Stability of Poly[Ni(Salen)]-Based Electrodes in the Presence of Halide Impurities: Coordination and Redox Contributions
Daniil A. Lukyanov, Ulyana M. Rodionova, Peixia Yang, Ruopeng Li, Bo Wang, Oleg V. Levin, Dmitrii V. Anishchenko, Elena V. Alekseeva

TL;DR
This paper studies how halide impurities affect the stability of nickel-based polymer electrodes used in batteries and supercapacitors.
Contribution
The study identifies two distinct degradation mechanisms caused by halide ions and evaluates their impact on polymer electrode stability.
Findings
Chloride ions cause significant degradation of polymer capacity over 50 cycles.
Bromide-containing electrolytes maintain polymer stability and capacity.
Fluoride ions coordinate without harming redox performance.
Abstract
The electrochemical stability of redox-active polymers based on Ni(II)–Salen complexes is of critical importance for their application as electrode materials for supercapacitors and lithium-ion batteries. This study presents a systematic analysis of the influence of fluoride, chloride, and bromide anions on the redox behavior of two polymeric films: poly[Ni(Salen)] and sterically protected poly[Ni(Saltmen)]. Using cyclic voltammetry (CV), electrochemical quartz crystal microbalance (EQCM), and X-ray photoelectron spectroscopy (XPS), we identify two distinct degradation mechanisms: (1) axial coordination of halide ions to the Ni(II) center followed by demetallation, which disrupts the conjugated system and reduces conductivity, and (2) oxidative halogenation of the ligand. In the presence of chloride ions, both poly[Ni(Salen)] and poly[Ni(Saltmen)] lose approximately 70% of their initial…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
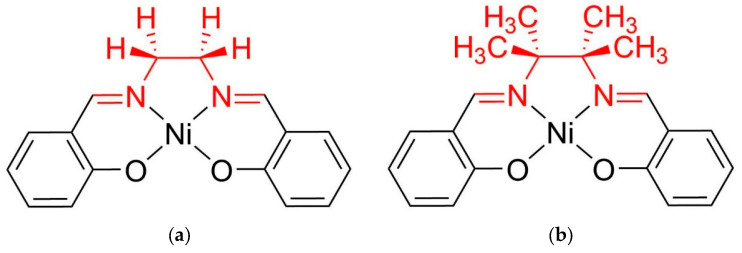 Figure 1
Figure 1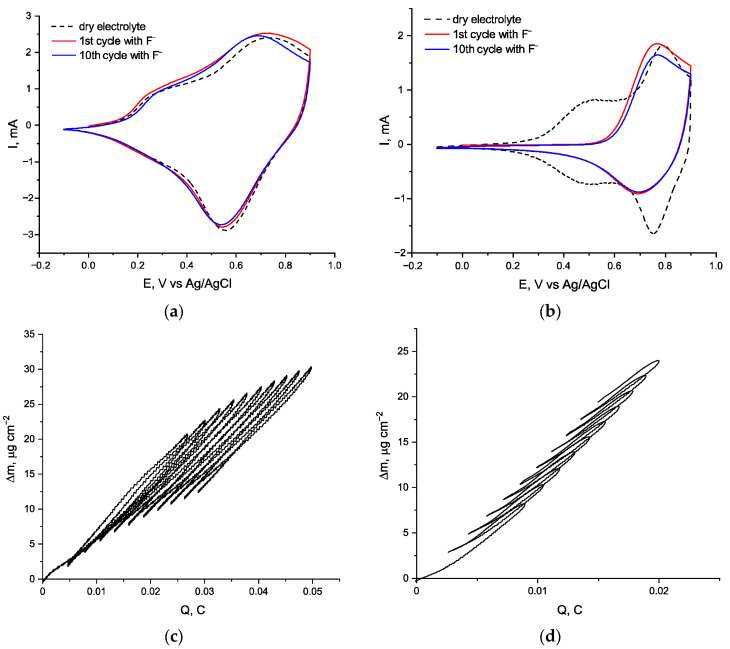 Figure 2
Figure 2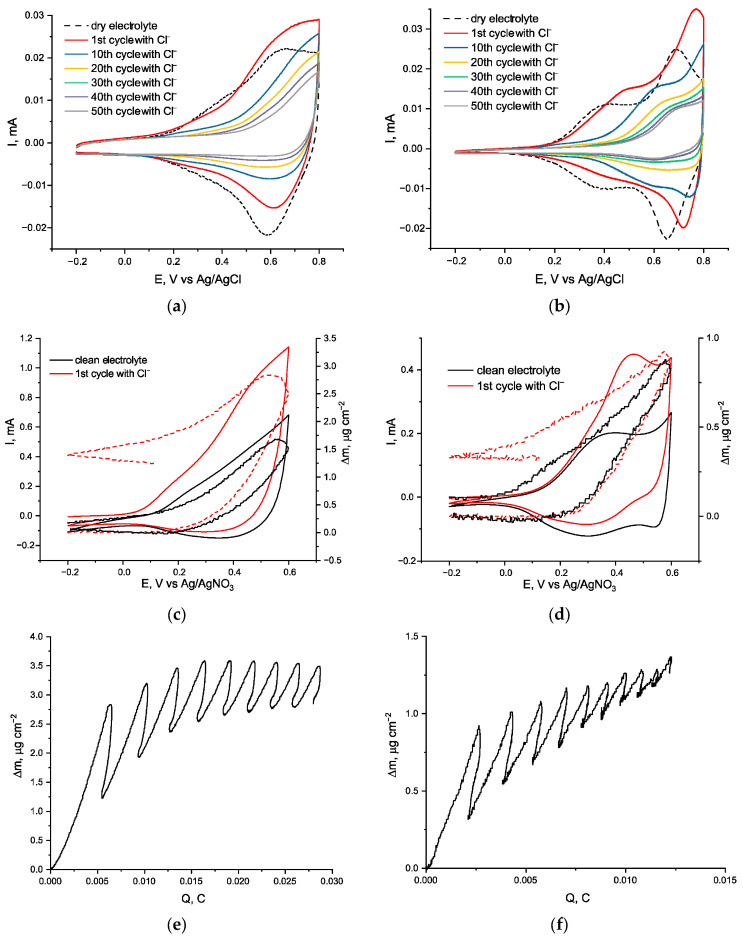 Figure 3
Figure 3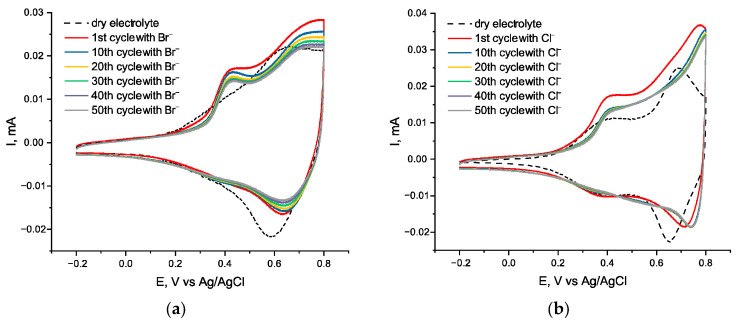 Figure 4
