Mucosal Remodeling in Chronic Rhinosinusitis with Nasal Polyps: The Role of Innate Lymphoid Cells and Reprogramming Under IL-4Rα Blockade
Giovanna Lucia Piazzetta, Nadia Lobello, Silvia Di Agostino, Isabella Coscarella, Corrado Pelaia, Anna Di Vito, Jessica Bria, Andrea Filardo, Annamaria Aloisio, Chiara Lupia, Nicola Lombardo, Emanuela Chiarella

TL;DR
This paper explores how innate lymphoid cells, especially ILC2s, contribute to nasal inflammation and how blocking IL-4Rα with dupilumab can reduce symptoms in chronic rhinosinusitis with nasal polyps.
Contribution
The paper highlights the role of ILC2s in nasal inflammation and introduces ILC profiling as a potential biomarker for predicting treatment response to dupilumab.
Findings
ILC2s drive type 2 inflammation in nasal polyps through IL-5 and IL-13 release.
Dupilumab reduces ILC2 activity and improves nasal mucosa health by blocking IL-4/IL-13 signaling.
Baseline ILC2 phenotypes may predict treatment responsiveness to dupilumab.
Abstract
The nasal mucosa functions as a highly specialized barrier that integrates epithelial, stromal, neuronal, and immune signals to maintain homeostasis and mount rapid responses to environmental challenges. Among its resident immune populations, innate lymphoid cells—particularly type 2 ILCs (ILC2s)—play a pivotal role in orchestrating type 2 inflammation driven by epithelial-derived alarmins such as IL-25, IL-33, and TSLP. Upon activation, ILC2s release IL-5 and IL-13, promoting eosinophilic inflammation, goblet cell hyperplasia, mucus hypersecretion, and tissue remodeling, all central features of chronic rhinosinusitis with nasal polyps (CRSwNP) and severe allergic rhinitis. Recent advances have revealed substantial ILC plasticity, the presence of nasal-resident ILC progenitors, and the influence of metabolic and neuroimmune cues in shaping ILC activation and persistence. Dupilumab, a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
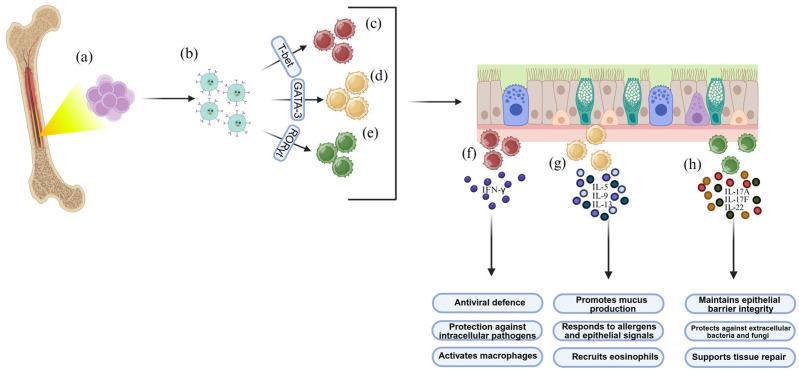 Figure 1
Figure 1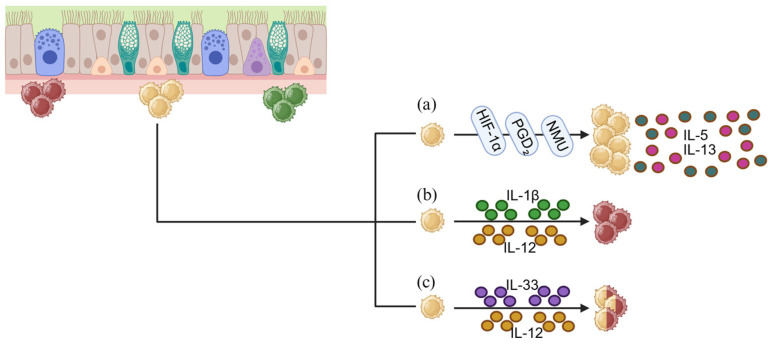 Figure 2
Figure 2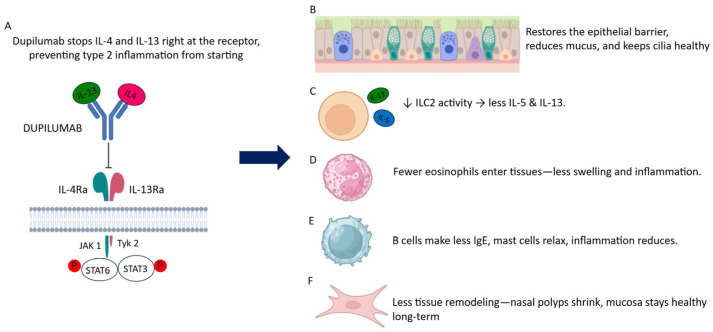 Figure 3
Figure 3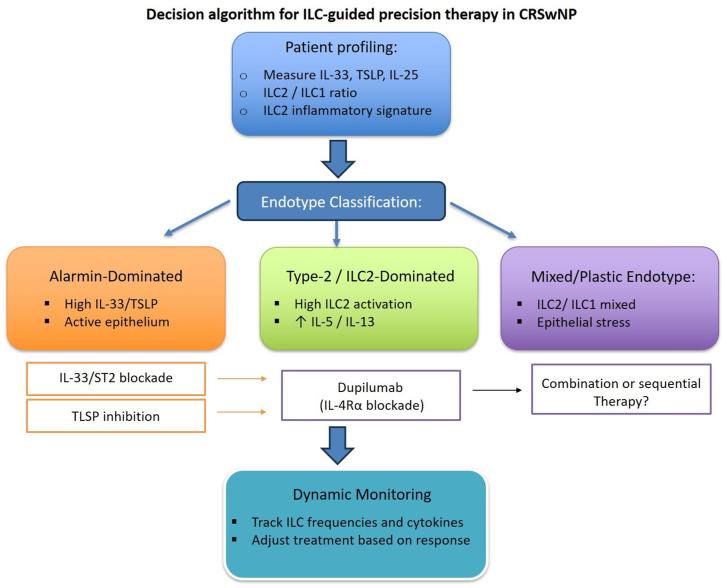 Figure 4
Figure 4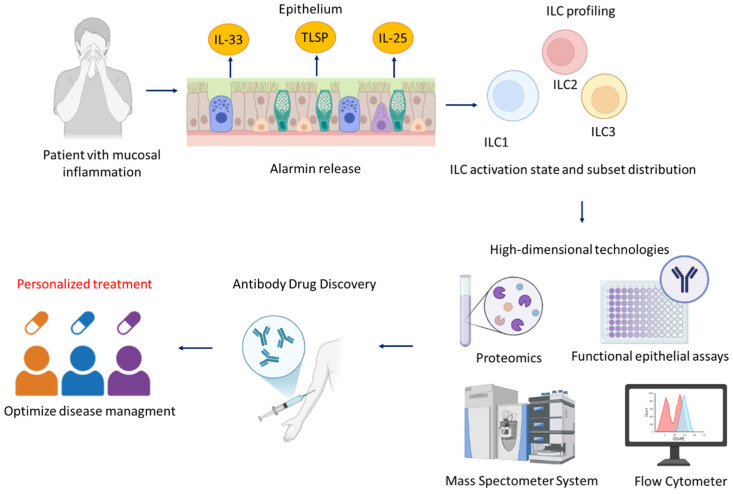 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIL-33, ST2, and ILC Pathways · Eosinophilic Esophagitis · Pediatric health and respiratory diseases
1. Introduction
The nasal cavity is the uppermost segment of the respiratory tract, acting as a sentinel site for airborne particles, allergens, and pathogens. It functions as a primary interface between the host and the external environment. Its mucosa is a specialized environment composed of epithelial, stromal, vascular, and immune components that cooperate to maintain tissue integrity and homeostasis [1,2]. Historically, mucosal immunity research has centered on adaptive lymphocytes, particularly T helper (Th) cells [3,4,5,6]. The identification of innate lymphoid cells (ILCs) has substantially reshaped this paradigm, revealing a rapid, tissue-resident immune compartment that operates upstream of adaptive immunity at barrier sites [7,8].
