Chloroplast Genome Evolution in Pleurothallidinae (Orchidaceae): Lineage-Specific Selection, Codon Usage Patterns, and Phylogenetic Implications
Yuxue Liu, Qiang Zhang, Zhenhua Wu, Zhenping Shi, Shuo Wang

TL;DR
This study analyzes chloroplast genomes in orchids to understand their evolution, selection patterns, and phylogenetic relationships.
Contribution
The study provides new insights into lineage-specific selection and identifies molecular markers for Pleurothallidinae orchids.
Findings
Chloroplast genomes in Pleurothallidinae show conserved gene content but lineage-specific positive selection in some genes.
Codon usage bias is shaped by natural selection, not mutation pressure, with geographic variation.
Eight hypervariable intergenic regions are identified as potential molecular markers for taxonomic studies.
Abstract
Background: The subtribe Pleurothallidinae is a diverse group within Orchidaceae with a complex taxonomic history. Comparative plastome analysis can provide insights into genome evolution and facilitate phylogenetic reconstruction. Methods: Here we analyzed 25 complete chloroplast genomes representing 15 genera, including 14 newly assembled genomes, to investigate plastome evolution in this subtribe. Results: All genomes exhibited the typical quadripartite structure (148, 246–158, 138 bp) with conserved gene content (128–134 genes). While most protein-coding genes were under purifying selection, we detected signatures of positive selection in specific lineages. Notably, ndhF in Lepanthes tachirensis showed a markedly elevated Ka/Ks ratio (3.65), which may be associated with adaptation to an extensive distributional range. ENC-plot analysis indicated that natural selection, rather than…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
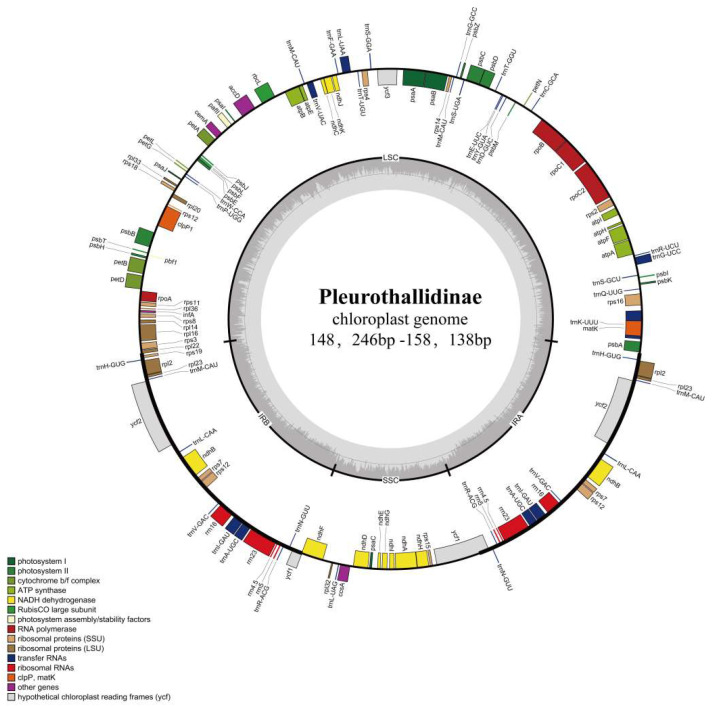 Figure 1
Figure 1 Figure 2
Figure 2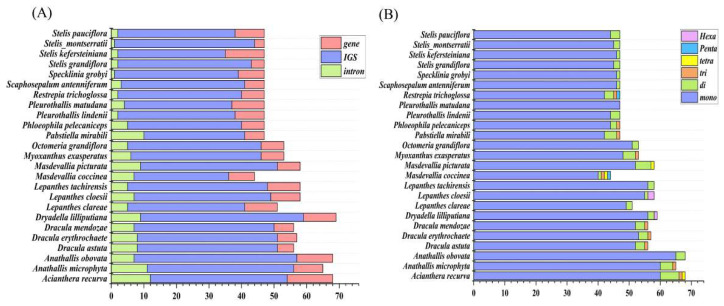 Figure 3
Figure 3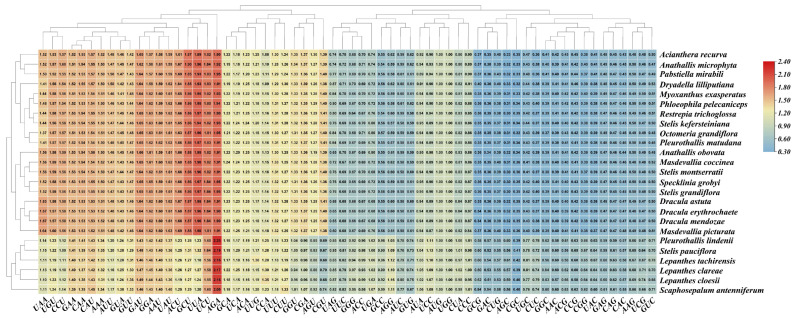 Figure 4
Figure 4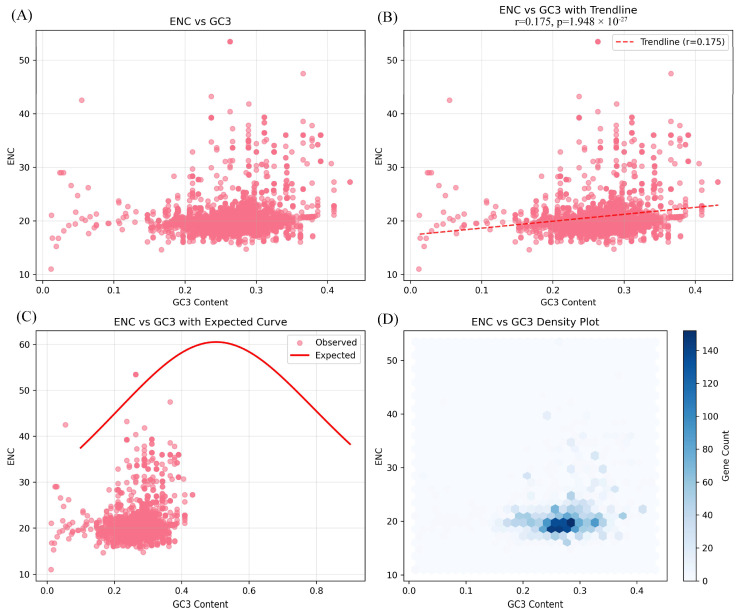 Figure 5
Figure 5 Figure 6
Figure 6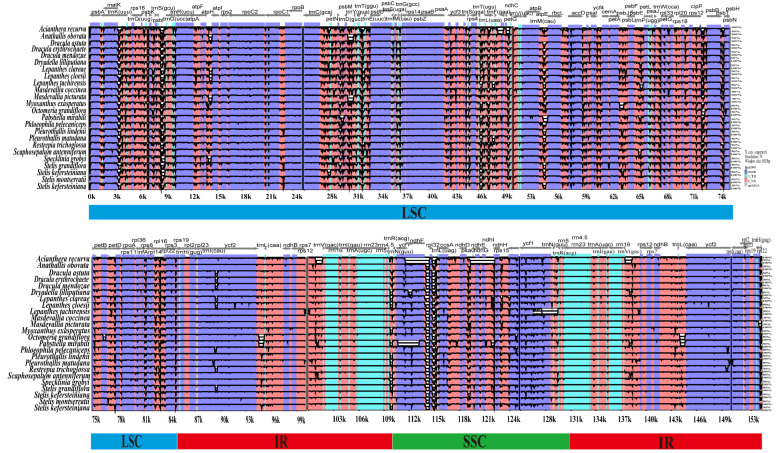 Figure 7
Figure 7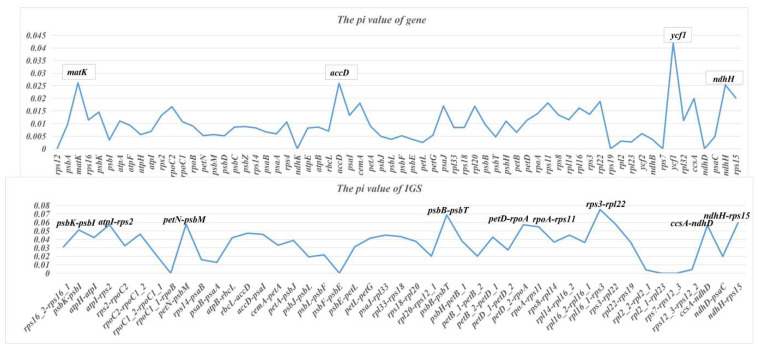 Figure 8