Figure 4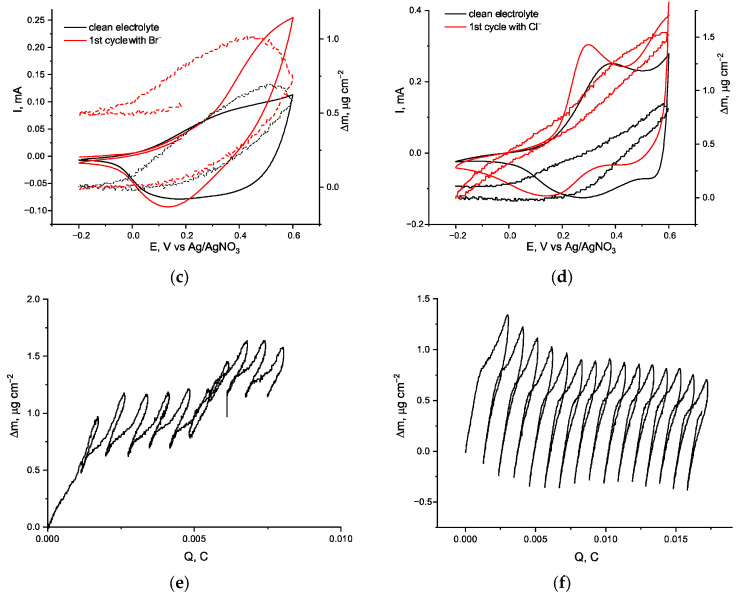 Figure 5
Figure 5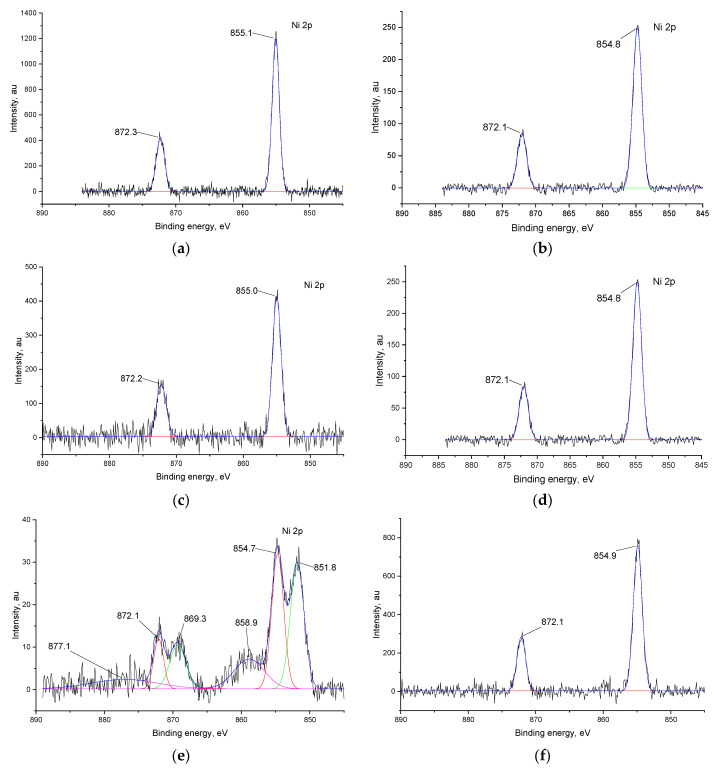 Figure 6
Figure 6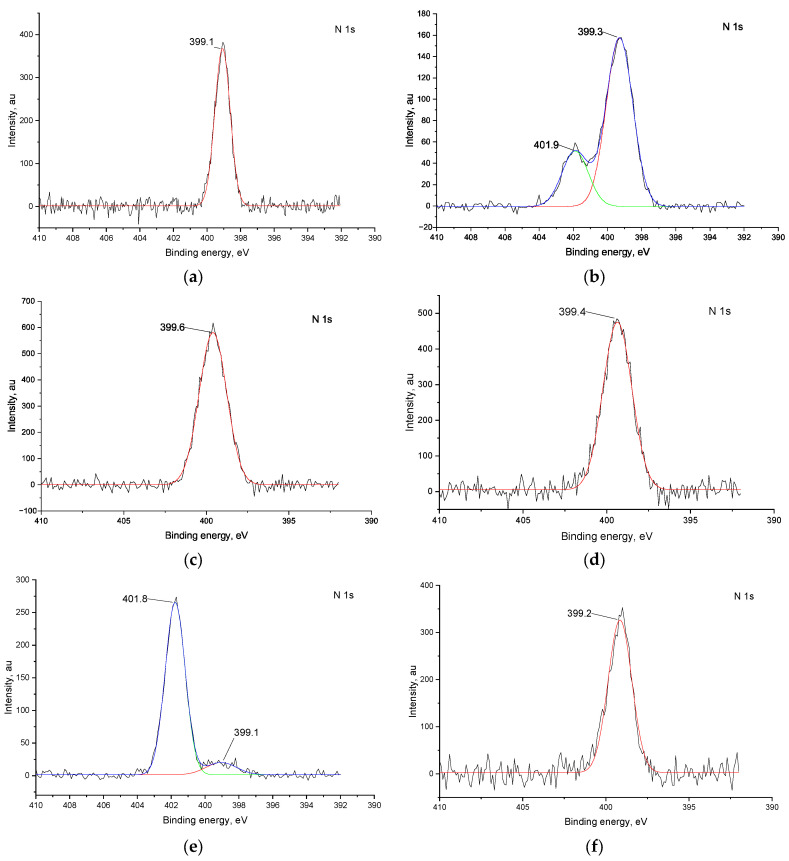 Figure 7
Figure 7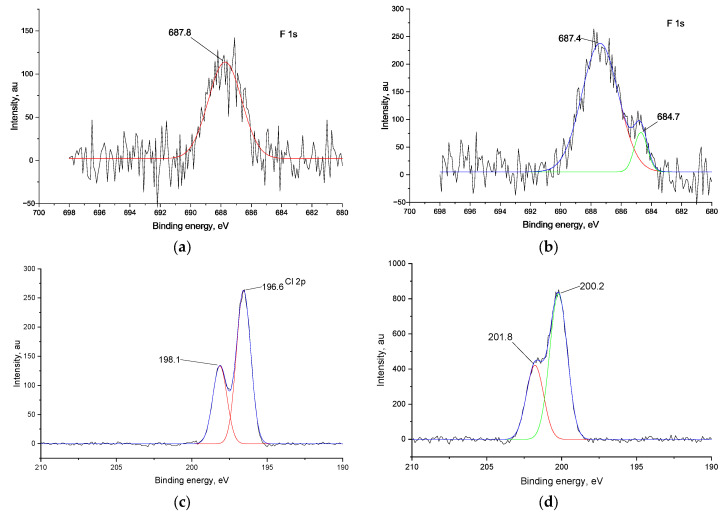 Figure 8
Figure 8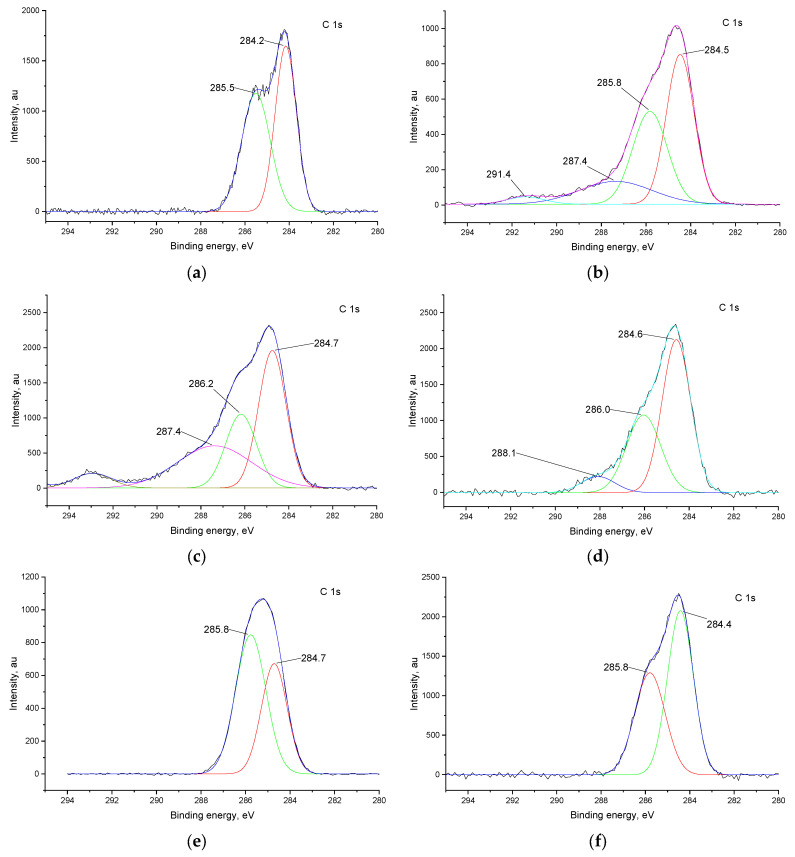 Figure 9
Figure 9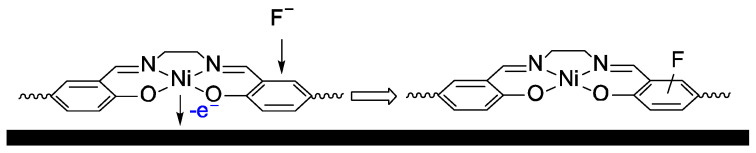 Figure 10
Figure 10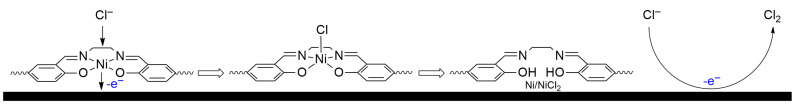 Figure 11
Figure 11- —ational Key R&D Program of China
- —Russian Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSupercapacitor Materials and Fabrication · Conducting polymers and applications · Advanced Battery Materials and Technologies
1. Introduction
The development of stable and efficient electrode materials is critical for advancing modern electrochemical energy storage systems, including Li-ion batteries and supercapacitors [1]. Electroactive polymers, such as polypyrrole, polyaniline, and polythiophene, have attracted attention due to their intrinsic conductivity and mechanical flexibility [2].
