SARS-CoV-2 Spike Protein XBB.1.5 Mutations Altered Four Conserved Antigenic Determinants
Ekrem Akbulut, Meltem Yildirim, Huseyin Kahraman

TL;DR
The XBB.1.5 variant of SARS-CoV-2 has mutations that change key parts of the spike protein, helping it evade antibodies and highlighting the need for updated vaccines.
Contribution
This study identifies four conserved antigenic determinants altered by XBB.1.5 mutations, providing insights into immune evasion mechanisms.
Findings
XBB.1.5 has 38 amino acid substitutions compared to the original Wuhan-Hu-1 strain.
Four conserved antigenic determinants are altered, reducing antibody recognition.
The spike protein maintains structural stability and ACE2 binding despite mutations.
Abstract
The continuous evolution of SARS-CoV-2 affects its infectivity and ability to evade the immune system. The XBB.1.5 subvariant carries numerous mutations compared to previous Omicron variants and exhibits significant evasion of polyclonal neutralizing antibodies. In this study, the mechanistic effects of mutations in the XBB.1.5 spike protein on structural stability, antigenic markers, and antibody epitopes were analyzed using homology modeling, epitope prediction, protein stability analysis, coarse-grained dynamic simulations, and chain-specific interface mapping. Thirty-eight amino acid substitutions were identified relative to Wuhan-Hu-1, including 22 in the receptor-binding region. The prefusion trimeric fold was conserved, with localized rearrangements in the N-terminal domain, receptor-binding domain, and S1/S2 region. Linear B-cell epitope prediction yielded similar epitope counts…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
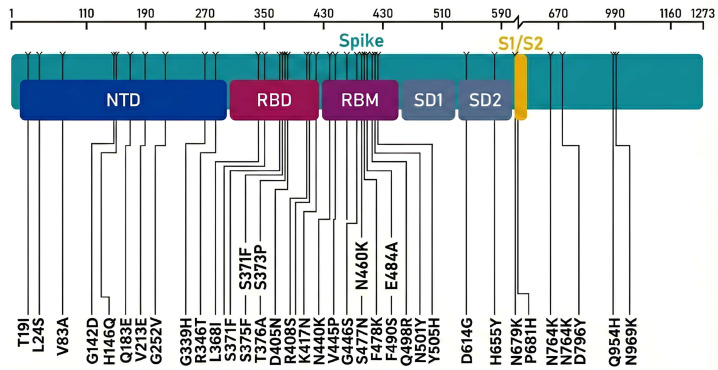 Figure 1
Figure 1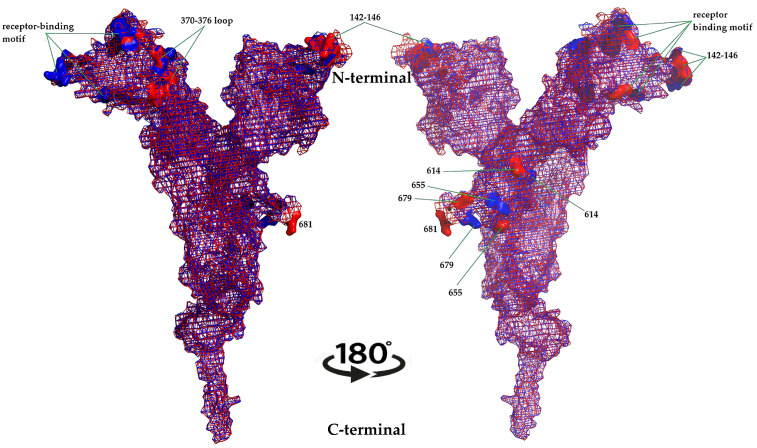 Figure 2
Figure 2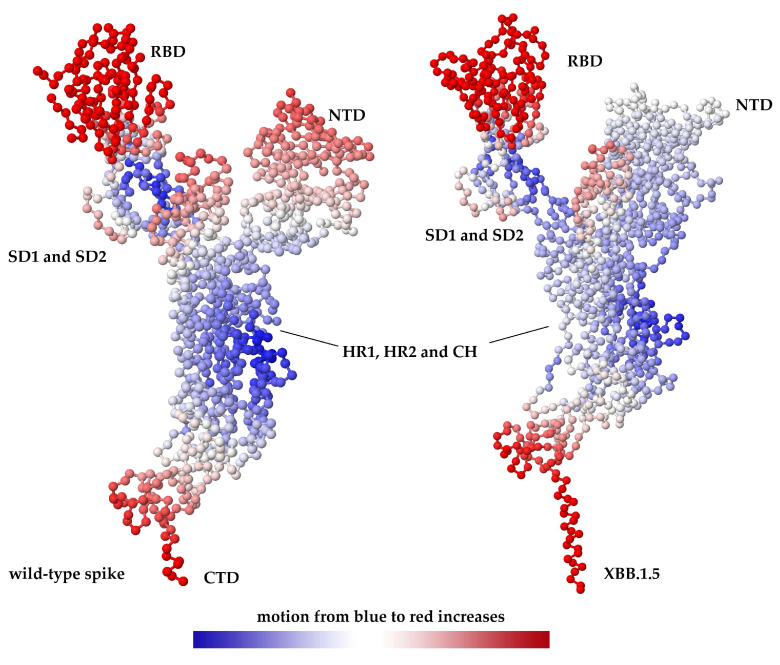 Figure 3
Figure 3- —Inonu University Scientific Research Projects Unit
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSARS-CoV-2 and COVID-19 Research · vaccines and immunoinformatics approaches · Monoclonal and Polyclonal Antibodies Research
1. Introduction
Coronavirus disease 2019 (COVID-19), the etiological agent of which is caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), is one of the most significant health problems of the twenty-first century. SARS-CoV-2, which has caused the death of more than 7 million people, is a positive-sense single-stranded RNA virus. The spike (S) protein, essential for the infectivity of SARS-CoV-2 and other coronaviruses, belongs to class I viral fusion proteins, similar to several proteins that have been extensively studied, such as HIV’s Gp41 and the influenza virus hemagglutinin. Like other fusogens in this category, the S-protein is trimeric and features a large ectodomain visible on the virion surface, an -helical transmembrane domain (TMD), and a small endodomain. The S protein is an important drug and vaccine target because it activates cell entry by binding to the angiotensin-converting enzyme-2 (ACE2) receptor during viral invasion [1,2,3,4]. Changes resulting from the continuous evolution of the SARS-CoV-2 genome have resulted in structural changes in the S protein. These structural changes have significant implications for SARS-CoV-2’s infectivity, immune evasion, and pathogenicity [5]. The physiological activities of the S protein make it a primary target for the development of neutralizing antibodies and vaccines.