Figure 8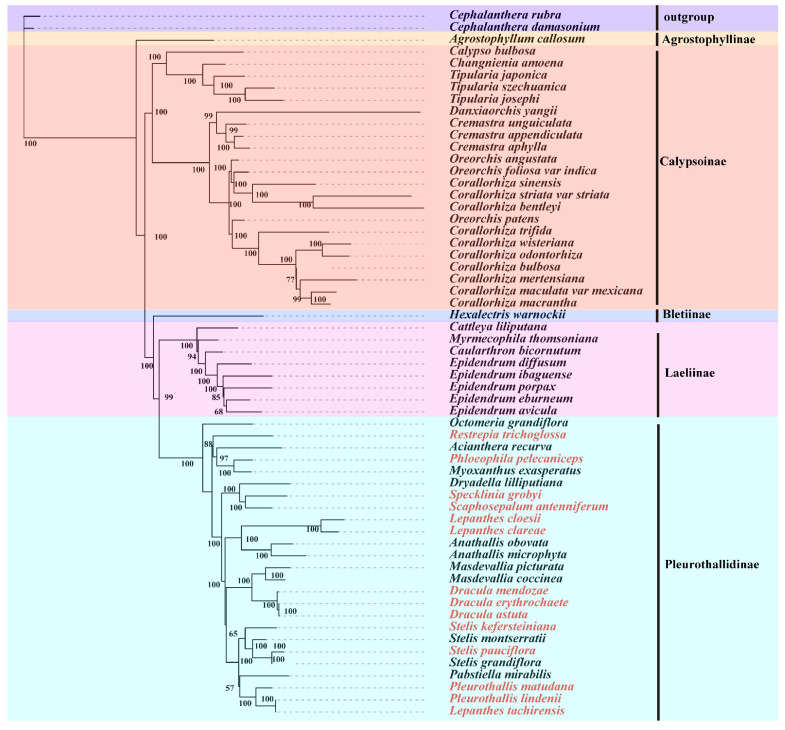 Figure 9
Figure 9- —Shandong Province Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Biological and pharmacological studies of plants · Plant and animal studies
1. Introduction
The subtribe Pleurothallidinae (Orchidaceae: Epidendroideae) comprises primarily epiphytic orchids distributed in Neotropical montane forests [1,2]. This subtribe has a complex taxonomic history, with generic boundaries repeatedly revised as molecular phylogenetic data have accumulated [2]. Many traditionally recognized genera have proven to be polyphyletic or paraphyletic, prompting ongoing efforts to establish classifications that reflect evolutionary relationships [1,3]. Molecular phylogenetic studies based on nuclear ITS and plastid matK sequences have confirmed the monophyly of Pleurothallidinae while revealing that several genera, including Lepanthes and Pleurothallis, are not monophyletic as traditionally circumscribed [1,2]. These findings highlight the need for additional molecular data to resolve phylogenetic relationships and facilitate taxonomic revision.