ILCs are lymphoid cells lacking antigen-specific receptors but capable of rapid cytokine production, mirroring Th subsets in function [9,10]. Within the nasal mucosa, ILCs localize beneath the epithelium, positioning them as key regulators of early immune activation and tissue responses [11,12]. ILC2s dominate this niche and orchestrate type 2 inflammation that is central to allergic rhinitis and CRSwNP [13,14]. By secreting IL-5 and IL-13, ILC2s promote eosinophilic infiltration, goblet cell hyperplasia, and mucus overproduction [15,16]. Beyond cytokine secretion, ILC2s engage in dynamic crosstalk with epithelial cells, fibroblasts, neurons, and eosinophils, forming integrated cellular networks that sustain chronic inflammation and drive tissue remodeling over time [17,18,19]. These observations position ILC biology at the interface between immune dysregulation and the architectural changes that underlie polyp growth and disease chronicity in CRSwNP [17,18,19].
The introduction of dupilumab, an IL-4Rα-blocking antibody, has improved clinical outcomes in patients with CRSwNP, offering novel research scenarios for the study of innate immune regulation. Dupilumab, a monoclonal antibody targeting the IL-4 receptor alpha (IL-4Rα) subunit shared by IL-4 and IL-13 receptors, has markedly improved clinical outcomes in patients with severe CRSwNP and has provided a unique opportunity to interrogate the role of type 2 immune pathways in mucosal disease [19,20,21,22,23]. Beyond its therapeutic efficacy, dupilumab treatment has revealed how modulation of epithelial–ILC interactions can recalibrate local immune responses and promote the restoration of tissue homeostasis [24]. Overall, dupilumab has demonstrated a favorable safety profile in both clinical trials and real-world studies [25,26,27]; however, rare cases of lymphoid neoplasms have been reported in patients treated for atopic dermatitis, based on isolated case reports, without evidence of a causal relationship [28,29,30,31]. Studies investigating ILC modulation under dupilumab therapy have highlighted how restoring epithelial–ILC balance recalibrates nasal immunity and supports tissue homeostasis.
In this context, mucosal remodeling is a central pathological hallmark of CRSwNP, encompassing epithelial barrier disruption, goblet cell hyperplasia, stromal edema, extracellular matrix reorganization, and polyp formation [32]. Disruption of this finely tuned mucosal system drives persistent inflammation and structural changes, with ILC2-derived cytokines—particularly IL-5 and IL-13—acting as key mediators by modulating epithelial differentiation, fibroblast activation, and eosinophil–stromal interactions [33,34]. Accordingly, ILC biology is not only central to inflammatory signaling but also to the mechanisms sustaining chronic tissue remodeling in the nasal mucosa [35].
This review aims to integrate recent insights from mucosal immunology and clinical studies to elucidate how ILC plasticity, epithelial–immune interactions, and dupilumab-mediated modulation converge to redefine nasal immune homeostasis. By framing ILCs within a remodeling-centered paradigm, we aim to provide a biologically grounded perspective on disease mechanisms and emerging therapeutic strategies.
2. Innate Lymphoid Cells and Progenitors in the Nasal Mucosa: Classification, Plasticity, and Functional Roles
2.1. ILC Subsets, Development, and Local Progenitors in the Nasal Mucosa
Innate lymphoid cells (ILCs) represent a heterogeneous family of lymphocyte-like innate immune cells that, despite lacking rearranged antigen receptors, exert key functions in early immune defense, tissue surveillance, and barrier protection [36,37]. Unlike T and B cells, they are equipped with pre-configured sensory programs enabling the rapid detection of cytokines, epithelial alarmins, and neuropeptides. This allows them to respond almost immediately to allergens, pathogens, or mechanical damage in mucosal tissues, including the nasal cavity, where they serve as first-line sentinels [35,38,39,40,41].
2.1.1. ILC Subsets in the Nasal Mucosa
In the nasal mucosa, ILCs are enriched beneath the epithelium and within the lamina propria, a location that affords them direct access to environmental signals. They are conventionally divided into three major functional groups—ILC1s, ILC2s, and ILC3s—mirroring the effector profiles of Th1, Th2, and Th17 cells, respectively [27]. ILC1s, characterized by T-bet expression, produce interferon-γ and contribute to antiviral defense and immunity against intracellular pathogens [42,43]. ILC2s depend on GATA-3 and generate IL-5, IL-9, and IL-13 in response to epithelial alarmins such as IL-25, IL-33, and TSLP, positioning them as primary drivers of allergic inflammation, mucus hypersecretion, and epithelial remodeling [44,45]. ILC3s require RORγt for their development and secrete IL-17A, IL-17F, and IL-22, thereby promoting epithelial repair, barrier stability, and protection against extracellular bacteria and fungi [44,46]. Although historically grouped within the type 1 ILC family, natural killer (NK) cells represent a distinct lineage specialized in cytotoxicity, relying on perforin- and granzyme-mediated killing and guided by transcription factors such as Eomes and T-bet [43,47]. Their presence in the upper airway complements the canonical ILC subsets by providing additional antiviral, antibacterial, and antitumor surveillance.
2.1.2. Developmental Pathways from Bone Marrow to Mucosal Surfaces
ILCs originate from common lymphoid progenitors (CLPs) in the bone marrow and progress through an intermediate stage known as the ILC progenitor (ILCp), defined by IL-7Rα, α4β7, and transcription factors such as PLZF, TOX, and ID2 (as shown in Figure 1a,b). Although already committed to the ILC lineage, these progenitors retain the capacity to differentiate into ILC1, ILC2, or ILC3 depending on local tissue cues [44,46,47,48,49]. The early innate lymphoid progenitor (EILP), marked by α4β7, Tcf7, Nfil3, Tox, Id2, Rora, and Gata3, gives rise to the common helper innate lymphoid progenitor (CHILP), which in turn generates the helper ILC subsets and lymphoid tissue inducer (LTi) cells [41,50,51,52].
From CHILP, two main pathways diverge: one leading to LTi progenitors, which express increasing levels of RORγt and participate in lymphoid tissue formation, and another giving rise to innate lymphoid cell progenitors (ILCPs), characterized by α4β7^+^ Thy1^+^ CD127^+^ PD-1^+^ and transcription factors such as PLZF (Zbtb16), Gata3, Id2, Tcf7, Tox, Runx, and Rora [53,54,55,56,57]. Depending on the cytokine milieu and local tissue cues, ILCPs then differentiate into ILC1s, ILC2s, or ILC3s (as shown in Figure 1c–e), with recent methodological advances—including efficient lentiviral gene-delivery systems—have further enabled detailed investigation of these developmental programs [41,58,59].