After receptor binding and proteolytic activation, spikes undergo major conformational rearrangements, in which S1 dissociates and S2 refolds to mediate membrane fusion via the conserved HR1/HR2 six-helix bundle (6-HB). The HR1 “fusion core” is central to 6-HB stabilization and is thus a plausible site where mutations could modulate fusion efficiency. Consistent with this, Oliva et al. (2021) reported recurrent substitutions in the HR1 fusion core (S929T, D936Y, S939F) and proposed that D936Y can weaken stabilizing interactions in the post-fusion assembly (via loss of an inter-monomer salt-bridge network), increasing flexibility [6]. Together, these observations suggest that spike evolution may tune not only S1-mediated receptor engagement/immune escape but also the downstream fusion machinery, providing a context for interpreting variant-specific effects.
To date, five variants of concern (VOCs) have been identified: Alpha, Beta, Gamma, Delta, and Omicron. Among these, Omicron exhibits the greatest divergence from the original Wuhan-Hu-1. The omicron variant and its sublineages are characterized by a high density of mutations in the S protein, particularly in the receptor-binding domain (RBD) and the N-terminal domain (NTD). This allows for the evasion of vaccines and therapeutic monoclonal antibodies while preserving effective ACE2 binding [7,8]. XBB, a lineage derived from two BA.2 sublineages (BJ.1 and BM.1.1.1), contains extensive receptor-binding domain (RBD) substitutions associated with antibody resistance. The XBB.1.5 sublineage differs from the parental gene, XBB.1, by the RBD mutation F486P in the receptor-binding motif [9].
Epidemiological and experimental evidence has demonstrated that XBB.1.5 possesses a substantial growth advantage over co-circulating variants, attributable to improved ACE2 binding and marked evasion of polyclonal neutralizing antibodies elicited by vaccination, infection, or hybrid immunity. Neutralization studies have shown markedly reduced serum neutralizing titers against XBB and XBB.1.5 across various immune systems, including during BA.5 breakthrough infections and after multiple vaccine doses [10,11,12]. This highlights the necessity of understanding the structural and antigenic foundations of this phenotype and revising vaccine formulations accordingly.
In this context, in silico approaches offer a powerful, rapid, and cost-effective strategy to study how complex mutations shape protein stability, epitope presentation, and conformational dynamics in the S protein [13]. The integration of homology modeling, epitope prediction, stability prediction, and coarse-grained dynamics can generate mechanistic hypotheses that complement experimental structural biology and neutralization data and aid in the prioritization of epitopes and regions for rational immunogen and antibody design [14,15].
This study systematically analyzed, using a comprehensive methodology, the changes in S protein structure, stability, and epitope presentation resulting from mutations observed in the XBB.1.5 variant of SARS-CoV-2. The resulting structural framework elucidates how the combined effects of multiple mutations in XBB.1.5 can predispose the virus to increased infectivity and evasion of the immune system and highlights suitable or unsuitable antigenic regions for next-generation vaccines and antibody targeting.
2. Results
2.1. Mutational Landscape of XBB.1.5 S Protein
Consensus sequence analysis of the SARS-CoV-2 XBB.1.5 S protein relative to the Wuhan-Hu-1 reference (NC_045512.2; YP_009724390.1) identified 38 amino acid substitutions distributed across S1 and S2 (T19I, L24S, V83A, G142D, H146Q, Q183E, V213E, G252V, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, N460K, S477N, T478K, E484A, F486P, F490S, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K) (Figure 1).
Twenty-two of these 38 substitutions were localized within the extended RBD, including many positions directly involved in ACE2 binding or targeted by potent class 1–3 neutralizing antibodies (G339H, R346T, S371F, S373P, S375F, T376A, N440K, V445P, G446S, N460K, S477N, T478K, E484A, F486P, F490S, Q498R, N501Y, and Y505H). Several additional mutations cluster in the NTD supersite and S1/S2 region (T19I, L24S, V83A, G142D, H146Q, G183E, V213E, G252V, D614G, H655Y, N679K, P681H), while S2-subunit mutations (N764K, D796Y, Q954H, N969K) localize near the heptad repeat and fusion machinery.
This dense mutational constellation places XBB.1.5 among the most heavily mutated S protein variants described to date, particularly within the RBD, and overlaps with many residues individually associated with increased ACE2 affinity and/or antibody escape in previous Omicron sublineages [9].
2.2. Homology Modeling and Overall Structural Impact
Wild-type (Wuhan-Hu-1) and XBB.1.5 S protein ectodomain trimers were modeled using both SWISS-MODEL and RoseTTAFold, followed by energy minimization and quality evaluation using the QMEAN scoring function. The best-scoring models for each variant displayed acceptable global QMEAN Z-scores and residue-level error profiles compatible with comparative modeling in regions with sufficient sequence conservation (QMEAN ≥ 0.7).
At the global level, the XBB.1.5 S protein models preserved the canonical pre-fusion trimer architecture, with three S1 heads atop S2 stalks. However, mapping the 38 mutations onto the three-dimensional structure revealed dense clustering along the RBD receptor-binding ridge, NTD antigenic supersite, and S1/S2 and S2′ cleavage-proximal regions [16]. The superimposed representation makes the changes in S protein topology caused by mutations visible. Changes in protein topology were particularly noticeable between residues 246–251, 436–441, and 675–683 (Figure 2).
Local structural rearrangements were observed around;
the 370–376 loop (S371F, S373P, S375F, T376A), which modulates RBD “up/down” transitions and is implicated in altering the conformational ensemble in Omicron sublineages;the receptor-binding motif core (G446S, N460K, S477N, T478K, E484A, F486P, Q498R, N501Y, Y505H), which directly contacts ACE2 and is a major target of class 1–3 neutralizing antibodies;the NTD supersite around positions 142–146, important for neutralizing NTD-directed antibodies;the D614G–H655Y–N679K–P681H region that influences S1/S2 cleavage, fusogenicity, and S protein stability.
The structural context of F486P and Q493 (revertant from R493) in the modeled XBB.1.5 RBD is consistent with experimental structural studies showing that XBB.1.5 maintains or slightly enhances ACE2 binding relative to XBB.1, with P486 contributing to compensatory stabilization of the ACE2–RBD interface despite the loss of hydrophobic interactions associated with F486 in earlier variants.
2.3. Predicted B-Cell Epitopes and Antigenic Determinants
B-cell epitope prediction was performed using BepiPred-2.0 for linear epitopes, Emini surface accessibility for solvent exposure, and the Kolaskar–Tongaonkar method for antigenicity, applied to the full S protein sequences of wild-type and XBB.1.5. BepiPred-2.0 identified 28 linear B-cell epitopes in the wild-type S protein and 29 epitopes in XBB.1.5, reflecting both the gain and loss of the predicted epitope segments (Table 1). The mean linear epitope score was calculated as 0.470 (0.183–0.695). Mean BepiPred scores were comparable between variants, but several epitopes shifted in position, length, or score, particularly within the NTD, RBD, and S1/S2 regions where most mutations reside.
