L. infantum MHOM/DZ/82/LIPA59-ISS182 from Algeria, and L. donovani MHOM/IN/80/DD8-ISS46 from India. A total of 72 out 88 strains, previously typed by MLEE, were representative of 15 L. infantum ZMONs and 3 L. donovani ZMONs [7,8]. In Table S1B, we summarized the analyses (MLEE, sequencing, RFLP, dendrogram, PCoA) performed for each strain.
2.3. DNA Extraction and Amplification
The genomic DNA from 10^6^ promastigote cultures in EMTM [44] was prepared by the DNA extraction protocol of the Maxwell^®^ 16 Cell DNA Purification Kit (Promega, Medison, WI, USA) following the manufacturer’s instructions. The DNA concentration was measured spectrophotometrically. The DNA was stored at −80 °C until use.
All the PCRs were performed in volumes of 50 µL of PCR mixture (GoTaq Green Master Mix, 2X Promega) using 2 µL of DNA (5–10 ng) and 1 µL of the appropriate primer pair for each of the three markers selected. The PCRs were performed in the C1000 Thermal Cycler (BIORAD, Hercules, CA, USA). A 2% agarose gel was used to verify the amplified product size.
The ITS1-5.8S-ITS2 cluster (here named ITS) was amplified by LITSV and LITSR primers, according to the amplification conditions described by El Tai et al., (2000) [45] with some modifications: initial denaturation at 95 °C for 5 min followed by 34 cycles consisting of denaturation at 95 °C for 20 s, annealing at 53 °C for 30 s, and extension at 72 °C for 1 min followed by a final extension cycle at 72 °C for 10 min in a single PCR reaction.
For all strains, the F-fragment of the hsp70 gene was amplified by using 20 pmoles of each primer F25 and R1310 [40]. The hsp70-F fragment RFLP assay was performed to discriminate L. donovani and L. infantum, as proposed by Montalvo et al. (2012) [40]. The digestion was performed in a total of 20 μL containing 5 μL of the PCR products in 1x buffer provided by the manufacturers, and 5U restriction enzyme MluI (Promega). The reactions were incubated at 37 °C overnight and analyzed by electrophoresis in 3% agarose gel (Promega). When uncertain, the PCR-RFLP assay was repeated to ensure the repeatability of the results.
Different copies of the cpb gene were amplified by using specific primers CpbEF-for and CpbEF-rev. This PCR was described as discriminative between the L. donovani and L. infantum species, amplifying the copies cpbF (741 bp) and cpbE (702 bp), respectively [46].
2.4. Sequencing and Genetic Analysis
The three markers (ITS, hsp70 and cpb) were amplified in triplicate from each strain and the independent PCR products were sequenced by using the same primers used in the amplification reactions in both directions. The final forward/reverse consensus was produced by CLC Main Workbench software package ver.23 (Qiagen Aarhus A/S). Only the sequences showing reproducible results were submitted in GenBank at NCBI.
The ambiguous nucleotides have been codified using IUPAC code. The heterozy-gous positions, identified by a double peak in the chromatogram, were distinguished from a poor sequence quality if the peaks were of similar height, or the lower peak was at least 50% of the highest or considerably higher than the background noise in the rest of the chromatogram, and if the double peak was present in sequencing results from both directions. All the hsp70 and cpb sequences were aligned using the multiple alignment tool implemented in CLC package and were manually adjusted to maximize identity in presence of microsatellites (ITS) and indels (cpb). Before analysis, the multiple alignments were trimmed at the 3’ and 5’ ends to the sequence fragment shared by all samples.
Each ITS sequence was typed by comparing it to an “ITS-types” set of L. infantum and L. donovani, previously classified by using the sequence polymorphisms of the 12 microsatellite regions, including four sites in ITS1 and eight sites in ITS2 [14,43]. The hsp70 and cpb sequences were compared to well-characterized reference sequences [29,47,48] retrieved from GenBank at NCBI.
A dendrogram of cpb was built to evaluate the genetic relationships among the L. donovani complex strains with respect to this molecular target. Sequences of cpbEF were retrieved from Genbank with BlastN, using as query the sequences obtained in this study. Sequences that showed a similarity of less than 95% with the query, containing frameshift mutations or several ambiguous nucleotides, were not selected for the analysis. Multiple alignment including 61 sequences from Genbank and the 23 from this study were used. The dendrogram was based on p-distances by using the Neighbor-joining method with MEGA version 7.0 (megasoftware.org). The bootstrap value for each node was computed using 2000 replicates.