IL-7 is indispensable for the development of all helper ILCs, especially ILC2s and ILC3s, whereas IL-15 primarily supports NK-cell and certain ILC1 subset maturation [60,61,62,63,64,65]. Terminal differentiation is marked by the acquisition of lineage-defining transcription factors: T-bet for ILC1s, GATA-3 and RORα for ILC2s, and sustained RORγt expression for ILC3s [66,67,68,69,70,71,72]. These transcriptional programs tightly dictate the production of IFN-γ by ILC1s, IL-5/IL-9/IL-13 by ILC2s, and IL-17A/IL-17F/IL-22 by ILC3s (as shown in Figure 1f–h) [10,73].
After maturation, ILCs migrate from the bone marrow to peripheral tissues, including the upper airway, where epithelial-derived IL-7, IL-25, IL-33, TSLP, and retinoic acid refine their phenotype and functional specialization.
2.1.3. Tissue Residency, Local Progenitors, and Homeostatic Roles in the Nasal Mucosa
Increasing evidence indicates that ILC progenitors are not restricted to the bone marrow. ILCPs have been identified in several peripheral tissues, including the nasal mucosa, where they respond to epithelial and stromal mediators—such as IL-7, IL-1β, and retinoic acid—to generate mature ILC subsets in situ [74,75,76]. This local differentiation capacity suggests that the nasal mucosa maintains partial autonomy in replenishing and shaping its innate immune compartment, enabling rapid adaptations to environmental challenges without relying solely on continual recruitment from central hematopoietic organs [65,76,77,78,79]. Under homeostatic conditions, ILC2s constitute the predominant subset in the nasal mucosa [80]. Their basal secretion of IL-5 and IL-13, along with amphiregulin release, supports epithelial integrity and promotes steady-state repair processes. ILC3s contribute to structural maintenance through IL-22-mediated enhancement of tight junctions, mucin synthesis, and antimicrobial peptide production. Although less abundant, ILC1s and NK cells remain crucial for early antiviral defense via IFN-γ production and cytotoxic activity [81,82,83].
Together, these populations form an integrated surveillance system that interprets epithelial, microbial, and neuronal cues to maintain epithelial barrier stability and coordinate with adaptive immunity when necessary. The presence of local ILCPs ensures that this network remains dynamic, resilient, and capable of adapting to both acute insults and ongoing tissue maintenance.
2.2. Plasticity and Functional Reprogramming of Nasal ILCs
ILCs within the nasal mucosa exhibit a high degree of phenotypic plasticity, allowing them to adopt new functional states or even undergo trans-differentiation in response to cytokine gradients, microbial products, or signals of tissue injury. This adaptability is essential for quickly responding to the fluctuating environmental demands of the upper airway; however, dysregulation of these reprogramming processes may promote chronic inflammation and disrupt mucosal homeostasis [84,85,86].
A well-characterized example is the conversion of ILC2s into ILC1-like cells under the influence of IL-12 and IL-18 [86,87]. These cytokines induce T-bet expression and enhance IFN-γ production, conferring type 1—like features on formerly type 2—polarized cells [42]. This shift frequently occurs during viral infections or under persistent epithelial stress, representing an adaptive mechanism for reinforcing antiviral immunity. Conversely, alarmins such as IL-33 and TSLP preserve GATA-3 expression and stabilize the classical ILC2 phenotype, counteracting the drift toward a type 1 profile [42,86,88].
ILC3s also display considerable plasticity and can acquire ILC1-like characteristics under inflammatory conditions rich in IL-1β, IL-12, or IL-23. Downregulation of RORγt accompanies their transition toward IFN-γ production, leading to the loss of epithelial-supportive functions and the promotion of pro-inflammatory responses that may impair tissue repair. Such changes are often implicated in the perpetuation of epithelial barrier dysfunction in chronic upper airway inflammatory diseases [42,89,90,91,92].
Single-cell RNA sequencing and high-dimensional cytometry uncovered transitional populations, including GATA-3^+^T-bet^+^ intermediate ILC2/ILC1 states. These hybrid populations often expand in severe or steroid-refractory airway inflammation, where the microenvironment imposes mixed effector pressures. Their ability to simultaneously produce type 1 and type 2 cytokines may exacerbate inflammation and contribute to treatment resistance [91,93,94,95].
At the mechanistic level, ILC plasticity relies on epigenetic accessibility at key transcription factor loci, enabling rapid and potentially reversible modulation of effector programs. In the nasal mucosa—a tissue continuously exposed to allergens, microbes, pollutants, and epithelial alarmins—this epigenetic flexibility provides a powerful means of adaptation while also creating a vulnerability in which persistent inflammatory signals can drive maladaptive reprogramming and chronic disease [96,97].
2.3. Metabolic and Neuro-Immune Regulation of ILCs in the Nasal Mucosa
ILC development and function in the nasal mucosa are deeply influenced by metabolic constraints and by intricate neuro-immune circuits that integrate epithelial, stromal, and sensory inputs. In inflamed or obstructed nasal tissues, edema and reduced airflow often create hypoxic niches that stabilize HIF-1α and HIF-2α. These transcription factors reprogram cellular metabolism toward glycolysis, a shift that supports rapid ILC2 activation and sustains high-level IL-5 and IL-13 production even under metabolic stress [98,99,100,101,102,103]. This facilitates the amplification of type 2 inflammation. On the other hand, metabolites such as retinoic acid and short-chain fatty acids can attenuate ILC activity by promoting regulatory pathways and enhancing tissue repair, acting as counterbalances to excessive inflammatory responses [104].
Furthermore, neuronal inputs modulate ILC behavior. Sensory fibers innervating the nasal mucosa release neuropeptides—including neuromedin U (NMU), vasoactive intestinal peptide (VIP), and calcitonin gene-related peptide (CGRP)—that directly influence ILC activation states. NMU engages NMUR1 on ILC2s and induces potent activation accompanied by robust type 2 cytokine production [105,106,107]. VIP exerts context-dependent effects, at times promoting ILC2-mediated inflammation and at others supporting epithelial repair, depending on the prevailing cytokine environment [108,109]. CGRP, primarily released by trigeminal sensory neurons, displays bidirectional activity: it can enhance or suppress ILC functions based on concomitant inflammatory cues [110,111].
Lipid mediators provide yet another regulatory layer. Prostaglandin D_2_ (PGD_2_), produced by mast cells and epithelial cells, binds CRTH2 on ILC2s to enhance their recruitment and augment IL-5 and IL-13 secretion. In contrast, specialized pro-resolving mediators such as resolvins limit ILC2 activation and promote the resolution of inflammation, highlighting the delicate equilibrium between pro-inflammatory and pro-resolving lipid pathways [112,113,114,115].
Collectively, the integration of metabolic, neuro-immune, and lipid-derived signals shapes ILC behavior, survival, and effector functions within the nasal microenvironment. This multilayered regulatory network ultimately determines whether ILCs promote epithelial protection and homeostasis or drive persistent type 2 inflammation, offering insights into therapeutic strategies aimed at modulating ILC activity in chronic nasal inflammatory diseases.