Surface accessibility analysis indicated that multiple mutations either increased or decreased the predicted solvent exposure of epitope segments (Table 2). The surface accessibility epitope score was calculated as 1.000 (0.042–6.051). Surface accessibility analysis revealed 30 different epitopes for the wild-type, whereas this value was calculated as 29 for the mutant type. For example, substitutions in the NTD loop region (T19I, L24S, V83A, G142D, H146Q, G183E, V213E, G252V) reshaped contiguous predicted epitopes and altered local accessibility scores, consistent with experimental observations that Omicron NTD mutations reconfigure the NTD supersite and abrogate binding of NTD-specific neutralizing antibodies.
Kolaskar–Tongaonkar antigenicity profiling showed that XBB.1.5 mutations altered the antigenic score distribution along the S protein sequence, with both increases and decreases in predicted antigenicity in key regions (Table 3). The Kolaskar and Tongaonkar antigenicity score was calculated as 1.041 (0.866–1.261). The analysis revealed 46 different epitopes for the wild-type, whereas this value was calculated as 45 for the mutant type. Notably, four antigenic determinants that were highly conserved and antigenic across many pre-Omicron and early Omicron variants showed marked changes in XBB.1.5, suggesting potential disruption of broadly conserved B-cell epitopes.
Using three independent linear B-cell epitope predictors (BepiPred-2.0, Emini surface accessibility, and Kolaskar–Tongaonkar antigenicity), we quantified the differences between wild-type and XBB.1.5 spikes in terms of epitope counts, length distributions, residue-level coverage, turnover, and cross-tool concordance. Overall, epitope counts were highly comparable between the variants across all tools. BepiPred-2.0 predicted 28 wild-type and 29 XBB.1.5 epitopes, Emini predicted 30 wild-type and 29 XBB.1.5 epitopes, and Kolaskar–Tongaonkar predicted 46 wild-type and 45 XBB.1.5 epitopes.
Epitope-length summaries also indicated broadly similar distributions between wild-type and XBB.1.5. For BepiPred-2.0, the mean length was 18.0 aa in wild-type and 16.6 aa in XBB.1.5 (medians 16 vs. 13; ranges 5–62 vs. 5–47). For Emini, the mean length was 9.0 aa in both variants (median 8; range 6–24). For Kolaskar–Tongaonkar, mean length was 9.33 aa (wild-type) versus 9.69 aa (XBB.1.5) (median 8; range 6–36). Consistent with these descriptive statistics, nonparametric comparisons of epitope-length distributions (two-sided Mann–Whitney U test) did not detect significant differences between wild-type and XBB.1.5 for any tool (BepiPred p = 0.57, Emini p = 0.96, Kolaskar p = 0.67), indicating that the mutation set in XBB.1.5 does not substantially shift the overall length profile of predicted linear epitopes. Although epitope counts and lengths were similar, residue-level “coverage” (unique residues falling within any predicted epitope interval) showed modest net changes but notable re-distribution between variants. For BepiPred-2.0, wild-type covered 504 unique residues versus 481 in XBB.1.5 (net: −23). For Emini, wild-type covered 270 residues versus 261 in XBB.1.5 (net −9). For Kolaskar–Tongaonkar, coverage slightly increased from 429 (wild-type) to 436 (XBB.1.5) (net +7).
Importantly, despite these small net differences, residue-level turnover was substantial, indicating marked epitope remodeling between wild-type and XBB.1.5. In BepiPred-2.0, XBB.1.5 gained 104 epitope residues not present in wild-type and lost 127 wild-type specific epitope residues (Jaccard similarity 0.62), with 74.8% of wild-type epitope residues retained in XBB.1.5. In Emini, 109 residues were gained and 118 were lost (Jaccard 0.40), corresponding to 56.3% retention. In Kolaskar–Tongaonkar, 166 residues were gained and 159 were lost (Jaccard 0.45), with 62.9% retention. Collectively, these results show that while the overall number of predicted linear epitope segments remains stable, the locations of the predicted epitope residues can change considerably between wild-type and XBB.1.5.
Next, we assessed internal consistency across tools by comparing the residue-level epitope sets within each variant. The pairwise overlaps were modest, reflecting the different underlying assumptions of propensity-based, accessibility-based, and antigenicity-based predictors. In wild-type, Jaccard overlaps were 0.319 (BepiPred∩Emini), 0.143 (BepiPred∩Kolaskar), and 0.037 (Emini∩Kolaskar). For XBB.1.5, the corresponding overlaps were 0.286, 0.151, and 0.033, respectively.
A “consensus” definition (residues supported by ≥2 tools) yielded 299 consensus residues for wild-type and 289 for XBB.1.5, whereas the strict three-tool intersection was small (fifteen residues in wild-type and nine residues in XBB.1.5). These findings indicate that most predicted epitope residues are tool-specific, whereas a smaller subset is recurrent across methods and may represent more robust candidates for downstream structural mapping and experimental prioritization.
2.4. Protein Stability and Dynamics
The impact of each XBB.1.5 mutation on S protein stability was evaluated using four complementary predictors: mCSM, DDMut, DUET, and DynaMut2, which estimate the change in folding free energy ΔΔG between wild-type and mutant (Table 4). The combined evaluation of ΔΔG values obtained from four independent estimating tools (mCSM, DDMut, DUET, and DynaMut2) revealed that the vast majority of mutations showed an average destabilizing trend. Of the 38 mutations, 29 had a negative average ΔΔG and 9 had an positive average ΔΔG; error bars (SD) indicated high inter-tool variability, particularly for some mutations (Supplementary S1). After a two-way one-sample t-test per mutation (H0: mean ΔΔG = 0) and Benjamini–Hochberg FDR correction, consistent and statistically significant inter-tool deviations were observed for eight mutations (q < 0.05). Six of these were associated with consistent destabilization: L24S (≈−2.10), V83A (≈−1.09), V213E (≈−1.51), S371F (≈−0.93), T376A (≈−0.50), and F490S (≈−1.89). In contrast, H655Y (≈+1.33) and D796Y (≈+0.45) mutations showed consistent signals of stabilization. While the largest negative mean ΔΔG values were evident for some mutations (L24S and G142D), the wider SD in this group indicated a lower consensus among the instruments. These results were interpreted considering that statistical significance reflects the consistency between predictive instruments, not experimental replicates.
When the functional effects of 38 mutations in the S protein were compared using three different bioinformatics tools (PROVEAN, SIFT, and PolyPhen-2), a significant difference was observed in the tendency to call them “damaging” (Table 5). PROVEAN classified only 2 out of 38 (5.3%) mutations as “deleterious (D)”, while SIFT predicted 4 out of 38 (10.5%) mutations as “affect of function (AF).” PolyPhen-2, on the other hand, evaluated 17 out of 36 (47.2%) mutations as “damaging (D)” (excluding two rows with n/c).