The Principal Coordinates Analysis (PCoA) was performed to investigate the rela-tionships among the Leishmania strains sub-set including the 23 strains for which ITS, hsp70 and cpb sequences were available. The PCoA does not need any a priori assumption on substitution model for nucleotide evolution and each nucleotide position is treated as a phenetic character with two characters states (presence “1” and absence “0”). The PCoA was performed by Past software version 4.14 [49] using the Jaccard’s index to compute the phenetic similarity matrix among strains. The default value of Transformation Exponent (c = 2) was applied. The input binary data table used to compute the PCoA was produced including as character each variable nucleotide position present in the multiple ITS, hsp70, and cpb alignment and codifying as presence/absence each alternative nucleotide (Table S3). The Indel in cpb, spanning up to 39 bp, was codified as a single character (i.e., single mutational event) and the same was for variable microsatellites in ITS (i.e., occurrence of alternative repeat was considered as a single event). The multidimensional space produced by the analysis and describing the relationships among the Leishmania strain was summarized by the two graphics defined by the three main coordinates (i.e., axes). The sequences of L. infantum MHOM/FR/78/LEM75 strain available in GenBank (accession numbers: AJ634339—ITS, LN907838—hsp70 and AY896781—cpbE) were also included in the analysis as reference.
3. Results
The genetic variability shown by the ITS, hsp70 and cpb sequences was used to address key diagnostic, epidemiological and population genetic issues regarding our 88 strains. The GenBank accession numbers are listed in Table S1A.
ITS displayed well-known L. infantum ITS-A, ITS-B and ITS-Lombardi genotypes, widespread in Mediterranean areas including North Africa, and on the other hand L. donovani ITS-E and ITS-H from East Africa and India [14,43]. Four ITS-B var, A/B var, E var sequence variants were observed (Table S1A).
Eighty-eight hsp70 sequences were obtained. The consensus sequences were identified as belonging to L. donovani complex species by a Blast search in GenBank at NCBI. The multiple sequence alignment of 1019 bp displayed a genetic variability due to nine polymorphic sites that gave 9 sequence variants with respect to the sequence of the reference strains L. donovani MHOM/KE/55/LRC-L53 (MN728785) and L. infantum MHOM/FR/78/LEM75 (LN907838) (Tables S1 and S2). The percentage of identity in intraspecific pairwise comparison was 100% in L. donovani and spanning from 99.75% to 100% in L. infantum. The sequence variants hsp70don(1) (n = 5/5, 100% of the L. donovani strains) and hsp70inf(2) (n = 67/83, 80.7% of the L. infantum strains) displayed 100% identity with L. donovani and L. infantum reference sequences, respectively. In contrast, the sequences inf(3–9) displayed different variant nucleotide positions (Table S2).
The most relevant sequence variants were inf(6–9), which identified 48% (n = 12/25) of the L. infantum ZMON-24 strains. They displayed variant nucleotides in 3 positions (266, 370, and 888) and were characterized by a heterozygous condition with Y (T/C) in the species-specific site 370, present in one of two MluI restriction sites (ACGCGT), proposed by Montalvo et al., (2012) [40] to discriminate L. infantum from L. donovani by RFLP assay (Table S2). Therefore, the reliability of this assay was evaluated on all the samples. The triple band pattern (117, 389, 780 bp), typical of L. infantum sp., was observed in 85.5% (n = 71/83) of the L. infantum strains. A double band pattern (117 and 1169 bp), typical of L. donovani sp., was observed in 100% (n = 5/5) of the L. donovani strains, as expected. In contrast, a L. infantum/L. donovani mixed pattern, with 4 restriction products (117, 389, 780 and 1169 bp) common to the two species, was found in 14.5% (n = 12/83) of L. infantum strains, referring to the sequence variants inf(6–9) (Figure S1). This pattern was due to the possible overlap of two hsp70 alleles, in the heterozygous position Y (Figure S2). For this reason, we referred to the sequence variants inf(6–9) as inf(Y).