3. Epithelial–ILC Crosstalk in the Nasal Mucosa
The nasal epithelium acts not only as a mechanical barrier but also as an active immunological sensor that continuously evaluates inhaled environmental stimuli and shapes local immune responses [116,117]. When exposed to allergens, pathogens, pollutants, or mechanical stress, epithelial cells rapidly release alarmins—including IL-33, IL-25, and TSLP—which represent the primary upstream signals activating ILC2s [118]. IL-33 engages the ST2 receptor on ILC2s, initiating MyD88-dependent NF-κB and MAPK pathways, whereas IL-25 and TSLP signal through IL-17RB and IL-7Rα receptor complexes, respectively. Together, these pathways induce rapid secretion of IL-5 and IL-13, cytokines that drive eosinophil recruitment, goblet cell hyperplasia, mucus hyperproduction, and tissue remodeling [119,120,121,122]. This establishes a self-reinforcing circuit that supports type 2 inflammation and contributes to diseases such as CRSwNP and severe allergic rhinitis [35,116,123,124].
Although IL-5 and IL-13 are the dominant effector cytokines produced by ILC2s, IL-4 plays a complementary and amplifying role in type 2 inflammation and nasal mucosal remodeling. Unlike IL-5 and IL-13, IL-4 is not primarily produced by ILC2s but is mainly derived from Th2 cells, basophils, and mast cells. IL-4 acts upstream in the type 2 cascade by promoting Th2 differentiation, IgE class switching, and sustained responsiveness of epithelial and stromal cells to IL-13. In the nasal mucosa, IL-4 enhances IL-13-dependent epithelial remodeling and indirectly contributes to mucus hypersecretion and barrier dysfunction by reinforcing type 2 immune polarization [118,125].
Beyond classical cytokine–receptor communication, epithelial–ILC interactions are influenced by neuroimmune signals and lipid mediators, which add complexity to the regulation of ILC activation. Neuropeptides, including NMU, VIP, and CGRP, can either amplify or restrain ILC2 activity depending on the microenvironmental context, thereby linking neuronal sensing to immune modulation. Lipid mediators such as PGD_2_ further enhance ILC2 chemotaxis and cytokine release through CRTH2 engagement, integrating environmental and neural inputs to fine-tune the magnitude and spatial distribution of type 2 responses [107,114,126,127].
ILCs also influence the adaptive immune system through multiple forms of crosstalk. IL-13 produced by activated ILC2s conditions dendritic cells to promote Th2 polarization, while IL-5 supports eosinophil maturation and survival, reinforcing a local inflammatory circuit [128,129]. In this context, IL-4 further sustains adaptive type 2 amplification by stabilizing Th2 identity and supporting IgE-mediated effector pathways, thereby indirectly contributing to chronic inflammation and tissue remodeling [130]. Moreover, ILCs directly interact with epithelial cells and fibroblasts to regulate epithelial barrier integrity and tissue remodeling, while regulatory mechanisms involving Tregs or IL-10-producing ILC subsets help limit excessive inflammation [83,88,131,132]. In chronic nasal diseases, however, these regulatory mechanisms often fail or become overwhelmed, contributing to persistent inflammation and polyp formation [118].
Recent findings suggest that nasal-resident ILC progenitors (ILCp) also participate in the dialogue between the epithelium and the innate lymphoid compartment. ILCp can sense epithelial-derived alarmins and other local cytokines, differentiating in situ into mature ILC2s or ILC3s. This localized differentiation provides the mucosa with the capacity to rapidly adapt its innate immune repertoire to environmental challenges, supporting both acute defense and chronic inflammatory remodeling [18,85,118].
Metabolic and microenvironmental cues further regulate epithelial–ILC communication. Hypoxia—commonly present in obstructed or inflamed nasal passages—induces HIF protein stabilization in ILC2s, favoring glycolysis and enhancing the effector functions. Nutrient availability, metabolite accumulation, and oxidative stress all influence cytokine production, survival, and plasticity. Importantly, plasticity enables ILC2s exposed to IL-12 or IL-18 to acquire ILC1-like features, while certain inflammatory contexts can drive ILC3-to-ILC1 transitions. These shifts help the tissue adapt to mixed inflammatory milieus but may also perpetuate chronic pathology [101,103,133,134].
In CRSwNP and allergic rhinitis, epithelial–ILC crosstalk becomes pathologically amplified. CRSwNP is characterized by marked expansion and activation of ILC2s beneath the epithelium and within the lamina propria, driven by persistent exposure to IL-33, IL-25, TSLP, and microbial signals. These activated ILC2s produce abundant IL-5 and IL-13, sustaining eosinophilia, mucus hypersecretion, epithelial remodeling, and ultimately polyp formation (as shown in Figure 2a) [35]. Within this inflammatory milieu, IL-4 contributes to the maintenance of a permissive type 2 environment by reinforcing Th2-driven immune circuits and amplifying IL-13-dependent remodeling responses [34]. In allergic rhinitis, particularly in severe or refractory cases, increased ILC2 frequencies and heightened type 2 cytokine production contribute to sustained mucosal edema and, in some patients, polypoid transformation [13]. Thus, ILC2s act as central regulators of type 2 inflammatory pathology across a spectrum of nasal diseases.
ILC plasticity further shapes disease severity. Hybrid ILC2/ILC1 populations—co-expressing GATA3 and T-bet and capable of producing both IL-13 and IFN-γ—have been identified in refractory CRSwNP and steroid-resistant airway disease [35]. These cells arise in contexts where type 2 alarmins coexist with IL-12 or other type 1—skewing cytokines and contribute to prolonged inflammation through combined type 1/2 effector profiles [86]. Likewise, chronic inflammation can promote ILC3-to-ILC1 transitions, exacerbating epithelial damage and hindering effective mucosal repair [135].
The nasal mucosa microenvironment strongly influences these processes. IL-12 and IL-1β promote ILC2-to-ILC1 conversion, IL-23 maintains ILC3 identity, and IL-33 and IL-25 drive ILC2 expansion. Simultaneously, neuropeptides, lipid mediators, stromal remodeling, and hypoxia-driven metabolic changes sustain a milieu that favors persistent ILC activation. Nasal-resident ILCp further reinforce chronic inflammation by continuously supplying new effector ILCs, highlighting why some patients experience recurrent or persistent disease despite therapy (as shown in Figure 2b,c) [89,136].
Overall, epithelial–ILC crosstalk in the nasal mucosa forms an intricate network in which alarmin signaling, neural inputs, metabolic cues, and progenitor dynamics converge to determine whether ILCs maintain tissue homeostasis or drive chronic inflammation and remodeling. Within this framework, IL-4 functions as an essential amplifier of type 2 immune polarization rather than a primary ILC2 effector cytokine, explaining its indirect yet biologically relevant contribution to nasal mucosal remodeling. These insights have clear translational implications. Therapeutic agents targeting upstream mediators of type 2 inflammation—particularly the IL-4Rα-blocking antibody dupilumab—can interrupt the epithelial–ILC amplification loop, reduce pathogenic ILC2 activity, and restore epithelial function. By recalibrating the inflammatory microenvironment, dupilumab not only alleviates symptoms but also modulates ILC behavior, underscoring the importance of understanding epithelial–ILC communication and ILC plasticity in chronic nasal disease [21,24,137].