This difference between the three tools was statistically significant using Cochran’s Q test (Q = 26.38, p = 1.87 × 10^−6^) and was supported by McNemar tests, particularly showing that PolyPhen-2 called them significantly more often than PROVEAN and SIFT. However, the consensus between the tools was limited. The number of mutations that at least two vehicles identified as “damaging” was 4/36, and this common signal was observed for V83A, Y505H, N764K, and N969K mutations. These results suggest that functional effect predictions can be vehicle-dependent and that the strongest interpretations should be made for mutations supported by a multi-vehicle consensus.
The virus binds to host cell receptors via the S1 subunit of the S proteins and initiates the membrane fusion process necessary for virion translocation to the host cell through S2 subunit activity [17,18]. Two conformational states are observed in the S protein receptor binding region. One is the down formation, where the receptor-binding region is hidden/masked, and the other is the up formation, where the receptor-binding region is accessible and exhibits a less stable structure [19]. In this study, six mutations (N460K, S477N, T478K, E484A, F486P, F490S) were observed in the region between residues 460 and 490, where this event occurs, which the virus uses to evade the body’s defense system, resulting in increased protein instability and mobility (Table 4). Changes in protein topology and atomic interactions in the mutation-related region are thought to contribute to the rapid transmission and increased affinity of the virus [20,21]. Functional impact predictions using PROVEAN, SIFT, and PolyPhen 2 indicated that approximately 44.7% of the analyzed mutations were likely to have deleterious or damaging effects on protein function, particularly those located at or near the ACE2 interface or within dominant neutralizing antibody epitopes. While “deleterious” in this context typically refers to perturbations of the ancestral S protein function, such perturbations may confer a fitness advantage by increasing ACE2 affinity, altering conformational dynamics, or enhancing immune escape.
Coarse-grained dynamics were investigated using elastic network models implemented in DynOmics. The cutoff value for the network model nodes was set at 7.3 Å. The correlation between the observed and predicted fluctuations was 0.57 Å^2^ for the wild-type and 0.41 Å^2^ for the variant. These analyses revealed that many XBB.1.5 mutations occur in dynamically active regions that contribute to the collective motions associated with RBD up/down transitions and S1 shedding (Figure 3). In particular, substitutions in the 370–376 loop, RBM core, and S1/S2 cleavage-proximal region were predicted to modulate low-frequency normal modes that control opening of the RBD and exposure of the ACE2-binding surface, consistent with structural and single-particle cryo-EM observations in related Omicron sublineages [22,23].
To move beyond global MD descriptors, we quantified chain-resolved non-covalent interactions at the A–B protomer interface using COCOMAPS 2.0. The overall interaction composition remained broadly similar between the wild-type and XBB.1.5, with proximal contacts remaining the dominant class (47.1% in wild-type vs. 44.8% in XBB.1.5), while small increases were observed for polar vdW contacts (17.6% → 18.8%), CH–O/N contacts (10.2% → 11.3%), hydrogen bonds (5.4% → 5.8%), and salt bridges (1.3% → 1.5%). In agreement with these distributions, the exported contact lists showed only minor changes in absolute interface contact counts (wild-type → XBB.1.5): proximal contacts 217 → 215, polar vdW 81 → 90, apolar vdW 69 → 71, hydrogen bonds 25 → 28, and salt bridges 6 → 7, consistent with subtle interface remodeling rather than gross disruption of the interface.
At the level of key electrostatic anchors, five major salt bridges were conserved across wild-type and XBB.1.5—ASP745–ARG319, GLU868–ARG646, ARG1019–GLU1017, GLU1031–ARG1039, and GLU1151–LYS1149—with shorter interatomic distances in XBB.1.5 (e.g., ARG1019–GLU1017: 4.18 → 3.68 Å; GLU868–ARG646: 4.38 → 3.96 Å), suggesting preserved and potentially strengthened inter-protomer electrostatic coupling. In addition, a new salt bridge was detected in XBB.1.5 (LYS790–GLU702), whereas one peripheral salt bridge displayed a residue-register shift (ARG44 → ARG45 paired with ASP571), consistent with a minor local rearrangement near the N-terminus of the modeled segment.
Hydrogen-bonding patterns were largely retained, with 21 of 25 wild-type residue-pair H-bonds preserved (≈84% conservation) and several showing shorter distances and improved geometry in XBB.1.5 (SER758–GLN965: 3.11 → 2.85 Å; ASN907–ARG1107: 3.06 → 2.81 Å). XBB.1.5 also introduced new H-bond contacts (ARG983–GLY381, LEU981–LYS386, SER982–GLY545), whereas a small number of wild-type H-bonds were lost (MET740–ARG319, TYR873–LEU699). Together, these data indicate that the XBB.1.5 A–B interface undergoes modest repacking, characterized by a slight reduction in non-specific proximity contacts but a compensatory gain in polar and directional interactions.
2.5. Integration with Experimental XBB.1.5 Data
Experimental studies have shown that XBB.1.5 combines strong ACE2 binding with substantial escape from neutralizing antibody activity. Surface plasmon resonance and biolayer interferometry measurements indicated that the XBB.1.5 RBD binds human ACE2 with nanomolar affinity, similar to or modestly higher than BA.2 and XBB.1, and higher than the ancestral D614G S protein. Structural analyses reveal that the F486P substitution in XBB.1.5 re-optimizes local packing and preserves ACE2 contacts despite the charge-reversing E484A and other nearby mutations, while the revertant Q493 residue restores favorable hydrogen-bonding interactions with ACE2 residues E35 and H34 [8,24].
Neutralization assays with sera from vaccinated and/or previously infected individuals consistently reported markedly reduced neutralizing titers against XBB and XBB.1.5 compared with earlier Omicron sublineages, even after recent BA.5 breakthrough infection or after updated booster vaccination. XBB.1.5 exhibits broad escape from most authorized therapeutic monoclonal antibodies, with only a limited subset (S309-derived antibodies such as sotrovimab) retaining partial activity, in line with the accumulation of mutations at key monoclonal antibody epitopes (G339H, R346T, G446S, F486P, N460K) [25,26].
The in silico findings of this study—dense mutation clustering at the ACE2 interface and antibody epitopes, reshaped B cell- epitope landscape, and altered stability/dynamics in functionally critical regions—are concordant with these experimental observations and provide a mechanistic structural interpretation of how XBB.1.5 achieves its phenotype.
3. Discussion
This study integrated sequence analysis, homology modeling, epitope prediction, stability estimation, and coarse-grained dynamics to characterize how the composite mutational pattern of the SARS-CoV-2 XBB.1.5 S protein variant remodels its structural and antigenic properties. The principal findings are as follows:
- XBB.1.5 harbors 38 S protein substitutions relative to Wuhan-Hu-1, 22 of which reside within the RBD, including many residues directly contacting ACE2 or targeted by potent neutralizing antibodies.
- Homology models indicate preservation of the global prefusion S protein architecture but substantial local remodeling of RBD and NTD loops and the S1/S2 region, consistent with experimental structural data for XBB-lineage spikes.