The cpb-PCR of the 88 samples evidenced the presence of the copy cpbE (702 bp) in 67 out of 83 (80.7%) L. infantum strains; the copy cpbF (741 bp) was detected, not only in 5 L. donovani strains, as expected, but also in 16 L. infantum strains (19.3%), classified as zy-modems MON-24 (n = 12 strains), MON-187 (n = 1), MON-189 (n = 1), and MON-190 (n = 2) (Table S1A).
By combining the ITS-hsp70 sequence variants and the copies cpbE and cpbF, 14 ITS-hsp70-cpb sequence variants were generated and here indicated as a “genotype” (Table 1).
Geographical distribution of the genotypes A–H was depicted on the MicroReact Map. The whole dataset can be downloaded and explored interactively in the MicroReact platform (https://microreact.org/project/iss, accessed on 30 November 2025) [50]. The genotype C was found in 69.9% (n = 58/83) of L. infantum strains belonging to 12 zymodemes from Mediterranean areas. Furthermore, the genotype H was found in ZMON-187, ZMON-189, and ZMON-190 zymodemes. Interestingly, at least 4 genotypes (C, E, F, and G) represented ZMON-24: F and G identified CL cases from Italy and North Africa, instead the genotypes C and E identified VL and CL cases from Mediterranean areas.
Consequently, in order to provide an accurate overview of the genetic relationships among the ZMON-24 strains, we sequenced cpbE and cpbF for 19 of them, along with one L. infantum ZMON-1 (ISS3176), ZMON-187, L. donovani ZMON-30 (ISS2440), and ZMON-18 (ISS50) strains (the sequence accession number in Table S1A). The alignment of 704 bp displayed a genetic variability due to sixteen polymorphic sites and one 39 bp gap that returned 7 sequence variants: cpbF(1–4) and cpbE(1–3), 5 of them detected in ZMON-24 strains (column CpbEF Seq(var) in Table S1A). Most of the variability was due to the presence of L. donovani strains (ISS50 and ISS2440), differing in identity percentage from all the other L. infantum strains from 98.15% to 98.43%. The identity percentage in the intraspecific pairwise comparison was 100% in L. donovani, while it was between 99.29% and 100% in L. infantum.
As shown in the dendrogram in Figure S3, our strains were grouped into seven clusters of identical sequences. The L. infantum cpbE(1–3) sequence variants clustered with the CL and VL L. infantum cluster (ZMON-1, ZMON-24, ZMON-80) from the Mediterranean area (France, Spain, Tunisia, and Algeria), previously described [47,48]. On the contrary, the sequences cpbF(1) clustered with L. donovani strains from East Africa, whereas the sequence variants cpbF(2–4) were grouped with L. infantum strains from North Africa, among which ZMON-24 CL strains (see Table S5).
By looking at multidimensional PCoA, we observed the genetic relationships among these 23 sequence variants of ITS-hsp70-cpb by studying three coordinates that accounted for 93.3% of the total variance (coordinate 1 = 54.3%, coordinate 2 = 33.9% and coordinate 3 = 5.1%) (Table S3). The most probable groupings are shown in Figure 1a,b. The analysis obtained by using the coordinates 1 vs. 2 (total variance 88.2%) (Figure 1a) grouped the strains into 3 main clusters: Cl 1 including L. donovani strains, characterized by ITS(E)-hsp70don(1)-cpbF(1); Cl 2, including only cutaneous L. infantum ZMON-24 strains from Italy and North Africa, characterized by hsp70inf(Y) and cpbF(3–4); Cl 3, in-cluding ZMON-24 VL, CL and DCL, and ZMON-1 VL cases (including MHOM/FR/78/LEM75 L. infantum reference strain, ZMON-1), characterized by hsp70inf(2) and cpbE(1–2). Finally, the ZMON-187 strain (Cl 4) was separated from the other ones, due to the unique combination of hsp70inf(2) with cpbF(2).
Furthermore, in the graphic obtained by the coordinates 1 vs. 3 (total variance 39.0%) (Figure 1b), Cl 2 was divided in 2 sub-clusters: I, represented by CL strains from Italy, exhibiting cpbF(3), II represented by strains from Italy and North Africa, exhibiting cpbF(4). Cl 3 resulted divided into 3 sub-clusters: I and II, including European L. infantum ZMON-1 and ZMON-24 from Italy and Spain, respectively, all strains with the cpbE(1), while the cluster III included all the L. infantum ZMON-24 from Italy and North Africa, with cpbE(2). Again, the Italian L. infantum ZMON-187 strain remained separated.