3.1. Targeted Therapy in Nasal Inflammation: Mechanism of Action of Dupilumab and Therapeutic Relevance
Biologic therapies have substantially advanced the management of chronic rhinosinusitis with nasal polyps (CRSwNP) by selectively targeting key mediators of type 2 inflammation. Among these, monoclonal antibodies directed against IL-5 or its receptor—such as mepolizumab and benralizumab—have demonstrated efficacy in reducing eosinophil differentiation, survival, and tissue infiltration [138,139]. Because IL-5 represents a major effector cytokine produced by activated ILC2s, IL-5 blockade effectively attenuates eosinophil-driven inflammation and improves clinical outcomes in patients with eosinophil-dominant CRSwNP endotypes [140]. Nevertheless, IL-5-targeted therapies primarily act downstream within the type 2 inflammatory cascade and do not directly interfere with upstream epithelial activation, alarmin release, or ILC2 priming. As a consequence, their effects on epithelial remodeling and barrier dysfunction may be indirect or incomplete. In contrast, therapies targeting IL-4Rα inhibit both IL-4- and IL-13-mediated signaling pathways, thereby exerting broader effects on epithelial cells, innate lymphoid cells, and adaptive Th2 responses [141]. This upstream mode of action provides a mechanistic framework to investigate how modulation of the epithelial–ILC axis can reshape mucosal inflammation and remodeling, which is the focus of the following sections.
3.1.1. Molecular Basis of IL-4Rα Blockade
Dupilumab is a fully human IgG4 monoclonal antibody that selectively binds the interleukin-4 receptor alpha subunit (IL-4Rα), thereby preventing its heterodimerization with the common γ chain (γc) or with IL-13Rα1 [142,143,144]. Through this single molecular interaction, dupilumab inhibits signaling mediated by both IL-4 and IL-13, two cytokines that form the central hub of type 2 inflammation across epithelial, innate, and adaptive compartments (as shown in Figure 3A). By neutralizing IL-4/IL-13 signaling at this shared receptor, dupilumab effectively disrupts one of the earliest checkpoints of type 2 immune activation, making its mechanism broader and more upstream than that of cytokine-specific biologics. By blocking IL-4Rα, dupilumab interrupts the activation of JAK1-dependent pathways and abolishes downstream STAT6 phosphorylation, ultimately suppressing the transcription of genes involved in eosinophil recruitment, mucus hypersecretion, IgE class switching, epithelial remodeling, and survival programs sustaining innate lymphoid cells. This upstream blockade not only prevents downstream inflammatory cascades but also dampens epithelial stress responses, contributing to more rapid clinical improvement [142,145].
3.1.2. Effects on Epithelial Cells
Epithelial cells in the nasal mucosa express IL-4Rα/IL-13Rα1 and are highly responsive to IL-4 and IL-13, which act as potent modulators of epithelial differentiation, mucus production, and epithelial barrier integrity. Under type 2 cytokine stimulation, epithelial cells undergo goblet cell metaplasia, increased mucin gene transcription, reduced ciliary function, and enhanced release of pro-inflammatory chemokines and alarmins such as IL-33, TSLP, and IL-25 [146]. Dupilumab counteracts these deregulated pathways by restoring STAT6-independent differentiation programs, re-establishing tight-junction integrity, and normalizing epithelial turnover. Furthermore, IL-4Rα inhibition helps stabilize epithelial–mesenchymal interactions, reducing mechanical cues that perpetuate chronic mucosal remodeling. As IL-4/IL-13-driven epithelial stress decreases, the production of alarmins diminishes, relieving continuous activation of tissue-resident ILC2s [21,147]. This epithelial re-equilibration is clinically reflected in reduced mucus hypersecretion, improved airflow, and restoration of olfactory epithelial function (as shown in Figure 3B). These early epithelial changes are considered among the strongest contributors to the rapid symptomatic relief observed during dupilumab therapy [148,149].
3.1.3. Effects on ILC2s and Innate Immunity
ILC2s are among the most responsive cell types to IL-4 and IL-13, which reinforce GATA-3 expression, maintain effector polarization, and enhance responsiveness to IL-33, IL-25, and TSLP. Within the nasal mucosa, these cytokines foster ILC2 survival, proliferation, and sustained production of IL-5 and IL-13, creating a feed-forward loop of eosinophilic inflammation and epithelial dysfunction [34,150]. Dupilumab interferes with this loop by withdrawing essential cytokine support for activated ILC2s (as shown in Figure 3C). As IL-4Rα-dependent signals diminish, ILC2s exhibit reduced activation marker expression, downregulated CRTH2 and CD69, and a marked decline in IL-5 and IL-13 secretion [34,138]. Notably, this reduction in ILC2 effector output occurs without triggering apoptosis, indicating a functional “quiescence” rather than depletion. Transcriptomic studies indicate that IL-4Rα blockade shifts ILC2s toward a less effector-polarized state without forcing transdifferentiation, thereby attenuating their pathogenic potential while preserving baseline homeostatic functions. This rapid modulation of the innate compartment aligns with the early clinical benefits reported in CRSwNP patients treated with dupilumab [34,138].
However, it remains unclear whether this quiescent state induced by IL-4Rα blockade is sustained after treatment cessation or represents a transient suppression of effector function. Understanding this distinction is pivotal in considering whether dupilumab may confer disease-modifying effects rather than solely symptomatic control.
Emerging evidence suggests that ILC2s can acquire features of “innate immune memory,” characterized by durable changes in functional responsiveness upon repeated stimulation. In both experimental models and human studies, subsets of ILC2s have been shown to persist and respond more vigorously upon secondary challenge, with associated changes in chromatin accessibility that underlie trained or memory-like states [151].
Mechanistically, repetitive inflammatory stimulation leads to epigenetic reprogramming in ILC2s, involving alterations in chromatin landscapes (e.g., changes in accessibility at key loci such as TLR4 and other memory-associated gene motifs), implicating transcriptional regulators and chromatin modifiers in establishing persistent functional states [152]. These findings raise the possibility that IL-4Rα blockade with dupilumab might not only suppress effector cytokine production acutely but could also influence the epigenetic “inflammatory memory” of ILC2s, potentially diminishing their propensity for reactivation after allergen exposure or treatment discontinuation. Although direct evidence for long-term epigenetic reprogramming of ILC2s in the context of IL-4Rα inhibition is not yet established, the concept aligns with broader observations of innate immune memory in ILC subsets and provides a possible framework for future investigation. Thus, modulation of ILC2s by dupilumab might extend beyond transient effector suppression toward a more durable recalibration of innate immune responsiveness, with implications for disease modification and tolerance.
3.1.4. Effects on Eosinophils and Granulocytes
Although IL-5 remains the principal cytokine responsible for eosinophil differentiation and survival, IL-4 and IL-13 indirectly regulate eosinophil biology by inducing epithelial and stromal production of chemokines such as eotaxins (CCL11, CCL24, CCL26). By neutralizing IL-4Rα signaling, dupilumab reduces eotaxin production, limits eosinophil recruitment, and diminishes tissue eosinophilia (as shown in Figure 3D). This attenuation of eosinophil trafficking contributes significantly to reductions in stromal edema and glandular hyperplasia. In nasal polyposis, this results in reduced epithelial edema and attenuation of eosinophil-driven tissue remodeling [21]. While peripheral eosinophil counts may transiently rise in some patients as a result of altered trafficking dynamics, tissue eosinophils consistently decline, correlating with structural and symptomatic improvement. This transient peripheral eosinophilia is generally considered a benign redistribution phenomenon rather than a marker of increased inflammation [20].