- B-cell epitope predictions reveal gains and losses of linear epitopes, altered surface accessibility, and changes in antigenicity scores, including disruption of previously conserved antigenic determinants, implying a qualitatively reshaped B-cell antigenic landscape.
- Stability and functional impact predictors show a mosaic of stabilizing and destabilizing mutations, with roughly half predicted to perturb the ancestral S protein function, and elastic-network models suggest that multiple substitutions tune collective motions associated with RBD opening and S1 shedding.
- These in silico observations align with experimental evidence that XBB.1.5 maintains a high ACE2 affinity comparable to BA.2, while exhibiting one of the most extreme neutralizing-antibody escape profiles observed to date.
3.1. Structural Basis of Enhanced Receptor Binding
Mutational combinations in XBB.1.5 appear to balance the trade-off between immune escape and receptor engagement, which has shaped SARS-CoV-2 evolution. In earlier Omicron sublineages, certain antibody-evading substitutions in the RBM (E484A and K417N) reduced ACE2 affinity and required compensatory mutations (N501Y and Q498R) to restore or enhance the binding. In XBB.1.5, the revertant Q493 residue reinstates favorable hydrogen bonding with ACE2, while F486P, although removing the aromatic F486 side chain, contributes to local conformational stabilization that supports the ACE2–RBD interface [9,27].
The modeling results highlight how clusters of mutations at 370–376 and 445–486 collectively modulate the conformation and dynamics of the receptor-binding ridge, a key determinant of both ACE2 affinity and antibody epitope exposure. Elastic network analyses suggest that mutations in these regions shift low-frequency normal modes that control the RBD up/down equilibrium and the degree of RBD opening, which in turn influences ACE2 accessibility and the probability of productive receptor engagement. Experimental cryo-EM structures of Omicron spikes, including XBB-lineage variants, support the notion that such mutations alter the distribution of RBD conformers and favor states with enhanced ACE2 occupancy [9,28].
3.2. Antigenic Remodeling and Immune Escape
The dense accumulation of mutations in the NTD supersite, RBD core, and RBM is expected to remodel B-cell epitope landscapes by altering the local sequence propensities, segment boundaries, and surface exposure. Accordingly, our linear B-cell epitope predictions indicate that XBB.1.5 does not necessarily reduce the overall number of predicted antigenic segments but rather redistributes antigenic residues are likely to occur along the S protein, implying a qualitative reshaping of antibody-accessible footprints.
To benchmark these immunoinformatics predictions against experimentally reported epitope knowledge, we cross-referenced our findings with IEDB-curated SARS-CoV-2 spike linear epitope datasets, as summarized in recent systematic analyses [29]. In an IEDB-based study of spike linear epitopes, XBB.1.5 signature mutations were reported to map onto 1395 B-cell linear epitopes (32.27% of the IEDB spike B-cell linear epitope set), indicating that roughly one-third of experimentally recorded linear antibody targets contain at least one XBB.1.5 change. Because “affected” here denotes positional overlap rather than direct loss of binding or neutralization, this comparison should be interpreted as a contextual cross-check that nevertheless supports substantial antigenic remodeling in XBB.1.5 [29,30].
Across three linear B-cell epitope prediction approaches (BepiPred-2.0, Emini surface accessibility, and Kolaskar–Tongaonkar antigenicity), wild-type and XBB.1.5 exhibited comparable numbers of predicted epitopes and broadly similar length distributions (Mann–Whitney U tests non-significant across tools), suggesting that the global “quantity” of predicted linear antigenic segments is not dramatically altered by the XBB.1.5 mutation set. Notably, however, the residue-level overlap between wild-type and XBB.1.5 epitope coverage was only moderate (Jaccard ≈ 0.40–0.62 across tools), indicating substantial epitope “turnover” (gain/loss of predicted epitope residues) despite similar global counts. This pattern supports a remodeling scenario in which substitutions shift linear epitope propensities and/or accessibility, potentially changing immunodominant segments without requiring a large change in the total number of predicted epitopes.
These findings are also relevant for interpreting immunoinformatics-driven multi-epitope vaccine designs. Although many efforts have prioritized S-derived epitopes to elicit broad immunity, the rapid evolution of the S protein raises concerns about cross-efficacy across emerging variants [31,32]. For example, epitopes reported as highly conserved across multiple lineages (ELLHAPATV, PYRVVVLSFELLHAP, NATRFASVYAWNRKR, and ERDISTEIYQAGNKP) localize to RBD segments that overlap XBB.1.5 mutational hotspots; thus, even when an epitope’s general region remains targeted, its precise sequence content and predicted segment boundaries can be altered (Table 1, Table 2 and Table 3), which may undermine the assumptions of conservation-based protection [33]. Importantly, inter-tool concordance was limited, reflecting algorithm-specific assumptions (sequence propensity, accessibility, and antigenicity). Therefore, residues supported by ≥2 tools may be viewed as more robust candidates, whereas tool-unique calls should be interpreted conservatively and prioritized for structure-based mapping and experimental validation studies. This qualitative reshaping is consistent with the dramatic loss of neutralization observed for XBB.1.5 against sera from individuals vaccinated with ancestral-strain vaccines, infected with earlier omicron variants, or experiencing hybrid immunity [34].
Many therapeutic monoclonal antibodies targeting class 1 (ACE2-overlapping), class 2 (adjacent to the ACE2 site), class 3 (lateral RBD epitopes), and NTD supersite epitopes lose activity against XBB.1.5 because of mutations at G339H, R346T, G446S, E484A, F486P, N460K, and in the NTD loops [35,36,37]. In this context, the in silico identification of altered antigenic determinants, including turnover within regions previously considered conserved, provides a structural rationale for broad antibody escape and highlights the challenge of designing universal S-based vaccines that rely exclusively on B-cell neutralization.
Simultaneously, experimental data indicate that T-cell recognition of XBB.1.5 S protein remains largely preserved despite extensive antibody escape, likely reflecting the broader and more conserved epitope repertoire of CD4+ and CD8+ responses. Although T-cell epitopes were not explicitly modeled in this study, the concentration of mutations in B-cell-dominated regions suggests that many T-cell epitopes outside the highly variable RBM may remain intact, helping to maintain protection against severe disease even as infection-blocking neutralization wanes [8,38].
3.3. Functional Implications of Stability and Dynamics Changes
The mixed pattern of stabilizing and destabilizing mutations in XBB.1.5 underscores that viral fitness does not necessarily correlate with maximal structural stability of the S protein. Instead, an optimal balance between stability and flexibility may favor efficient conformational transitions required for receptor binding, fusion activation, and immune evasion [13,39,40].
Early work on spike S2 evolution provides a useful framework for interpreting XBB.1.5 results beyond antibody escape. Oliva et al. highlighted that mutations can arise in the HR1 “fusion core” a key element of the S2 fusion machinery that refolds and associates with HR2 to form the post-fusion six-helix bundle (6-HB), a conserved architecture essential for membrane fusion and infectivity. In that analysis, the frequent HR1 substitution D936Y was predicted to be structurally consequential because D936 participates in an inter-monomer salt bridge in the post-fusion assembly, motivating comparative dynamics analyses to assess how HR1 mutations may weaken stabilizing interactions in the 6-HB and alter fusion-core behavior [6].