In the doped state, reported conductivities include from 17.1 to 60.9 S·cm^−1^ for one-dimensional polypyrrole nanostructures and 2–10 S·cm^−1^ for polyaniline in pressed-pellet form, while polar polythiophenes can reach conductivity up to 1173.9 S·cm^−1^ upon strong-acid vapor doping [3,4,5,6]. Mechanical robustness has been demonstrated in flexible conducting-polymer devices; for example, electropolymerized PEDOT:PSS supercapacitors operate at bending radius of 4.0–0.6 mm and retain ~94% of their capacitance after 1000 bending cycles [7].
These materials exhibit a unique combination of high electrical conductivity, reasonable stability, and favorable mechanical, optical, and electronic properties, which makes them highly promising for applications in sustainable energy technologies. Their high charge transfer rates and useful adhesion properties have long attracted scientific interest, even extending to their use as conductive binders in conventional lithium-ion batteries [8,9,10].
Among these materials, nickel complexes with polymeric Salen-type ligands have emerged as functional materials for a wide range of electrochemical applications [11]. Electropolymerized polymeric films based on nickel Schiff base complexes have been widely investigated as functional electrode materials due to their direct formation on conductive substrates, high interfacial adhesion, and stable redox activity. In particular, NiSalen-type polymer films have demonstrated pronounced pseudocapacitive behavior in supercapacitor electrodes with high capacity, depending on the complex structure and electrolyte composition [11,12,13,14]. Beyond supercapacitors, polymeric NiSalen derivatives have also been explored as cathode materials in rechargeable lithium-ion batteries, exhibiting stable cycling performance and efficient utilization of metal–ligand redox centers [15,16,17,18,19].
The electrochemical capacity of NiSalen-type polymer films strongly depends on the ligand structure and steric environment of the nickel coordination center. For simple Salen-based systems, relatively moderate capacities have been reported, for example 28 mAh·g^−1^ for unsubstituted poly[Ni(Salen)] measured by galvanostatic charge–discharge at 1 C, and 50 mAh·g^−1^ for poly[Ni(CH_3_Salen)] under similar conditions [16]. In contrast, structurally expanded ligands such as Saltmen lead to significantly enhanced pseudocapacitive performance, with poly[Ni(Saltmen)] exhibiting specific capacity around 70 F·g^−1^ from cyclic voltammetry [20]. Further structural modification also affects charge storage behavior. For instance, poly[Ni(CH_3_O-Saltmen)] films demonstrate maximum capacitances of approximately 130 F·g^−1^ [20]. Similarly, extended conjugation in poly[Ni(salphen)] results in specific capacitances around 85 F·g^−1^ measured by galvanostatic methods [21]. More recently, NiSalen-based polymer films have been implemented as functional switchable-resistance protective interlayers in lithium-ion battery systems to suppress overcharge-induced failure [16]. In addition, NiSalen-based polymer films have been successfully applied in photoelectrochemical and electrochromic systems [22,23], as well as in chemical and biosensors [24,25], further highlighting the multifunctional nature of these materials. These results demonstrate that the polymers represent a highly versatile and promising class of electroactive materials, since their molecular architecture can be systematically tailored to achieve targeted electrochemical properties through rational ligand design.
Despite exhibiting high cycling stability in rigorously anhydrous electrolytes [26,27,28], NiSalen-based polymers are often highly susceptible to moisture, such that even trace amounts of residual water in sealed electrochemical cells can result in significant performance degradation over extended cycling periods [29,30,31]. To mitigate this, structural modifications introducing steric groups—such as methyl—are helpful. Using a combination of cyclic voltammetry, EQCM, and XPS, the authors demonstrated that water coordination to the Ni(II) center leads to irreversible structural disruption and a gradual loss of electrochemical activity [30,31]. These works confirmed that steric hindrance around the metal center effectively suppresses water coordination and delays the onset of electrochemical degradation. This approach to structural stabilization has proven to be a viable strategy for enhancing the performance of electroactive polymers in humid environments.
In addition to water, halide anions may be considered as a relevant class of potential degradation agents for polymeric Ni complexes due to their nucleophilicity and coordination ability. They are frequently encountered in commercial electrolyte formulations as impurities or counter-ions in supporting salts such as LiPF_6_, LiBF_4_, and tetraalkylammonium salts. Furthermore, halides may originate from various stages of materials synthesis, including incomplete purification of starting reagents, residuals from synthetic precursors, or degradation of halogenated solvents under electrochemical conditions. According to our experience in the electrochemistry of NiSalen-type polymers, even at trace levels, halides can cause visible effects on the electrochemical properties of NiSalen-type polymers, leading to deterioration of electrode performance. However, the influence of the structural features of NiSalen complexes on the tolerance of the electroactive polymers to halide impurities in the electrolyte has not been discussed yet.
This study aims to fill this gap by evaluating the effects of fluoride, chloride, and bromide anions on the redox stability of two Ni-based polymers: one with minimal steric hindrance, poly[Ni(Salen)] (Figure 1a), and one with enhanced steric protection, poly[Ni(Saltmen)] (Figure 1b). Electrochemical stability is characterized by using CV, EQCM, and XPS. The results reveal distinct pathways of improving cycling stability of Ni-Salen based polymer electrodes by regulating the nature of the anion and the steric structure of the ligand, offering new insights into the design of stable redox-active polymers for electrochemical energy storage applications.