Complete chloroplast genome (plastome) sequences provide rich information for phylogenetic reconstruction and molecular evolutionary analysis [4,5,6]. Beyond phylogenetic signal, plastomes can reveal patterns of structural evolution, selection pressure on protein-coding genes, and codon usage bias that may reflect adaptation to environmental conditions [7]. The typical angiosperm plastome comprises a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeat (IR) regions [8,9]. Comparative plastome analysis has proven valuable for identifying hypervariable regions useful as molecular markers and for detecting signatures of adaptive evolution [10,11].
Although plastome sequences from several Pleurothallidinae genera have been reported [12], comprehensive comparative analyses examining selection pressures and codon usage patterns across multiple genera remain limited. In particular, whether plastome evolution in Pleurothallidinae shows lineage-specific patterns potentially associated with ecological adaptation has not been systematically investigated.
In this study, we assembled 14 new plastomes and analyzed them together with 11 previously published genomes, representing 15 genera and 25 species of Pleurothallidinae. Our objectives were to: (1) characterize plastome structural features and identify hypervariable regions suitable as molecular markers; (2) evaluate selective pressures on protein-coding genes and identify genes potentially under positive selection; (3) analyze codon usage patterns and assess potential factors shaping codon bias; and (4) reconstruct phylogenetic relationships and evaluate congruence with traditional classifications. This study aims to expand genomic resources for Pleurothallidinae and provide insights into plastome evolution in this taxonomically challenging subtribe.
2. Materials and Methods
2.1. Chloroplast Genome Assembly and Annotation
Raw sequencing reads for 30 Pleurothallidinae accessions were retrieved from the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/). De novo chloroplast genome assembly was performed using GetOrganelle v1.7.7.0 [13] with default parameters. Fourteen complete plastomes were successfully assembled and annotated using GeSeq v1.42 [14]. Annotations were manually curated in Geneious v9.0.2 [15], with particular attention to intron boundaries and start/stop codon positions. Annotated genomes were formatted using GB2Sequin v16 [16] and deposited in GenBank (accession numbers: OR713737–OR713740, OR909686–OR909688, PP094516, PX828354–PX828359). Circular genome maps were generated using OGDRAW v1.3.1 [17].
2.2. Genome Structural Analysis
Plastome characteristics including region lengths, GC content, and gene composition were calculated using Geneious v9.0.2 [15,18]. Simple sequence repeats (SSRs) were identified using MISA [19] with minimum repeat thresholds of 9, 4, 3, 3, 3, and 3 for mono- through hexanucleotide motifs, respectively [20]. Tandem repeats were detected using Tandem Repeats Finder [21]. IR boundary positions were visualized using IRscope [22] with Cephalanthera damasonium (MH590345) as reference. Whole-genome alignments were performed using mVISTA [23] in Shuffle-LAGAN mode with Anathallis microphyta as reference.
2.3. Selection Pressure and Codon Usage Analysis
The ratio of non-synonymous to synonymous substitutions (Ka/Ks) was calculated for all shared protein-coding genes using C. damasonium as reference. Genes with Ka/Ks > 1 were identified as candidates potentially under positive selection. Relative synonymous codon usage (RSCU) values and effective number of codons (ENCs) were calculated using MEGA v11 [24]. Effective number of codons–GC content at the third codon position (ENC-GC3) plots were constructed following [25] to assess the relative contributions of mutation pressure and selection to codon usage bias. RSCU heatmaps were generated using TBtools v1.1047 [26].
2.4. Nucleotide Diversity and Marker Identification
Nucleotide diversity (π) was calculated using DnaSP v6.12 [27] with a sliding window approach (window length = 600 bp; step size = 50 bp). Regions with π > 0.025 for coding sequences or π > 0.05 for intergenic spacers were designated as hypervariable regions.