In our XBB.1.5 models, while antigenic remodeling and immune escape are dominated by S1 (NTD/RBD), mutation mapping also indicates changes in the S1/S2 and cleavage-proximal axis, extending into S2-annotated regions (including heptad repeats). Notably, the multi-tool functional effect consensus in our study flagged S2-localized substitutions (N764K and N969K) among the strongest shared “damaging” signals, underscoring that S2 changes may contribute to the phenotype even when most attention is on the RBM. Consistent with this broader view, our elastic network analyses suggest that XBB.1.5 mutations reshape collective motions that couple RBD opening and S1 shedding with S1/S2 dynamics, supporting a model in which variant fitness may reflect coordinated tuning of receptor engagement, immune evasion, and downstream fusion activation rather than S1 changes alone.
Destabilizing mutations in S1 and at the S1/S2 interface can promote S1 shedding and the exposure of the S2 fusion machinery, potentially enhancing fusogenicity; however excessive destabilization risks premature activation and loss of infectivity [41]. Conversely, stabilizing substitutions in the RBD and S2 can preserve the overall integrity of the prefusion trimer while fine-tuning local flexibility [42,43]. Elastic network modeling suggests that XBB.1.5 mutations reshape the collective motions that couple RBD opening with S1/S2 dynamics, potentially optimizing the energetic landscape for ACE2 engagement while minimizing the exposure of vulnerable epitopes.
Global post-MD descriptors (e.g., rmsd/Rg) provide an overall view of structural stability but do not localize how mutations redistribute inter-protomer coupling. By applying COCOMAPS 2.0 to the A–B interface, we showed that XBB.1.5 preserves the core electrostatic “anchor” network connecting neighboring protomers (multiple conserved salt bridges with shorter distances), while simultaneously exhibiting a small shift from bulk proximity packing toward more polar and directional interactions (increased polar vdW/CH–O/N contacts and a slightly larger H-bond and salt-bridge inventory). This pattern is consistent with interface repacking rather than global destabilization: the trimeric architecture remains supported by conserved S2/S1 cross-protomer contacts, yet local rearrangements can tune the mechanical coupling between protomers, potentially influencing conformational transitions linked to receptor engagement, S1 shedding propensity, or exposure of antigenic surfaces.
Notably, the appearance of variant-specific contacts and the gain/loss of selected hydrogen bonds suggest that even modest mutation-driven rearrangements can rewire the local interaction geometry without large changes in the total contact abundance. Such “fine-tuning” is mechanistically plausible for Omicron-descended lineages, where immune escape is often achieved by reshaping accessible surfaces while maintaining the fusion competence. Importantly, COCOMAPS-derived contacts are geometric and threshold-dependent and reflect the analyzed representative structures; therefore, contact differences should be interpreted as structural rationales for altered protomer coupling and prioritized for time-resolved MD contact persistence analysis and experimental validation.
3.4. Implications for Vaccine and Antibody Design
The structural and epitope analyses presented here have several implications for the development of next-generation vaccines and therapeutic antibodies:
- Reliance on ancestral strain S protein immunogens is increasingly inadequate in the face of variants such as XBB.1.5, which profoundly remodel key B-cell epitopes. Updated vaccines incorporating XBB-lineage or other highly evolved Omicron spikes are likely necessary to restore robust neutralization breadth.
- Conserved antigenic determinants that remain stable across diverse Omicron sublineages, including XBB.1.5, are attractive targets for broadly neutralizing antibodies and vaccine design. In silico epitope mapping can help identify such regions by excluding highly variable and structurally plastic sites.
- Vaccine strategies that emphasize T-cell epitopes, particularly in more conserved regions of S2 or non-spike proteins, may provide durable protection against severe disease even as S protein-focused neutralization is eroded by antigenic drift.
- Structural insights into how specific mutations, such as F486P and Q493, modulate ACE2 binding and antibody recognition can guide engineering of stabilized immunogens and design of antibody cocktails that target complementary, less mutable epitopes.
3.5. Limitations
This study was purely in silico and therefore generated mechanistic hypotheses that require experimental confirmation. First, we did not perform wet-lab validation (e.g., antibody binding assays such as ELISA or SPR/BLI, or authentic/pseudovirus neutralization); therefore, the predicted impacts of XBB.1.5 mutations on antigenicity and antibody escape should be interpreted as putative. Second, our dynamics analysis relies on coarse-grained elastic-network normal-mode models (GNM/ANM) implemented in DynOmics, which are well-suited to capture low-frequency collective motions but do not explicitly represent atomistic side-chain interactions, glycosylation, membrane environment, or solvent, and may therefore miss local energetic effects and transient contacts. Third, the conclusions were derived from the XBB.1.5 consensus sequence and the specific structural templates and parameters used. Because SARS-CoV-2 antigenic evolution is ongoing and epistatic interactions can shift mutational effects across variant backgrounds, generalization to other (especially newly emerging) variants should be made with caution and revisited as new sequences and structures are discovered. Finally, epitope mapping and stability/function predictions depend on the selected algorithms and their training datasets; linear B-cell epitope predictors have limited accuracy, and stability predictors can show inter-method variability. Future studies integrating glycosylated all-atom simulations, structure-based epitope prediction, and targeted experimental assays will be important for validating and refining these findings.
4. Materials and Methods
4.1. Sequence Retrieval and Variant Definition
The reference S protein sequence for SARS-CoV-2 Wuhan-Hu-1 was obtained from the NCBI database (genomic accession NC_045512.2; S protein accession YP_009724390.1). Consensus S protein sequences representing the XBB.1.5 lineage were retrieved from the NCBI Virus database and aligned to the reference to define a set of characteristic amino acid substitutions [44]. Only high-quality sequences without frameshifts or premature stop codons in the S protein coding region were included. The protein sequence data used in the bioinformatic analyses of wild-type and mutant S proteins are presented in Supplementary S2.
4.2. Multiple Sequence Alignment and Domain Mapping
Wild-type and XBB.1.5 S protein sequences were aligned with MAFFT (version 7) using an appropriate scoring matrix (BLOSUM80 and 1PAM) and gap opening penalties (=2), and visualized with Mega11 [45,46]. Mutations were annotated according to standard S protein domain boundaries: signal peptide, NTD, RBD (including RBM), subdomains 1 and 2 (SD1/SD2), S1/S2 cleavage site, S2 fusion peptide, heptad repeats, central helix, connector domain, transmembrane segment, and cytoplasmic tail.