2. Results
The poly[Ni(Salen)] and poly[Ni(Saltmen)] complexes differ in the structure of the ligand backbone: the Saltmen ligand contains four methyl groups in the ethylenediamine bridge, resulting in increased steric hindrance around the coordination center and a modified electron density at the nickel atom. Thus, the changes in effect of the halide ions on the electrochemical properties moving from unhindered poly[Ni(Salen)] to highly hindered poly[Ni(Saltmen)] may provide information on the mechanisms of their interactions. We have implemented a series of halide ions, namely F^−^, Cl^−^ and Br^−^, which differ with their size, nucleophilicity/coordination ability and redox activity, to distinguish the possible mechanisms of interaction of the polymers with halide ions.
2.1. Effect of Fluoride Ions
Surprisingly, the addition of the F^−^ ion significantly affects the electroactivity of poly[Ni(Saltmen)] (Figure 2b), while poly[Ni(Salen)] remains nearly intact in the same conditions (Figure 2a). CV of poly[Ni(Saltmen)], which initially contains two distinct reversible peak pairs at 0.5 V and 0.77 V, loses the first peak immediately after the addition of fluoride, while the second peak pair becomes less reversible. At the same time, overall charge capacity of the film drops only by around 35%, which indicates that F^−^ ions cause redistribution of the peak currents rather than the degradation of electroactivity. Further cycling in fluoride-containing electrolytes only results in a minor degradation of the oxidation peak, while its reduction counterpart remains stable.
The mass of both films increases irreversibly cycle to cycle, which indicates the intake of the fluoride ions. However, poly[Ni(Salen)] gains 11 µg of excessive mass after 10 cycles (Figure 2c), which is about 50% of the mass change amplitude, and intake per cycle significantly decreases at this point, while poly[Ni(Saltmen)] gains 19.5 µg (Figure 2d) in the same conditions, which comprises more than 230% of the mass change amplitude, and the intake per cycle remains nearly the same with that observed at the first cycle. On the first slope of the inbound ionic flux upon oxidation, the m/z value comprises ca. 57 g·mol^−1^ for both poly[Ni(Salen)] and poly[Ni(Saltmen)], which is close to the mass of the F^−^•CH_3_CN particle. For the second slope of flux, the m/z value increases to 71 and 85 g·mol^−1^, respectively, which reflects that the BF_4_^−^ transport prevails on this stage. Upon reduction, exhaust BF_4_^−^ transport also prevails.
2.2. Effect of Chloride Ions
The addition of Cl^−^ had a pronounced effect on the electrochemical behavior of both polymers over the course of 50 cycles (Figure 3a,b). Inherent redox activity of both polymers gradually degrades with comparable rates until the disappearance of the reduction peaks, leaving only an irreversible shoulder of Cl^−^ oxidation. However, in case of poly[Ni(Saltmen)], some minor polymer-based electroactivity remains after 50 cycles. Similarly to for fluoride-promoted degradation, the low-potential peak pair disappears rapidly, while the high-potential peak remains for much longer.
In the case of poly[Ni(Salen)] (Figure 3a), the capacity of the film decreased by 65% upon addition of Et_4_NCl (Cl^−^). In the case of the addition of Cl^−^ to electrolyte poly[Ni(Saltmen)], it exhibited around 70% loss in capacity (Figure 3b).
After adding Cl^−^ ions, the oxidation ion flux for both polymers becomes linear with a single slope of 34 g·mol^−1^, in contrast to the bilinear curves seen in the control experiment without an additive, suggesting [31] a single dominant ion transport process with the ion flux of 34 g·mol^−1^, which corresponds to the Cl^−^ ion (Figure 3e,f). Although the m/z of the reduction mass flux for both polymers is much higher than m/z of the oxidation mass flux and correspond to the exit of the BF_4_^−^ anions, both polymers irreversibly gain mass during each CV cycle, which indicates that the Cl^−^ ions do not only substitute the BF_4_^−^ as a dopant, but irreversibly incorporate in the structure of the polymers. In the case of poly[Ni(Salen)], the mass gain stops after five cycles and turns to a slight decrease in the subsequent cycles, while poly[Ni(Saltmen)] gains mass with nearly constant rate all 10 cycles, which indicates lower rate of degradation.
2.3. Effect of Bromide Ions
Upon the addition of Br^−^ ions, two peak pairs emerge at 0.4 and ca. 0.8 V, corresponding to the two-step oxidation of Br^−^ to Br_2_ via an intermediate Br_3_^−^. Since the electroactivity of bromide ions massively overlap the peaks of the polymers, it is impossible to make a conclusion regarding their electrochemical stability, but since the overall CV curves show only minor shrinkage after 50 cycles, the films are likely to be nearly stable in the presence of bromide. However, the stability of poly[Ni(Salen)] (Figure 4a) seems to be slightly worse than poly[Ni(Saltmen)] (Figure 4b).
During EQCM, poly[Ni(Saltmen)] (Figure 4d,f) exhibits good behavior with slow cycle-to-cycle mass decrease, which may be due to the ion exchange of BF_4_^−^ to Br^−^, and stable mass amplitude. The ion flux is approximately 78 g·mol^−1^ on the first slope, which corresponds to the inbound flux of Br^−^ ions, and 27 on the second slope, which can be explained by an inbound flux of Br^−^ ions, accompanied by an exhaust flux of neutral Br_2_. The EQCM of poly[Ni(Salen)] (Figure 4c,e) is much more complicated, but apparently, the poly[Ni(Salen)] film gains significant mass from cycle to cycle, showing that bromide incorporates in the structure of this polymer to some extent, which agrees with its lower electrochemical stability.
To further assess the morphological and elemental stability of the polymer films after electrochemical cycling in chloride-containing electrolytes, SEM imaging and EDX mapping were performed for both poly[Ni(SalEn)] and poly[Ni(Saltmen)] electrodes (Figures S1 and S2). The SEM images revealed that the morphology of the films was preserved after 50 CV cycles, indicating the absence of mechanical degradation or delamination. Elemental mapping of nickel by EDX confirmed that the surface distribution of Ni remained largely uniform, supporting the structural stability of the films.