2.5. Phylogenetic Analysis
Phylogenetic relationships were inferred using plastome sequences from 57 species of tribe Epidendreae (Table S3), including representatives of subtribes Agrostophyllinae, Bletiinae, Calypsoinae, Laeliinae, and Pleurothallidinae. Based on previous phylogenetic studies, C. damasonium (MH590345) and Cephalanthera rubra (NC_041181) were selected as outgroups, as they belong to a clade phylogenetically distinct from Epidendreae within Epidendroideae, ensuring an appropriate evolutionary distance for rooting the phylogenetic tree [28]. For phylogenetic reconstruction, protein-coding sequences were extracted following strict criteria: (1) non-pseudogenized protein-coding genes shared across all 57 species were included to ensure data consistency; (2) duplicate gene copies from the two inverted repeat (IR) regions (IRA and IRB) were excluded. They were aligned using PhyloSuite [29,30] and trimmed using trimAl [31]. Maximum likelihood analysis was conducted in IQ-TREE v2.1.3 [32] with the GTR + I + G model selected by BIC. Branch support was assessed using 1000 ultrafast bootstrap replicates.
3. Results
3.1. General Features of Pleurothallidinae Chloroplast Genomes
All 25 Pleurothallidinae plastomes exhibited the typical angiosperm quadripartite structure (Figure 1; Table 1). Total length ranged from 148,246 bp (Acianthera recurva) to 158,138 bp (Restrepia trichoglossa). The LSC region ranged from 83,902 bp (Anathallis obovata) to 86,291 bp (R. trichoglossa), while the SSC region showed greater variation from 10,573 bp (Ac.n recurva) to 21,966 bp (L. tachirensis).
Gene content was generally conserved, with 128–134 genes identified including 81–88 protein-coding genes, 37–39 tRNAs, and 8 rRNAs (Table 2). These genes can be categorized into photosynthesis-related genes, self-replication genes, other functional genes, and genes of unknown function. GC content was stable across species (36.7–37.1%), with IR regions more GC-rich (43.1–43.2%) than LSC (34.3–34.8%) or SSC (29.6–30.1%) regions. Ac. recurva possessed the smallest plastome with apparent gene losses compared to other species [33,34]. This reduction may be associated with its particular ecological niche, though additional sampling would be needed to determine whether this pattern is species-specific.
3.2. IR Boundary Variation
Analysis of IR/SC junction positions revealed variation among species (Figure 2). The JLB junction was relatively conserved, with rpl22 and rps19 genes consistently present in most species. However, Masdevallia picturata showed a distinctive arrangement with trnH-GUG and rps19 at this junction, representing a potential species-specific marker.
The JSB and JSA junctions exhibited greater variation. At the JSB junction, Ac. recurva contained ycf1 and rpl32, M. coccinea contained trnN and ndhF, while most other species possessed ycf1 and ndhF. At the JLA junction, species could be grouped into three categories: those with rpl22 and psbA, those with rps19 and psbA, and those with trnH and psbA.
3.3. Repeat Sequence Analysis
Six SSR types were identified across the 25 plastomes (Figure 3). Mononucleotide repeats (predominantly A/T) were most abundant, consistent with patterns in other vascular plants [35,36]. SSRs were mainly distributed in intergenic spacers (29–50 SSRs), followed by coding regions (1–12 SSRs) and introns (3–14 SSRs). Tandem repeat analysis identified 20–60 repeats per genome. Two tandem repeats were located adjacent to functional genes (rbcL, psaI, rpl14, rps3) at distances of 133–851 bp (Table 3).
3.4. Codon Usage Patterns
RSCU analysis revealed consistent patterns across species (Figure 4) [37]. Thirty codons exhibited RSCU > 1.0, with codons ending in A or U generally showing higher values than those ending in G or C, reflecting the typical A/T preference of angiosperm plastomes.
Interestingly, we detected differences in codon preferences for CGU, AGU, GGU, CGA, GCU, and UUA among species from different geographic regions. Pleurothallis and Stelis species from different habitats showed some divergent patterns. These observations suggest that geographic or environmental factors may contribute to codon usage variation. ENC-GC3 analysis showed that most genes fell below the expected neutral curve (Figure 5), which is indicative of a potential role for natural selection in shaping codon usage bias in Pleurothallidinae, beyond the effects of mutational bias alone. However, it should be noted that factors such as mutation bias, GC content, and tRNA abundance could also contribute to the observed patterns, and their relative influences warrant further investigation. This finding is consistent with studies in other orchid lineages showing selection-driven codon bias [38].