4.3. Homology and Deep-Learning-Based Modeling
Homology models for wild-type and variant S proteins were generated using SWISS-MODEL [47]. Templates satisfying threshold values of sequence identity > 97%, sequence similarity > 85, and coverage > 85 were searched using BLAST+ [48] and HHblits [49] for evolutionarily related constructs matching the target sequence in the SWISS-MODEL template library. In protein modeling studies, 6VXX was chosen as the template [50]. Models are built based on the target-template alignment using ProMod3.3.0 [51]. Coordinates that are conserved between the target and template are copied from the template to the model. A fragment library was used to remodel the insertions and deletions, followed by the reconstruction of the side chains. The geometry of the final model was regularized using a force field. The annotation of the template quaternary structure was employed to model the target sequence in its oligomeric state. This approach utilizes a supervised machine learning algorithm, specifically Support Vector Machines (SVM), which integrates interface conservation, structural clustering, and other template characteristics to estimate quaternary structure quality (QSQE) [52]. In parallel, single-stranded SARS-CoV-2 S protein sequences (wild-type and XBB.1.5) were modeled using the RoseTTAFold option of the Robetta structure prediction server [53]. Default settings were used, including automated multiple sequence alignment (MSA) generation and the standard refinement pipeline of the server. Five models were generated for each target (some models were template-dependent), and the final model was selected based on the global confidence/quality outputs (>0.7) reported by the server. To enable direct comparison, the same modeling protocol and settings were applied to the wild-type and XBB.1.5 sequences.
For each variant, multiple models were generated and evaluated, and the best-scoring models based on global quality scores and stereochemical parameters were selected for further analysis.
4.4. Model Quality Assessment
The structural quality of the resulting models was assessed using the QMEAN scoring function, which combines residue-level potentials for torsion angles, solvation, and long-range interactions into a normalized Z-score that reflects model agreement with high-resolution experimental structures of similar size [54]. Local QMEAN scores were inspected to identify poorly modeled regions that might limit the reliability of detailed structural interpretations.
4.5. B-Cell Epitope and Antigenicity Prediction
Linear B-cell epitopes were predicted using BepiPred-2.0, which applies a random-forest classifier trained on experimentally determined antibody–antigen complexes to sequence-derived features [55]. Default thresholds (=0.5) were used to define epitope residues. Linear epitopes that met the threshold of ≥5 amino acid residues were considered.
Surface accessibility was evaluated using the Emini method, which estimates the probability of each residue being solvent-exposed based on normalized surface accessibility scales derived from known protein structures [56]. Residues with high surface accessibility scores were considered more likely to be part of antibody-accessible epitopes.
Antigenicity profiles were calculated using the Kolaskar–Tongaonkar semi-empirical method, which integrates physicochemical properties and residue frequencies in experimentally defined antigenic regions to predict the antigenic determinants [57]. Regions with antigenicity scores above the method-specific threshold (=0.5) were considered putative antigenic sites.
Linear B-cell epitopes were predicted independently for the wild-type and XBB.1.5 S protein sequences using three tools (BepiPred-2.0, Emini surface accessibility, and Kolaskar–Tongaonkar antigenicity). For each tool and variant, we summarized (i) the number of predicted epitope intervals and (ii) epitope length (amino acids), reporting the mean, median, and range. Differences in epitope length distributions between variants were tested using the two-sided Mann–Whitney U test (α = 0.05). To quantify epitope “coverage”, each epitope interval was expanded to residue-level sets (all positions between start and end), merged to remove overlaps, and the number of unique covered residues was computed per tool/variant. Epitope remodeling (“turnover”) between wild-type and XBB.1.5 was assessed at the residue level by calculating gained residues (XBB-only), lost residues (wild-type-only), and similarity using the Jaccard index , where A and B denote the residue sets covered by epitopes in wild-type and XBB.1.5, respectively; retention was additionally reported as . Finally, within each variant, inter-tool concordance was quantified by pairwise Jaccard indices between residue-level sets and by counting the “consensus” residues supported by ≥2 tools (and the three-tool intersection).
To contextualize immunoinformatics-based epitope predictions with experimental evidence, we consulted IEDB-curated SARS-CoV-2 spike linear epitope resources and recent IEDB-wide analyses that quantify the fraction of curated linear epitopes intersected by XBB.1.5-characterizing mutations [29,30].
4.6. Stability and Functional Impact Prediction
The impact of individual amino acid substitutions on S protein stability was assessed using:
- mCSM, which encodes the wild-type structural environment of a residue as graph-based signatures and uses machine learning to predict ΔΔG [58],
- DDMut, a deep-learning model trained on a large dataset of experimentally measured stability changes [59],
- DUET, which combines mCSM and the SDM (site-directed mutator) potential to provide consensus ΔΔG estimates [60],
- DynaMut2, which integrates normal-mode analysis with machine learning to estimate both stability changes and alterations in protein flexibility upon mutation [61].
To quantitatively compare the effect of mutations on protein stability, ΔΔG (kcal·mol^−1^) values obtained from four independent predictor tools (mCSM, DDMut, DUET, and DynaMut2) were evaluated for each mutation. The mean ΔΔG between tools was calculated for each mutation, and the standard deviation (SD) and standard error (SEM = SD/√n, n = 4) were derived to show uncertainty and inter-method variability. To test whether the stability change deviated significantly from zero, a two-way one-sample t-test was applied for each mutation to test the hypothesis H0: mean ΔΔG = 0, and the corresponding p-values were reported. To control for false positives from multiple comparisons, p-values were corrected using the Benjamini–Hochberg false discovery rate (FDR) method, and q-values were calculated for all tests. In the graphs, error bars are shown as SD (where appropriate) to represent the variability between predictor tools; negative mean ΔΔG values are interpreted as destabilization, while positive values are interpreted as stabilization.
To quantitatively compare the potential effects of mutations on protein function, the outputs of three different bioinformatics tools (PROVEAN, SIFT, and PolyPhen-2) were evaluated together for each mutation [62,63,64]. First, the scores and classes obtained from each instrument were standardized and converted into binary results. For PROVEAN, the “deleterious (D)” category and for SIFT, the “affect of function (AF)” category were coded as “damaging,” while other classes were coded as “non-damaging.” For PolyPhen-2, the “damaging (D)” result was considered “damaging.” Rows with “n/c” were treated as missing data in relevant analyses. The rates of “damaging” calls made by the instruments were calculated as percentages based on valid observations, and 95% confidence intervals for these rates were reported using the Wilson method. Whether these binary calls, which are repeated/dependent measurements on the same mutations, differed among the instruments was evaluated using Cochran’s Q test in rows where the three instruments had common data. Systematic differences between pairs of instruments were tested using the McNemar exact test. Inter-instrument agreement was summarized using the raw agreement rate and Cohen’s kappa coefficient for binary classes. To ensure a directional comparison of continuous scores, PROVEAN scores were converted to a “severity” indicator (-score), SIFT scores were inverted to 1-score, and PolyPhen-2 scores were used directly. Inter-instrumental ranking consistency was examined using Spearman’s correlation. Finally, to evaluate consensus based on mutations, mutations that were identified as “damaging” by at least two instruments were defined as “consensus-damaging”.