3.1.5. Effects on B Cells and IgE Pathways
IL-4 is essential for immunoglobulin class switching to IgE, while IL-13 supports IgE amplification and influences stromal and epithelial IgE-dependent responses. Dupilumab inhibits these signaling pathways, leading to a progressive reduction in total and local IgE production (as shown in Figure 3E) [21]. Although IgE levels decrease more slowly than other markers of type 2 inflammation, their gradual decline reflects a sustained inhibition of IL-4-driven humoral immunity [21,145]. Although IgE normalization proceeds more slowly compared to cytokine suppression, the reduction in IgE-dependent mast-cell activation contributes to lower mediator release, reduced neurogenic inflammation, and diminished amplification of type 2 effector circuits within the nasal mucosa. This long-term modulation of IgE biology adds an additional layer of therapeutic benefit beyond the immediate suppression of type 2 cytokine activity [20].
3.1.6. Effects on Stromal Cells, Fibroblasts, and Tissue Remodeling
Beyond immune regulation, IL-4 and IL-13 are key profibrotic cytokines that act on fibroblasts and structural cells to promote collagen deposition, extracellular matrix remodeling, periostin upregulation, and subepithelial fibrosis (as shown in Figure 3F) [34]. These remodeling pathways are central to polyp formation and recurrence in CRSwNP [34,153,154]. By interrupting IL-4Rα signaling in stromal cells, dupilumab attenuates fibroblast activation and matrix gene transcription, progressively reducing the structural drivers of nasal polyp growth [142]. This effect is particularly relevant for long-standing disease, in which fibrotic and stromal changes are major contributors to symptom persistence. Imaging studies and histopathology confirm that polyp regression during therapy correlates with molecular signatures of reduced type 2-driven remodeling and normalization of epithelial–stromal interactions. Over time, these modifications support a more stable and less relapse-prone mucosal architecture [155].
Clinical trials and translational studies have consistently shown that IL-4Rα blockade leads to a significant reduction in type 2 inflammatory biomarkers—including total IgE, eotaxin-3, IL-5, and IL-13—in nasal secretions, serum, and polyp tissue. These molecular changes occur early during treatment and are associated with reductions in polyp size, mucosal edema, and radiologic disease burden, indirectly reflecting improved epithelial–stromal interactions and tissue architecture [20,143].
From a histopathological perspective, direct quantitative assessments of classical remodeling features—such as basement membrane thickening, subepithelial fibrosis, or collagen deposition—before and after dupilumab therapy remain limited. However, emerging real-world and biopsy-based observations indicate that modulation of the IL-4/IL-13 axis influences stromal remodeling pathways. In particular, residual or treatment-refractory polyp tissues have been reported to display increased deposition of extracellular matrix components and enhanced expression of periostin, a matricellular protein strongly induced by IL-13 and implicated in fibrotic remodeling, suggesting that structural remodeling may evolve more slowly than inflammatory suppression and may differ according to individual response patterns [155].
The involvement of periostin in CRSwNP remodeling is biologically plausible and well supported. Elevated periostin expression in nasal polyp tissue correlates with epithelial barrier disruption, increased basement membrane thickness, goblet cell hyperplasia, and eosinophilic infiltration—hallmark features of chronic mucosal remodeling [156]. Because periostin is produced mainly by activated fibroblasts in response to IL-4 and IL-13 signaling, its modulation represents a key link between immune dysregulation and structural tissue changes.
In parallel, transforming growth factor-β (TGF-β) signaling plays a central role in regulating extracellular matrix turnover and fibrosis in CRSwNP. Histological studies have demonstrated altered spatial and cellular patterns of TGF-β expression in nasal polyps, particularly within the subepithelial stroma, contributing to abnormal collagen deposition and tissue stiffness. By inhibiting upstream IL-4/IL-13-dependent fibroblast activation, dupilumab may indirectly influence TGF-β-driven remodeling pathways, thereby promoting gradual normalization of stromal architecture over time [157].
Overall, while dupilumab clearly induces rapid immunological reprogramming and clinical improvement, robust evidence of complete reversal of established histological remodeling remains limited. Current data support a model in which IL-4Rα blockade initiates early epithelial and immune normalization, followed by slower, progressive modulation of fibrosis-related pathways, highlighting the need for longitudinal biopsy studies incorporating standardized histological scoring systems to better define the extent and durability of mucosal remodeling under biologic therapy.
3.1.7. Integrated Clinical and Biological Implications
The combined effects of IL-4Rα blockade on epithelial cells, ILC2s, eosinophils, B cells, and stromal elements create a coordinated reprogramming of the nasal mucosal microenvironment. Early clinical improvements largely mirror rapid suppression of epithelial dysfunction and ILC2 effector activity, whereas long-term benefits reflect deeper inhibition of remodeling pathways, reduced IgE-mediated amplification, and restoration of immune homeostasis [34].
This layered temporal pattern with a rapid symptomatic relief followed by slower structural normalization, helps explain the durability of dupilumab’s clinical response, as highlighted in the SINUS-24 and SINUS-52 trials [20,34]. Moreover, the dual impact on epithelial and innate compartments provides a mechanistic rationale for investigating baseline ILC2 phenotypes as potential biomarkers of therapeutic responsiveness. Altogether, IL-4Rα blockade emerges as a uniquely comprehensive strategy capable of reshaping both immune and structural components of type 2 nasal inflammation [158].
3.2. Dupilumab Effects on Nasal ILCs
Dupilumab exerts a coordinated immunoregulatory effect on the nasal mucosa by interrupting IL-4/IL-13–dependent pathways that sustain chronic ILC2 activation. In nasal tissue and peripheral blood, treatment is associated with a rapid decline in activated ILC2s, reflected by reduced expression of CRTH2, CD69 and GATA-3 and by diminished secretion of IL-5 and IL-13—key mediators of eosinophilic inflammation and mucus hyperproduction [19,21,137]. These early immunological shifts mirror the prompt disruption of the epithelial–ILC2 feedback loop that drives persistent type 2 inflammation. A comprehensive overview of dupilumab-mediated effects on nasal innate lymphoid cells, including mechanisms and clinical consequences, is provided in Table 1.
At the epithelial level, transcriptomic analyses demonstrate that IL-4Rα blockade promotes epithelial barrier restoration by upregulating genes involved in junctional integrity and ciliogenesis, while simultaneously reducing epithelial alarmins and chemokines that fuel innate activation [24,146]. This early stabilization of the epithelial barrier attenuates upstream inflammatory stimuli and contributes to the progressive functional quiescence of local ILC2s.
Beyond immediate effector suppression, dupilumab also influences the transcriptional and epigenetic programs governing ILC2 identity. Studies on ILC2 plasticity show that chronic exposure to inflammatory cytokines can drive the emergence of unstable intermediate phenotypes—hybrid ILC2/ILC1 states—associated with disease severity and reduced responsiveness to therapy. IL-4/IL-13 signaling plays a central role in maintaining chromatin accessibility at type 2 cytokine loci and in reinforcing effector polarization. Thus, IL-4Rα blockade reduces accessibility to these loci and stabilizes GATA-3-driven transcriptional programs. By limiting stress-induced plasticity, dupilumab helps preserve a more homeostatic and less pathogenic ILC2 phenotype [42,91,96,97].