3.5. Selection Pressure Analysis
Ka/Ks analysis revealed that most protein-coding genes across all species exhibited values <1.0, indicating purifying selection (Figure 6). This pattern reflects functional constraints on plastome-encoded genes essential for photosynthesis and gene expression.
However, we identified several genes with Ka/Ks > 1 in specific lineages. Most notably, ndhF in Lepanthes tachirensis exhibited Ka/Ks = 3.65, markedly higher than congeners L. cloesii and L. clareae. The ndhF gene encodes a subunit of the NADH dehydrogenase complex involved in cyclic electron transport. Given that L. tachirensis is an epiphytic orchid, its life-history strategy inherently imposes stringent demands on photosynthetic regulation. The broad distribution across heterogeneous environments in the Andes further suggests that elevated Ka/Ks ratios in key photosynthetic genes ndhF—may signal adaptive evolution in response to heterogeneous combinations of light intensity, drought frequency, and thermal stress across its range. However, Ka/Ks > 1 can also arise from relaxed purifying selection. Elevated Ka/Ks in ndh genes have been associated with environmental adaptation in other plant lineages [39].
Additional genes showing Ka/Ks > 1 included: rpl14 in L. tachirensis, Sc. antenniferum, and Pleurothallis lindenii; rps14 in Sp. grobyi, St. grandiflora, Stelis pauciflora, Pl. matudana; rps15 in Ac. recurva, Dra. mendozae, and Dracula astuta; rps16 in Ph. pelecaniceps and Dracula astuta; rps19 in M. coccinea; psbH in Stelis kefersteiniana; psbK in Pleurothallis lindenii; and ycf2 in Dracula erythrochaete and Dry. lilliputiana. These lineage-specific patterns suggest that selection pressures on plastome genes may vary among Pleurothallidinae species.
3.6. Genome Comparison and Sequence Divergence
Whole-genome alignment revealed overall high sequence similarity among the 25 plastomes (Figure 7). Sequence variation was concentrated in LSC and SSC regions, with IR regions more conserved. Non-coding regions showed higher divergence than coding regions. Nine divergent intergenic regions were identified by mVISTA: matK-trnR-UCU, atpF-atpH, rpoB-psbD, ycf3-trnV-UAC, ndhK-atpE, atpB-rbcL, accD-psaI, psbJ-psbB, and ndhF-ccsA. Notable variation was also detected in ycf1 [40].
Sliding window analysis (window length = 600 bp; step size = 50 bp) identified hypervariable regions suitable for marker development (Figure 8). For coding regions, matK, accD, ycf1, and ndhH showed π > 0.025 (more than twice the mean: 0.01). For intergenic spacers, eight regions showed π > 0.05 (exceeding 1.4 times the mean: 0.035): psbK-psbI, atpI-rps2, petN-psbM, psbB-psbT, petD-rpoA, rpoA-rps11, rps3-rpl22, and ccsA-ndhD. Therefore, the regions are robust candidate markers identified under the current analytical framework. The identification of hypervariable genomic regions is contingent upon analytical parameters. Excessively large window sizes may dilute localized selective signatures, whereas overly small windows introduce stochastic noise due to limited site sampling. A large step length can overlook narrow peaks of diversity, while a small step increases redundancy without enhancing biological resolution. Moreover, restricted taxon sampling may yield biased diversity estimates due to increased susceptibility to sampling error. In future research, the practical utility in species differentiation or population studies should be further validated by expanding the taxonomic sampling scope and testing under different parameter settings where relevant. Nonetheless, these regions provide candidate markers for species identification and population genetic studies in this taxonomically challenging subtribe [41].