4. Discussion
Our study focused on integrating genetic variability data by sequencing diagnostic targets that have been extensively studied in Leishmania, such as ITS, hsp70, and cpb, to identify strains from Mediterranean areas. Moreover, we described different Leishmania populations with a multi-targets-based approach by combining the sequence variants of ITS-hsp70-cpb and obtaining 8 “genotypes” (A–H) (Table 1).
By examining each marker individually, we can highlight some aspects. Our sequence analysis of ITS target allowed a Leishmania species classification of our strains, showing well known geographic L. infantum populations (genotypes ITS-A, ITS-B, and ITS-Lombardi), widespread in the Mediterranean area including North Africa, and L. donovani populations (genotypes ITS-E and ITS-H) from East Africa and India [14,43]. There was no evidence of a clear correlation between ITS genotypes and either enzyme populations, hosts, or clinic.
The hsp70 gene sequence is widely accepted as a tool for discrimination of medically important Leishmania species from the Old and New World, due to the high conservation observed within L. donovani complex genomes from different geographic regions [40,41,51]. In our study, we found an unexpected intraspecific variability in the Mediterranean L. infantum strains, proven by 8 hsp70 F-fragment sequence variants, while confirming hsp70 its validity as a genotyping target of Leishmania species genotyping target. The most represented variant hsp70inf(2), displaying 100% identity with the L. infantum LEM75 reference strain, identified 80.7% of L. infantum strains, including all the 15 zymodemes studied. However, we detected 4 sequences (i.e., inf(6–9)), referred to as inf(Y), that are characterized by their common heterozygous position (Y = C/T) at the species-specific site distinguishing L. donovani from L. infantum. Standard conditions have been applied to perform reliable sequences and exclude sequencing artefacts (see Section 2.4); moreover, the RFPL results supported the proof that this position could highlight the presence of two hsp70 alleles, as already reported in several Leishmania species [32,52]. However, heterozygosity should be further investigated. Therefore, these strains will be analyzed by a low-coverage whole-genome sequencing (WGS) to evaluate the possible hybrid status.
A BLAST search in GenBank at NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 28 July 2023) showed the originality of this hsp70-F fragment sequence variants that, according to our analysis, was specific of a ZMON-24 sub-cluster of Italian and African strains (n = 12) with cutaneous feature. Therefore, the sequence hsp70inf(Y) and/or the RFLP assay, which showed a reproducible and specific pattern, can represent possible rapid typing assays able to identify a L. infantum ZMON-24 sub-cluster. The correlation between this hsp70inf(Y) sequence variant, the cutaneous presentation, and geographic origin should be evaluated with additional samples. Meanwhile, we found, from previous works by Gritti et al., (2023, 2025) [53,54] in the Emilia-Romagna region (Italy), some original L. infantum hsp70 fragment sequence variants from CL cases, displaying heterozygous position Y, consistent with ZMON-24, historically present in this area [7,8].
The cpb gene is considered an efficient marker to discriminate between Leishmania species [38,54], based on PCR-size polymorphism. However, we highlighted intraspecific polymorphism (cpbE and cpbF) within our L. infantum strains. In fact, in some L. infantum zymodemes (i.e., ZMON-24, ZMON-187, ZMON-189, ZMON-190) we detected (by cpb-PCR assay) the copy cpbF, known to be specific of L. donovani, as also described in some L. infantum populations in Tunisia, Algeria, and recently in North Italy [48,54,55,56,57]. Therefore, the cpb-PCR size polymorphism is not a useful assay to distinguish L. donovani from L. infantum. The sequencing of 23 L. infantum and L. donovani strains displayed 7 cpbEF sequence variants (Table S1A). As we showed in the cpb dendrogram (Figure S3), the sequences variants cpbE(1–3) indicated an L. infantum cluster of strains from Mediterranean areas, whereas cpbF was not exclusive of L. donovani sp., but indicated a L. donovani complex cluster, with the sequence variants cpbF(1) and cpbF(2–4) identifying L. donovani and L. infantum strains, respectively (Table S5). Therefore, the validity of cpb sequencing as a L. infantum population genetic marker should be evaluated by a more comprehensive study.