A recent translational study by Golebski and colleagues [40] provided critical insight into how baseline innate lymphoid cell composition influences dupilumab responsiveness in CRSwNP. In a cohort of 38 CRSwNP patients treated with dupilumab, high-dimensional flow cytometry revealed that individuals with elevated baseline frequencies of inflammatory ILC2s (CD45RO^+^, CD62L^−^) were fast clinical responders, whereas slow responders showed increased proportions of ILC3s. Inflammatory ILC2s, derived from resting CD45RA^+^ ILC2s under mucosal cytokine stimulation, were enriched in patients who rapidly improved, suggesting that a strongly T2-skewed yet targetable immune landscape predicts greater treatment sensitivity. Functional assays confirmed that dupilumab directly suppressed IL-5 and IL-13 production by ILC2s in vitro without affecting their activation or differentiation state, demonstrating that IL-4Rα blockade primarily curtails cytokine effector output rather than cellular conversion. The authors proposed that baseline frequencies of inflammatory ILC2s may serve as predictive biomarkers of dupilumab efficacy and response speed, linking the therapy’s clinical benefits to its capacity to limit IL-13-mediated epithelial and eosinophilic inflammation. This study is particularly important because it connects clinical trajectories to precise innate immune phenotypes, underscoring the possibility of stratifying CRSwNP patients according to ILC-based biomarkers.
These findings reinforce the concept that dupilumab acts not only as a cytokine inhibitor but also as a modulator of innate immune dynamics, recalibrating the epithelial–ILC axis toward homeostasis. The early symptomatic improvements observed in fast responders likely reflect the rapid reduction of IL-13-driven inflammation and epithelial dysfunction. Moreover, by attenuating IL-33 and TSLP release from epithelial cells, dupilumab disrupts the amplification loop that perpetuates ILC2 activation, allowing restoration of mucosal integrity. This break in feedback signaling appears to be one of the pivotal steps in shifting the nasal environment from a perpetually activated state toward a more quiescent tissue profile [40].
Importantly, the profound reduction of epithelial alarmin release and type 2 inflammatory mediators also affects the behavior of nasal ILC progenitors (ILCp). Emerging evidence suggests that IL-4/IL-13 blockade decreases local differentiation signals that favor the continuous generation of inflammatory ILC2s from resident ILCp, thereby limiting the renewal of pathogenic effector subsets within the mucosa. This observation provides an explanation for the sustained benefit of dupilumab even in long-standing disease, where constant replenishment of inflammatory ILC2s contributes to chronicity and recurrence [40].
Further evidence of dupilumab’s immunomodulatory role at the systemic level was provided by Matsuyama and colleagues [19], who evaluated circulating immune profiles in CRSwNP patients before and after 24 weeks of therapy. Their study demonstrated a significant and sustained decrease in peripheral ILC2 frequencies following dupilumab treatment, paralleled by reductions in Th2 cells and serum IL-5 and IL-13 concentrations. The Th2/Th1 ratio markedly declined, while regulatory T cells showed a mild compensatory increase, suggesting a shift toward immune equilibrium. Notably, the decline in ILC2s correlated with both Th2 suppression and clinical improvement, indicating that IL-4Rα blockade exerts coordinated inhibition of innate and adaptive type 2 immunity. These systemic data reinforce the concept that dupilumab’s impact extends beyond local mucosal lesions, contributing to a global rebalancing of type 2 immunity [19].
Collectively, these mechanistic studies position dupilumab as a dual-level immunologic modulator, one that rapidly neutralizes type 2 cytokine signaling while progressively reprogramming both innate and adaptive immune compartments. By targeting the shared IL-4/IL-13 receptor, dupilumab interrupts the epithelial–ILC–Th2 feedback loop central to CRSwNP pathogenesis, leading to durable disease control and mucosal homeostasis. The identification of inflammatory ILC2s as both effectors and biomarkers of treatment response opens the way for personalized therapeutic strategies and real-time monitoring of biologic efficacy in type 2 inflammatory diseases of the upper airway. This perspective aligns with a broader shift in the field toward precision immunology, where innate immune profiling may soon guide both treatment selection and prediction of response dynamics.
Taken together, these findings highlight the central importance of ILC2s, along with their progenitors and transitional states, as both therapeutic targets and potential biomarkers of treatment responsiveness, opening the way for increasingly personalized approaches to biologic therapy in nasal type 2 inflammatory disease. As research continues to refine our understanding of ILC behavior, dupilumab may represent not only an effective therapeutic option but also a model for how targeted cytokine blockade can reset dysregulated innate immune networks in chronic airway disease.
4. Toward Precision Medicine: Integrating ILC Biology into Personalized Treatment Strategies
The rapidly expanding understanding of ILC biology and epithelial–immune crosstalk in the nasal mucosa is reshaping the way type 2 inflammatory diseases are defined, stratified, and treated [40,42,145]. Rather than viewing CRSwNP and severe allergic rhinitis as uniform clinical entities, current evidence reveals a constellation of distinct immunological endotypes, each driven by a characteristic pattern of epithelial activation, alarmin release, ILC subset composition, and downstream Th2 polarization [42,91,96,135,159]. This evolving framework is accelerating the transition from conventional symptom-oriented care toward precision immunomodulation, in which biologic therapy is tailored to the dominant inflammatory circuits operating in each patient [40,145].
A growing body of translational research indicates that the baseline architecture of the innate immune compartment profoundly shapes therapeutic responsiveness. Patients with a high burden of activated or inflammatory ILC2s, or with a strongly type-2-skewed ILC profile, frequently experience faster and more robust responses to IL-4Rα blockade. By contrast, individuals with higher proportions of ILC3s, mixed ILC2/ILC1 states, or signatures of ILC plasticity may require longer treatment durations or may benefit from alternative upstream strategies, particularly in cases where epithelium-derived alarmins remain the primary drivers of inflammation. These findings underscore the potential of ILC-centric immune profiling—including inflammatory vs. resting ILC2 states, ILC3-to-ILC1 ratios, and global ILC abundance—as a biologically grounded tool for meaningful patient stratification [42,91,96].
In parallel, epithelial biology is emerging as an equally powerful determinant of treatment choice. Elevated IL-33 or TSLP expression, persistent epithelial stress responses, and structural barrier defects define patients with alarmin-dominated disease, a phenotype that may be optimally addressed through targeted IL-33/ST2 or TSLP inhibition. Conversely, patients with modest alarmin elevation but strong type-2 cytokine output may benefit most from dupilumab as a first-line biologic, reflecting a downstream blockade approach within the epithelial–ILC–Th2 axis. In this context, an algorithmic strategy integrating epithelial alarmin levels, ILC subset composition (including ILC2/ILC1 balance and inflammatory ILC2 signatures), and downstream inflammatory markers may help translate immunological complexity into clinically actionable treatment selection, providing a framework for precision medicine in type 2 upper airway disease (as shown in Figure 4).
This concept of endotype-directed therapeutic sequencing—intervening upstream or downstream according to the predominant inflammatory pathway—represents a rational and increasingly evidence-supported strategy for optimizing clinical outcomes [137,145,160].