However, semi-quantitative surface analysis indicated a detectable change in composition: both systems showed the presence of chlorine on the surface after cycling, suggesting that Cl^−^ anions are coordinated to the nickel centers or embedded in the film matrix. The observed behavior was consistent for both poly[Ni(SalEn)] and poly[Ni(Saltmen)] films, demonstrating that although the polymer frameworks remain morphologically stable, the surface undergoes chemical transformation due to anion uptake. These findings are in agreement with the hypothesis that a loss of electrochemical activity is not driven by mechanical damage, but rather by changes in the redox conductivity of the conjugated system induced by halide coordination.
2.4. XPS Analysis
To confirm the incorporation of external ligands into the polymer films, X-ray photoelectron spectroscopy of the polymeric films before and after 50 cycles in halide-containing electrolytes was performed (Figure S3). The Ni 2p, C 1s, N 1s and F 1p or Cl 2p peaks were compared and analyzed.
In the pristine state, before the addition of halide ions, the Ni 2p_3/2_ spectra of both polymers (Figure 5a,b) display characteristic peaks at ca. 855 and 872 eV. These peaks correspond to Ni(II) in a square-planar N,N,O,O-coordination environment, which is typical for nickel complexes with Salen-type ligands [30]. The same pattern is observed for all films with halide ions (Figure 5c,d,f), except the ion of poly[Ni(Salen)] after the addition of Cl^−^ ions (Figure 5e). In this case, there are two additional peaks at 851.8 and 858.9 eV. The first peak corresponds to the metallic Ni, ref. [32], while the second one can be attributed to nickel oxide or chloride [33].
N 1s spectra (Figure 6) of the pristine films contain one peak at ca. 399 eV, corresponding to the imine nitrogen, coordinated to the Ni atom, ref. [34], while there is an additional peak at ca. 402 eV due to the presence of the Et_4_N^+^ ions from the electrolyte [34]. The only sample that shows changes after CV is again poly[Ni(Salen)] after Cl^−^ ions. The initial peak at ca. 399 eV nearly disappears, replaced by a new peak at ca. 402 eV, which can be attributed to the Et_4_N^+^ ions.
After CV in fluoride-containing electrolytes, a peak at ca. 687.5 eV emerges in the F 1s spectra of both films (Figure 7b), which is attributed to the fluorination of the aromatic rings of the ligand [35]. In the same manner, cycling of poly[Ni(Saltmen)] in a chloride-containing electrolyte results in the chlorination of the aromatic rings of the ligand, since the Cl 2p peak at 200.2 eV is typical for chlorobenzenes [36] (Figure 7d). In the case of poly[Ni(Salen)], a peak at 196.6 eV emerges after CV with Cl^−^ ions, which can be attributed to the Cl^−^ ions [37] (Figure 7c).
In the C 1s spectra of all films (Figure 8), there are two predominant peaks at ca. 284.5 and 286.0 eV, which correspond to the carbon atoms of the ligand backbone. In addition, in F^−^-treated samples, a new peak emerges at 287–288 eV, which may be attributed to the fluorinated carbon atoms. In contrast, no additional peaks are present in the spectra after Cl^−^ treatment, which is due to the fact that the peaks of chlorinated carbons usually situate near 286 eV and overlap with existing peaks.
Concluding the results of XPS, both polymers undergo fluorination of the ligand upon CV with F^−^ ions. With Cl^−^ ions, poly[Ni(Saltmen)] undergoes chlorination as well, while poly[Ni(Salen)] suffers destruction of the complex, while in other cases the coordination cavity remains intact.
3. Discussion
A comparative analysis of the obtained CV and EQCM data shows the following trend: relative electrochemical stability of poly[Ni(Saltmen)] to poly[Ni(Salen)] increases in the row F^−^ < Cl^−^ < Br^−^, or, in other words, small, hard and nucleophilic anions better attack poly[Ni(Saltmen)], while large, soft and weekly nucleophilic anions better attack poly[Ni(Salen)]. Such an effect of the size of the attacking particle can be easily explained by the higher steric hindrance of poly[Ni(Saltmen)], but the opposite effect observed in the case of poly[Ni(Salen)] needs particular consideration regarding the possibility of interaction mechanisms.
According to the EQCM, both polymers uptake the fluoride anion in oxidized form. However, this process does not affect the electroactivity of poly[Ni(Salen)], while the low-potential peak of poly[Ni(Saltmen)] totally disappears after the first cycle. Such behavior may be explained by the fluorination of the ligand (Figure 9), because F^−^ is a much stronger nucleophile in aprotic and moderately polar CH_3_CN, in comparison with other halide ions. F^−^ is expected to fluorinate the benzene ring of the radical cationic species localized on the Salen ligands after oxidation, since fluorination of the electrochemically generated radical cationic species is widely described in the literature [38]. In contrast to the coordination on the Ni atom, which disables the conductivity and electroactivity of the polymer, benzene ring fluorination does not interrupt the conductivity pathway along the polymer chain. In case of poly[Ni(Saltmen)], the fluorination of the benzene rings may cause the distortion of the complex plane due to the repulsion of F^−^ substituents with methyl groups of the imine bridge, and thus prevent the polaron delocalization, which is known to be responsible for the low-potential peak of poly[Ni(Saltmen)] [39].