The multi-target-based diagnostic approach, obtained by combining the ITS-hsp70 sequence variants and the copies cpbE and cpbF, allowed a more accurate identification of Leishmania populations than the single-target-based approach. We found that the geno-type C was the most represented in our samples, identifying 68.2% of L. infantum strains, which belonged to 12 zymodemes from the Mediterranean area (see Microreact Map at https://microreact.org/project/iss) (accessed on 30 November 2025) [50]. On the contrary, cpbF, in combination with the sequence variant hsp70inf(Y) (genotypes F and G), detected a ZMON-24 sub-cluster of CL cases, not ITS(Lombardi). In addition, cpbF, in combination with the reference sequence hsp70inf(2) (genotype H), identified a non-ZMON-1 cluster (i.e., ZMON-187, ZMON-189, ZMON-190). Due to the rarity of these latter zymodemes [7,8,9,10], it is not easy to increase the number of samples for further analyses in an effort to correlate this genotype to specific zymodemes or clusters of zymodemes. However, as part of collaborations with the Leishmaniasis centers of the Mediterranean Countries, we hope to further explore this issue.
With respect to the correlation of ZMONs with genotypes ITS-hsp70-cpb, we ob-served that L. infantum ZMON-1 was not very heterogeneous, being represented by gen-otype C, detected in 89.3% (n = 25/28) of strains (identical to the reference strain LEM75). In contrast, ZMON-24 (n = 25 strains) seems to be a polymorphic zymodeme, being identified by 6 genotypes, of which the most represented were C, E, F, and G (Table 1).
Moreover, the multi-target approach unveiled additional genetic populations in ZMON-24, whose relationships, also with respect to ZMON-1 and L. donovani, were inferred by PCoA analysis. Heterogeneous clinical, geographic and genetic aspects of the ZMON-24 sub-populations were evaluated: ITS(A)-hsp70inf(2)-cpbE(1, 2) identified all the most serious clinical cases, such as VL (with concomitant CL in HIV co-infection) and DCL, along with ITS(Lombardi)-hsp70inf(2)-cpbE(1), including both CL and VL cases, from all over the Mediterranean area. These genotypes were closer, if not identical, to those of ZMON-1. In contrast, ITS(A, B)-hsp70inf(Y)-cpbF(3, 4) included only ZMON-24 CL cases. The cpbF sequence variants further differentiated the exclusively Italian L. infantum strains, cpbF(3), from those in cluster Italy and North, cpbF(4) (Figure 1). A low-coverage whole-genome sequencing (WGS) of our strains, in particular of ZMON-24, is currently being performed to characterize these L. infantum populations and to evaluate its consistency with our typing by ITS-hsp70-cpb in a more comprehensive study.
Regarding the clinical correlation of ZMON-24 findings, different zymodemes are related to changes in biological features, such as clinical manifestation and virulence [58,59]. ZMON-24, widely spread in South Europe and North Africa, has been considered for a long time an isoenzymatic population with a dermotropic character, causing VL only in HIV-co-infected patients, and sporadically in children [59,60,61,62,63]. Researchers in previous studies, showed that some VL and CL ZMON-24 strains from Tunisia and Algeria, ana-lyzed by MLMT and cpb sequencing [47,48,64], could be divided into two genetic pop-ulations with clinical correlation. They also discussed whether this was a general trait of these populations, or a spurious association caused by geographical sampling bias. Our study, by different molecular targets, seems to confirm these observations. We also draw some conclusions from a speculative point of view.
First, in our sample set, L. infantum ZMON-24 includes two main genetic populations correlated to clinical characteristics. Hsp70inf(Y)-cpbF population could be a distinct evolutionary lineage within the L. donovani complex, originated in Africa and spread across the Mediterranean region (e.g., Algeria, Tunisia, Morocco, and Italy). This population could be a dermotropic variant of a normally viscerotropic species, although it inevitably causes VL in patients with HIV/Leishmania co-infections, such as ISS417 strain [7,57,60,64]. In contrast, the ZMON-24 population hsp70inf(2)-cpbE, closer to ZMON-1, appears virulent, causing VL in adult and children, immunocompetent and, more frequently, HIV-positive patients (8 strains) (Table S1A). These conclusions lead to the hypothesis that the clinical manifestation of ZMON-24 could also depend on its genetics, in addition to the host’s immunological state. Additional strains of different ZMON-24 CL and VL genotypes need to be investigated in order to better understand the role of parasite fitness and host susceptibility.