Precision medicine also extends beyond baseline stratification to include longitudinal immune monitoring. Peripheral and tissue-resident ILC2 frequencies, nasal cytokine patterns, epithelial barrier biomarkers, transcriptional states, and even epigenetic signatures of ILC activation are emerging as dynamic readouts of treatment efficacy. Integrating these markers with high-dimensional technologies such as mass cytometry, single-cell transcriptomics, proteomics, and functional epithelial assays may ultimately enable real-time adjustment of biologic therapy, ensuring durable disease control while avoiding unnecessary exposure to immunomodulation.
Future personalization strategies may further incorporate metabolic, neuroimmune, and microenvironmental determinants of ILC2 activation. Because ILC2s are highly sensitive to oxygen availability, nutrient gradients, neuronal mediators, and tissue-derived lipids, these cues may help refine dosing intervals, identify windows of heightened responsiveness, or guide the use of combination approaches that synergize with IL-4Rα blockade. Within this emerging paradigm, therapy may extend beyond monotherapy to coordinated regimens that simultaneously or sequentially target epithelial alarmins, ILC effector pathways, and the microenvironmental context that sustains type 2 inflammation [42,91,96].
Taken together, these insights mark a decisive shift toward a biologically anchored, patient-specific model of care. By integrating epithelial profiling, ILC subset analysis, and dynamic biomarker monitoring, the field moves closer to truly personalized therapy for CRSwNP and severe allergic rhinitis. Within this broader perspective, Figure 5 illustrates how current and emerging biologic therapies may be positioned along the epithelial–ILC–Th2 axis, emphasizing the continuum and plasticity of inflammatory pathways rather than a fixed treatment hierarchy. In this landscape, dupilumab represents not only a highly effective treatment but also a proof-of-concept model demonstrating how mechanistic understanding of the epithelial–ILC–Th2 axis can translate into precision interventions. Importantly, this conceptual framework is not limited to IL-4Rα-targeted therapy. Emerging biologic agents directed against epithelial-derived alarmins, particularly thymic stromal lymphopoietin (TSLP), may further expand precision medicine strategies by acting upstream at the level of epithelial–immune crosstalk. In patients with alarmin-dominated disease signatures, such approaches may complement or, in selected endotypes, represent alternatives to downstream cytokine blockade [161,162,163]. Ultimately, the expanding knowledge of ILC biology positions innate lymphoid cells at the center of next-generation, endotype-driven therapeutic strategies for type 2 inflammatory diseases of the upper airway.
5. Conclusions
Innate lymphoid cells, particularly ILC2s, emerge as central conductors of type 2 inflammation in the nasal mucosa, integrating signals from epithelial alarmins, neuronal pathways, metabolic cues, and stromal interactions into coordinated effector programs. Their strategic localization at epithelial barrier surfaces, rapid responsiveness to danger signals, and remarkable functional plasticity place them at the crossroads of tissue immunity, where they drive eosinophilic inflammation, mucus hypersecretion, and the progressive remodeling characteristic of chronic disease. Recent insights further reveal that both mature ILC2s and their resident progenitors contribute not only to disease initiation but also to its persistence and chronicity, sustaining inflammatory cycles that define CRSwNP and severe allergic rhinitis.
Dupilumab, through targeted IL-4Rα blockade, exemplifies how a biologic therapy grounded in mechanistic understanding can modulate interconnected innate and adaptive pathways. Clinical and translational studies consistently show that dupilumab reduces activated ILC2 populations, suppresses IL-5 and IL-13 secretion, restores epithelial barrier integrity, and disrupts the epithelial–ILC–Th2 amplification loop that underlies chronic type 2 inflammation. Meanwhile, emerging upstream therapies targeting IL-33 or TSLP provide complementary opportunities to intervene even earlier in the cascade of ILC activation, broadening the therapeutic armamentarium.
Looking ahead, the integration of high-dimensional immune profiling, epithelial barrier assessment, metabolic characterization, and ILC-focused biomarkers holds promise for a new era of deeply personalized immunomodulation. By uniting fundamental mechanistic insight with clinical precision, advances in nasal ILC biology offer a transformative framework for long-term management of type 2 inflammatory diseases of the upper airway.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alfi O. Yakirevitch A. Wald O. Wandel O. Izhar U. Oiknine-Djian E. Nevo Y. Elgavish S. Dagan E. Madgar O. Human Nasal and Lung Tissues Infected Ex Vivo with SARS-Co V-2 Provide Insights into Differential Tissue-Specific and Virus-Specific Innate Immune Responses in the Upper and Lower Respiratory Tract J. Virol.20219510112810.1128/JVI.00130-21PMC 822392033893170 · doi ↗ · pubmed ↗
- 2Zhang R. Zhang L. Li P. Pang K. Liu H. Tian L. Epithelial Barrier in the Nasal Mucosa, Related Risk Factors and Diseases Int. Arch. Allergy Immunol.202318448150110.1159/00052896936724763 PMC 10137320 · doi ↗ · pubmed ↗
- 3Jong R.M. Van Dis E. Berry S.B. Nguyenla X. Baltodano A. Pastenkos G. Xu C. Fox D. Yosef N. Mc Whirter S.M. Mucosal Vaccination with Cyclic Dinucleotide Adjuvants Induces Effective T Cell Homing and IL-17–Dependent Protection against Mycobacterium Tuberculosis Infection J. Immunol.202220840741910.4049/jimmunol.210002934965963 PMC 8755605 · doi ↗ · pubmed ↗
- 4Li M. Gan C. Zhang R. Wang J. Wang Y. Zhu W. Liu L. Shang J. Zhao Q. TRAF 5 Regulates Intestinal Mucosal Th 1/Th 17 Cell Immune Responses via Runx 1 in Colitis Mice Immunology 202317049550910.1111/imm.1368537575027 · doi ↗ · pubmed ↗
- 5Brockmann L. Tran A. Huang Y. Edwards M. Ronda C. Wang H.H. Ivanov I.I. Intestinal Microbiota-Specific Th 17 Cells Possess Regulatory Properties and Suppress Effector T Cells via c-MAF and IL-10Immunity 20235627192735.e 710.1016/j.immuni.2023.11.00338039966 PMC 10964950 · doi ↗ · pubmed ↗
- 6Piazzetta G.L. Lobello N. Pelaia C. Mariaimmacolata P. Lombardo N. Chiarella E. Modulating Nasal Barrier Function and Tissue Remodeling in Inflammatory Diseases: The Role of Ginseng and Its Bioactive Compounds Tissue Barriers 202513247047710.1080/21688370.2025.247047739988791 PMC 12667642 · doi ↗ · pubmed ↗
- 7Ryu S. Lim M.Y. Kim J. Kim H.Y. Versatile Roles of Innate Lymphoid Cells at the Mucosal Barrier: From Homeostasis to Pathological Inflammation Exp. Mol. Med.2023551845185710.1038/s 12276-023-01022-z 37696896 PMC 10545731 · doi ↗ · pubmed ↗
- 8Fan H. Wang A. Wang Y. Sun Y. Han J. Chen W. Wang S. Wu Y. Lu Y. Innate Lymphoid Cells: Regulators of Gut Barrier Function and Immune Homeostasis J. Immunol. Res.20192019252598410.1155/2019/252598431930146 PMC 6942837 · doi ↗ · pubmed ↗