Upon oxidation of poly[Ni(Salen)] in a chloride-containing electrolyte, chloride anions readily coordinate to the oxidized nickel centers within the polymer’s backbone (Figure 10). This axial coordination leads to a marked decrease in redox activity, analogous to the demetallation of the NiSalen complex, as previously observed in aqueous environments. At the same time, the Ni atom in the poly[Ni(Saltmen)] complex is sufficiently hindered by four bulky methyl substituents on both sides of the complex plane. Instead, oxidative chlorination of the aryl rings of the ligand occurs, analogous to a previously discussed fluorination reaction. In parallel, chloride ions may also undergo electrochemical oxidation at the electrode surface, producing molecular chlorine, which subsequently evolves from the system. This additional process contributes to the CV of both poly[Ni(Salen)] and poly[Ni(Saltmen)] films.
The bromide ion, due to its lower oxidation potential, undergoes electrochemical oxidation via a two-step process involving the intermediate formation of tribromide (Br_3_^−^). This behavior is evidenced by the appearance of two well-defined bromide-related peaks in the voltammogram, while the polymer retains its intrinsic redox activity [40]. Poly[Ni(Salen)] still suffers slow degradation, which is apparently caused by the coordination of the bromide ion to the Ni center, while poly[Ni(Saltmen)] is completely resistant to this process.
It should be noted that in electrolytes containing Cl^−^ and especially Br^−^, partial oxidation of halide anions may occur in the same potential window as the redox transitions of the Ni–Salen polymer. This overlap could potentially complicate the direct attribution of current responses to polymer redox activity. However, control experiments with blank electrodes and the absence of irreversible oxidative signals in pristine films suggest that the majority of the changes observed during cycling are due to polymer degradation rather than halide oxidation.
While the observed mass changes in EQCM experiments may partly originate from swelling or partial entrapment of electrolyte species, the complementary XPS and EDX data confirm the presence of halide ions on the polymer surface after cycling. This supports the interpretation that specific halide coordination contributes to the observed mass increase and redox behavior.
To place the obtained results into a broader research context and to better rationalize the observed degradation trends, we summarized and compared the electrochemical performance of the present poly[Ni(Salen)] and poly[Ni(Saltmen)] films with the representative literature on structurally related nickel–Salen and nickel–salphen polymers (Table 1). Due to the variety of nickel–Salen-type polymer structures and their applications, the collected data span different device concepts (supercapacitors vs. Li-ion batteries), electrolytes (carbonate- and acetonitrile-based aprotic media, as well as alkaline aqueous electrolytes), film architectures (pristine polymers vs. carbon-based composites), and electrochemical protocols (CV, GCD, and EIS/spectroelectrochemistry). Therefore, Table 1 is not intended to provide a strictly quantitative benchmark, but rather to illustrate the diversity of reported performances and the factors most frequently discussed in the literature.
Importantly, the cycling stability reported for Ni–Salen-type polymer electrodes shows pronounced scatter across studies, even for closely related ligand frameworks. Depending on the electrolyte formulation, testing protocol, and film morphology, retention values range from nearly quantitative stability in rigorously controlled “dry” aprotic systems to rapid losses in the presence of nucleophilic or redox-active species (e.g., H_2_O and halides). Notably, although ligand design and steric shielding are often emphasized as key stabilization strategies, systematic investigations that isolate the role of electrolyte composition—particularly the impact of trace impurities—remain limited. The broad dispersion of stability metrics reported in the literature is consistent with the notion that minor contaminants can disproportionately influence degradation pathways and may partially explain why comparable Ni–Salen polymer electrodes sometimes exhibit markedly different durability in nominally similar experiments.
In this context, the present work provides a controlled and mechanistically oriented assessment of electrolyte impurity effects by deliberately introducing F^−^, Cl^−^, and Br^−^ (1 mM) into a well-defined acetonitrile-based supporting electrolyte and interrogating the films by complementary CV, EQCM, and XPS/EDX analyses. Our results show that chloride causes substantial pseudocapacitance loss for both protected and unprotected polymers over prolonged cycling, whereas the bromide-containing electrolyte preserves the redox response to a much greater extent, particularly for the sterically protected poly[Ni(Saltmen)]. These findings emphasize that the apparent stability of Ni–Salen polymers cannot be interpreted independently of electrolyte purity and impurity chemistry, and rational ligand steric design should be considered together with electrolyte impurity control when targeting durable polymer electrodes for practical electrochemical energy storage environments.
4. Materials and Methods
4.1. Materials and Synthesis
All reagents were purchased from commercial suppliers and used as received unless otherwise stated. Anhydrous acetonitrile (99.9%) was further dried over activated 3 Å molecular sieves for at least 20 days prior to use. The final water content was controlled by Karl Fischer titration and did not exceed 10 ppm. Supporting electrolytes were prepared by dissolving in a solution containing 0.1 M Et_4_NBF_4_ in CH_3_CN. Halide additives (tetraethylammonium chloride (Et_4_NCl), tetraethylammonium bromide (Et_4_NBr), or tetraethylammonium fluoride (Et_4_NF)) were introduced at concentrations of 1 mM (10^−3^ M). Nickel(II) complexes of Schiff base ligands (Figure 1) were synthesized as previously described [43].
The ligands were obtained via condensation of the corresponding salicylaldehyde derivatives with ethylenediamine or 2,2,3,3-tetramethylethylenediamine under reflux in ethanol. The resulting complexes were purified by recrystallization.
4.2. Electrochemical Characterization
Electrochemical measurements were performed using Autolab PGSTAT30 and PGSTAT302N potentiostats (Metrohm Autolab B.V., Utrecht, The Netherlands). A conventional three-electrode cell was employed with a glassy carbon disk (0.07 cm^2^) as the working electrode, a platinum foil as the counter electrode, and a Ag|Ag^+^ reference electrode (acetonitrile solution of 0.1 M Et_4_NBF_4_ + 5 mM AgNO_3_). All potentials are reported versus this pseudo-reference electrode. Polymer films were deposited electrochemically on glassy carbon or platinum substrates from 1 mM monomer solutions in acetonitrile containing 0.1 M Et_4_NBF_4_. Potentiostatic electropolymerization was carried out at 0.8 V vs. Ag|Ag^+^ until a charge density of 0.04 C·cm^−2^ was reached. To assess the influence of polymer film thickness on degradation behavior, all samples in this study were synthesized using the same electropolymerization protocol to ensure comparable thicknesses. This approach allowed us to isolate the effect of halide coordination on electrochemical stability without introducing variability from morphological or thickness-related gradients.