Second, it must be considered that discriminatory power of MLEE is limited, because genotypes are assayed indirectly, since synonymous nucleotide substitutions may not be observed and different allozymes may have coincident mobility. In contrast, despite identical genotypes, post-translational modifications may influence the electrophoretic mobility of the proteins [65]. Consequently, the ZMON-24 strains with genotype ITS(A, Lombardi)-hsp70inf(2)-cpbE(1) could be considered closer to L. infantum ZMON-1 viscerotropic strains, as highlighted by sequence analysis.
This study has some limitations. As already mentioned, it should be noted that our interpretation of the clinical–genetic correlations of the different molecular Leishmania populations was somehow speculative. Indeed, the correlation of specific molecular traits with zymodemes or clinical features requires more robust evidence, in terms of number of strains, and molecular approaches. More robust approaches will also be needed to assess the heterozygous status of some ZMON-24 strains. Moreover, due to the possible geographical sampling bias of our clinical samples, which are a retrospective collection (not representative of all the Mediterranean endemic areas), the actual spread of the described genotypes is not comprehensive. Nevertheless, this study can provide an overview of different genotypes, which were obtained by a multiple-target-based approach, and address the significance of some L. infantum sequence variants for each target detected in Mediterranean areas, linking them with specific zymodemes.
5. Conclusions
The wide heterogeneity of the Mediterranean L. infantum strains returned a molecular polymorphism through well validated species-specific molecular markers. Our results have confirmed the ITS and hsp70 validity as markers in Leishmania species genotyping; however, new L. infantum hsp70 variants have been described, and their correlation with specific L. infantum populations should be further investigated. On the contrary, PCR-cpb size polymorphism was found to be an ineffective species-specific marker in the Mediterranean area for the L. donovani complex species, showing an L. infantum intra-specific size polymorphism due to the detection of both copies cpbE and cpbF. However, a more comprehensive study will test whether the cpb sequencing can be used as a geographic marker for L. infantum. Although our multi-target approach does not allow for in-depth identification compared to other molecular approaches, it can easily and quickly distinguish several Leishmania populations. Finally, due to the Leishmania genetic heterogeneity detected by hsp70 and cpb, the leishmaniasis typing protocols should, in our view, provide a combination of these markers, associated with ITS, in the Mediterranean areas.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization Unveiling the Neglect of Leishmaniasis Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis(accessed on 24 May 2025)
- 2Ruiz-Postigo J.A. Jain S. Madjou S. Maia-Elkhoury A.N. Valadas S. Warusavithana S. Osman M. Yajim A. Lin A. Beshah A. Global Leishmaniasis Surveillance: 2019–2020, a Baseline for the 2030 Roadmap World Health Organization Geneva, Switzerland 2022575590
- 3Gradoni L. Lopez-Velez R. Mokni M. Manual on Case Management and Surveillance of the Leishmaniases in the WHO European Region WHO Regional Office for Europe Copenhagen, Denmark 2017
- 4European Centre for Disease Prevention and Control (ECDC) Surveillance, Prevention and Control of Leishmaniases in the European Union and Its Neighbouring Countries ECDC Technical Report ECDC Stockholm, Sweden 2022
- 5Maia C. Sand fly-borne diseases in Europe: Epidemiological overview and potential triggers for their emergence and re-emergence J. Comp. Pathol.202420961210.1016/j.jcpa.2024.01.00138320331 · doi ↗ · pubmed ↗
- 6Rioux J.A. Lanotte G. Serres E. Pratlong F. Bastien P. Perieres J. Taxonomy of Leishmania. Use of isoenzymes. Suggestions for a new classification Ann. Parasitol. Hum. Comp.19906511112510.1051/parasite/19906531112080829 · doi ↗ · pubmed ↗
- 7Gramiccia M. The identification and variability of the parasites causing leishmaniasis in HIV-positive patients in Italy Ann. Trop. Med. Parasitol.200397657310.1179/00034980322500254314678634 · doi ↗ · pubmed ↗
- 8Gramiccia M. Scalone A. Di Muccio T. Orsini S. Fiorentino E. Gradoni L. The burden of visceral leishmaniasis in Italy from 1982 to 2012: A retrospective analysis of the multi-annual epidemic that occurred from 1989 to 2009 Eurosurveillance 201318 e 2053510.2807/1560-7917.ES 2013.18.29.2053523929120 · doi ↗ · pubmed ↗