4.7. Coarse-Grained Dynamics: Elastic-Network Models
To examine how clusters of mutations affect collective protein motions, coarse-grained dynamics simulations were conducted using DynOmics, which implements the Gaussian Network Model (GNM) and Anisotropic Network Model (ANM) elastic network approaches [65]. In these models, Cα atoms are connected by springs within a cutoff distance, and normal-mode analysis is used to compute low-frequency modes that describe large-scale motions around the equilibrium structure. In the context of ANM, the threshold for internode interactions was established at 15.0 Å, with a γ value of 1.0. Conversely, for the GNM, a threshold of 10.0 Å and a γ value of 1.0 were employed.
The slowest modes were analyzed for mean-square fluctuations (MSFs), computed as , where denotes the k-th eigenvector component for residue i, the eigenvalue, m the mode cutoff, kcal·mol^−1^ at RT, and individual residue collectivity quantified via . Vibrational entropy contributions per mode were evaluated as , summed across modes for total configurational entropy.
For the mutants, identical parameters ensured direct comparability, with ΔMSF profiles highlighting mutation-induced dynamic perturbations in functional regions, such as binding interfaces. All computations used the default server settings without membrane or environmental adjustments, processing PDB structures post-hydrogen addition, and energy minimization.
4.8. Chain-Specific İnter-Protomer İnteraction Profiling
Chain-specific intermolecular contacts between spike protomers were characterized using the COCOMAPS 2.0 web server [66] by analyzing the A–B chain interface for both wild-type and XBB.1.5 models. Interacting residues were selected using a 5.0 Å cut-off. The contact definitions and thresholds were set as follows: H-bond distance 3.9 Å with minimum DHA angle 90°; salt-bridge distance 4.5 Å; water-mediated distance 3.9 Å; disulfide distance 3.6 Å; π–π distance 5.5 Å (θ = 80°, γ = 90°); cation–π, anion–π, lone pair–π, and amino–π distances 5.0 Å; CH–O/N distance 3.6 Å with CCH–O/N angle 110°; CH–π distance 4.5 Å (θ1 = 120°, θ2 = 30°); N/O/SH–π distance 4.5 Å (θ1 = 120°, θ2 = 30°); apolar and polar tolerance values 0.5.
5. Conclusions
The SARS-CoV-2 Omicron XBB.1.5 S protein illustrates how extensive sequence diversification can be leveraged to preserve receptor engagement while reshaping antigenicity. Our integrated computational workflow indicates that the prefusion trimeric architecture is globally maintained, yet mutations are concentrated in the NTD and RBD and drive local structural rearrangements in regions that govern ACE2 binding, antibody recognition and S1/S2 activation.
Across three independent linear B-cell epitope predictors, XBB.1.5 retained broadly similar epitope counts and length distributions compared with the wild-type but showed only moderate residue-level overlap, consistent with substantial epitope “turnover” rather than a simple net loss of antigenic segments. This remodeling is further supported by an IEDB-based cross-check indicating that a sizable fraction of curated spike linear B-cell epitopes overlap with XBB.1.5 signature mutation sites, underscoring the likelihood that antibody footprints are redistributed across the spike surface. Together, these results support a model in which immune evasion can arise through qualitative re-patterning of accessible and antigenic residues, even when the global epitope totals remain comparable.
Beyond global stability descriptors, chain-specific interface analysis with COCOMAPS suggests that XBB.1.5 preserves the core A–B inter-protomer electrostatic “anchor” network while exhibiting modest repacking, including slight enrichment of polar/directional contacts and variant-specific gains/losses of selected hydrogen bonds and/or salt bridges. This pattern argues against gross destabilization of the trimer and instead supports fine-tuning of inter-protomer coupling that may influence conformational transitions linked to receptor engagement and S1 shedding. Overall, our findings align with the reported experimental observations of strong ACE2 binding coupled with profound neutralizing-antibody escape in XBB-lineage variants and reinforce the need for updated vaccine antigens and therapeutic antibodies explicitly considering the structural and antigenic landscape of highly evolved Omicron sublineages. The proposed framework can be readily applied to newly emerging variants to rapidly generate mechanistic hypotheses and prioritize conserved targets for downstream structure-guided design and experimental validation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shang J. Wan Y. Luo C. Ye G. Geng Q. Auerbach A. Li F. Cell Entry Mechanisms of SARS-Co V-2Proc. Natl. Acad. Sci. USA 2020117117271173410.1073/pnas.200313811732376634 PMC 7260975 · doi ↗ · pubmed ↗
- 2Akbulut E. Mutations in the SARS Co V-2 Spike Protein May Cause Functional Changes in the Protein Quaternary Structure Turk. J. Biochem.20214613714410.1515/tjb-2020-0290 · doi ↗
- 3COVID—Coronavirus Statistics—Worldometer Available online: https://www.worldometers.info/coronavirus/(accessed on 4 December 2025)
- 4Aliper E.T. Krylov N.A. Nolde D.E. Polyansky A.A. Efremov R.G. A Uniquely Stable Trimeric Model of SARS-Co V-2 Spike Transmembrane Domain Int. J. Mol. Sci.202223922110.3390/ijms 2316922136012488 PMC 9409440 · doi ↗ · pubmed ↗
- 5Yajima H. Nomai T. Okumura K. Maenaka K. Genotype to Phenotype Japan (G 2P-Japan) Consortium Ito J. Hashiguchi T. Sato K. Molecular and Structural Insights into SARS-Co V-2 Evolution: From BA. 2 to XBB Subvariantsm Bio 202415 e 032202310.1128/mbio.03220-2339283095 PMC 11481514 · doi ↗ · pubmed ↗
- 6Oliva R. Shaikh A.R. Petta A. Vangone A. Cavallo L. D 936Y and Other Mutations in the Fusion Core of the SARS-Co V-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect Molecules 202126262210.3390/molecules 2609262233946306 PMC 8124767 · doi ↗ · pubmed ↗
- 7Yue C. Song W. Wang L. Jian F. Chen X. Gao F. Shen Z. Wang Y. Wang X. Cao Y. ACE 2 Binding and Antibody Evasion in Enhanced Transmissibility of XBB. 1.5Lancet Infect. Dis.20232327828010.1016/S 1473-3099(23)00010-536746173 PMC 9897732 · doi ↗ · pubmed ↗
- 8Mannar D. Saville J.W. Poloni C. Zhu X. Bezeruk A. Tidey K. Ahmed S. Tuttle K.S. Vahdatihassani F. Cholak S. Altered Receptor Binding, Antibody Evasion and Retention of T Cell Recognition by the SARS-Co V-2 XBB. 1.5 Spike Protein Nat. Commun.202415185410.1038/s 41467-024-46104-238424106 PMC 10904792 · doi ↗ · pubmed ↗