Cyclic voltammetry was conducted in the potential window from –0.2 V to +0.6 V (or +0.8 V for Saltmen-type films) at a scan rate of 50 mV·s^−1^. After electropolymerizing, the films were first tested in a pure supporting electrolyte for 5 cycles, followed by an additional 50 scans after the introduction of halide anions. For each condition, at least three independent electrodes were tested to ensure reproducibility.
EQCM measurements were performed using a QCM200 quartz microbalance system (Stanford Research Systems, Sunnyvale, CA, USA) equipped with 5 MHz AT-cut quartz crystals coated with Ti/Pt (active area: 1.37 cm^2^). The film was electropolymerized under potentiostatic conditions at 0.8 V vs. Ag|Ag^+^ until a charge density of 0.95 C·cm^−2^. After electropolymerizing, the first 5 cycles were tested in a pure supporting electrolyte and then 50 scans were conducted after the introduction of halide anions. Mass changes were monitored during potential cycling under identical electrochemical conditions. Ion flux m/z values were calculated using the Sauerbrey equation [44].
4.3. Physicochemical Characterization
X-ray photoelectron spectroscopy was performed using a Thermo Fisher Scientific Escalab 250Xi (Waltham, MA, USA) instrument equipped with a non-monochromatized Al Kα X-ray source (1486.6 eV). Spectra were collected with a pass energy of 20 eV and an energy resolution better than 0.8 eV. The pressure in the analysis chamber was maintained below 10^−8^ mbar during measurements.
Polymer films for XPS analysis were electropolymerized on platinum foil substrates using the same conditions as for electrochemical experiments. Prior to XPS measurements, the samples were subjected to 50 CV cycles in a halide-containing electrolyte to simulate typical degradation conditions.
5. Conclusions
As a result of the comprehensive analysis of the influence of halide anions (Cl^−^, Br^−^, and F^−^) on the electrochemical stability of nickel–Schiff base polymer films, we established that the sterically hindered complex poly[Ni(Saltmen)] is vulnerable to strong nucleophiles like F^−^ through the oxidative ligand halogenation, while the less hindered poly[Ni(Salen)] suffers from the destruction of the coordination cavity by Cl^−^ ions. We found the bromide ion to be the most harmless halide ion due to its bulkiness and low oxidation potential, although the steric protection increased the stability of the complexes against the attack of this ion. We identified two principal degradation mechanisms: (1) axial coordination of external ligands, which disrupts the conjugated system and reduces charge transport, and (2) ligand-centered halogenation of the ligand. The interplay between these factors governs the long-term stability or degradation of the electroactive polymer films.
These findings underscore the critical importance of both electrolyte composition and ligand molecular architecture in ensuring the durability of Ni-based redox polymers. Strategies focused on metal center shielding and the exclusion of electrochemically active impurities offer a promising route toward the rational design of stable materials for supercapacitor applications.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sharma R. Kumar H. Kumar G. Sharma S. Aneja R. Sharma A.K. Kumar R. Kumar P. Progress and challenges in electrochemical energy storage devices: Fabrication, electrode material, and economic aspects Chem. Eng. J.202346814370610.1016/j.cej.2023.143706 · doi ↗
- 2Rahman M.H. Werth H. Goldman A. Hida Y. Diesner C. Lane L. Menezes P.L. Recent Progress on Electroactive Polymers: Synthesis, Properties and Applications Ceramics 2021451654110.3390/ceramics 4030038 · doi ↗
- 3Beygisangchin M. Abdul Rashid S. Shafie S. Sadrolhosseini A.R. Lim H.N. Preparations, Properties, and Applications of Polyaniline and Polyaniline Thin Films-A Review Polymers 202113200310.3390/polym 1312200334207392 PMC 8234317 · doi ↗ · pubmed ↗
- 4Moucka R. Sedlacik M. Kasparyan H. Prokes J. Trchova M. Hassouna F. Kopecky D. One-Dimensional Nanostructures of Polypyrrole for Shielding of Electromagnetic Interference in the Microwave Region Int. J. Mol. Sci.202021881410.3390/ijms 2122881433233379 PMC 7700242 · doi ↗ · pubmed ↗
- 5Qiu Y. Zhong F. Xu Z. Song J. Shen K. Li H. Chen L. Acid vapor doping of polar polythiophenes for high electrical conductivity RSC Adv.202515263472635210.1039/D 5RA 03453 A 40703071 PMC 12284755 · doi ↗ · pubmed ↗
- 6Song E. Choi J.W. Conducting Polyaniline Nanowire and Its Applications in Chemiresistive Sensing Nanomaterials 2013349852310.3390/nano 303049828348347 PMC 5304646 · doi ↗ · pubmed ↗
- 7Suh S. Kim K. Park J. Kim W. Ultrafast flexible PEDOT:PSS supercapacitor with outstanding volumetric capacitance for AC line filtering Chem. Eng. J.202346314237710.1016/j.cej.2023.142377 · doi ↗
- 8Anishchenko D.V. Kolesnik S.S. Valova S.F. Wang B. Levin O.V. Phase Transition-Governed Asymmetry in Poly(3-alkylthiophenes) Redox Kinetics: An Electrochemical Study J. Phys. Chem. C 2025129162441626010.1021/acs.jpcc.5c 02840 · doi ↗