3.7. Phylogenetic Relationships
Maximum likelihood analysis of 57 Epidendreae species yielded a well-resolved phylogeny with strong bootstrap support (>95% at most nodes; Figure 9). All five subtribes were recovered as monophyletic. Within Pleurothallidinae, 25 species were resolved into six lineages. Consistent with previous studies [2,3], Lepanthes and Pleurothallis were not monophyletic. L. tachirensis—traditionally placed in Lepanthes subgenus Lepanthes subsection Breves [42]—was embedded within a clade containing Pleurothallis species. The phylogenetic tree based on the inverted repeat (IR) regions was consistent with this result (Figure S1). These results further support the need for taxonomic revision of these genera based on molecular evidence.
4. Discussion
4.1. Plastome Features and Structural Variation
The 25 Pleurothallidinae plastomes analyzed here showed the conserved quadripartite structure typical of angiosperms. Plastome sizes (148,246–158,138 bp) and gene numbers (128–134) are within the range reported for other orchids [11,43]. The strong A/T preference in SSRs is consistent with patterns across vascular plants [34]. IR boundary variation, particularly at JSB and JSA junctions involving ycf1 and ndhF, is common in angiosperms and may provide phylogenetically informative characters [44]. The unique arrangement in Masdevallia picturata at the JLB junction could serve as a species-specific marker pending confirmation with additional sampling.
4.2. Lineage-Specific Positive Selection and Potential Environmental Adaptation
While most protein-coding genes showed Ka/Ks < 1 indicating purifying selection, we detected signatures of positive selection in specific lineages [45,46,47]. The most striking finding was the elevated Ka/Ks (3.65) for ndhF in L. tachirensis, which contrasts sharply with values in congeners from restricted distribution. The ndhF gene product participates in the NDH complex mediating cyclic electron flow around photosystem I, which is 7important for photoprotection and adaptation to fluctuating light conditions [39]. The Andean mountain range, extending from northwestern Venezuela to Peru, encompasses highly heterogeneous habitats with pronounced latitudinal and altitudinal gradients. This environmental diversity results in variable light conditions, UV radiation, and temperature fluctuations, which may exert unique selective pressures on the photosynthetic machinery of resident species [48,49].
This finding is consistent with studies reporting elevated evolutionary rates in ndh genes in plant lineages experiencing environmental stress [39]. Although the geographic and ecological context of L. tachirensis is consistent with the hypothesis that its elevated Ka/Ks may reflect environmental adaptation, positing a direct adaptive link based on a single species comparison is speculative. Functional validation is therefore required to confirm the adaptive significance of these genetic changes. Future studies with expanded sampling across altitudinal gradients could test whether similar patterns occur in other high-altitude Pleurothallidinae. Therefore, while this anomalous ndhF value is noteworthy and merits further investigation, we do not consider habitat heterogeneity conclusive evidence for positive selection.
4.3. Codon Usage Bias: Evidence for Selection
ENC-plot analysis indicated that codon usage in Pleurothallidinae deviates from a model governed solely by mutation pressure, indicating a probable contribution of natural selection (e.g., translational efficiency) alongside other factors such as genomic nucleotide composition and mutational spectra [38,50]. This finding has implications for understanding plastome evolution in this subtribe. Selection on codon usage can reflect optimization of translational efficiency, particularly for highly expressed genes.
The observation that species from different geographic regions showed variation in codon preferences for specific codons (CGU, AGU, GGU, CGA, GCU, UUA) is intriguing. While these patterns suggest a potential influence of geographic or environmental factors on codon usage variation, this interpretation should be treated with caution [51]. Equally plausible alternatives for these differences include stochastic evolutionary processes specific to certain lineages, spatially heterogeneous mutation pressures, or the influence of other factors that our phylogenetic framework lacks the power to resolve. If confirmed with broader sampling, this would indicate that plastome evolution in Pleurothallidinae responds to local environmental conditions, potentially through selection for translational efficiency under different temperature or light regimes [52,53,54].
4.4. Molecular Markers and Taxonomic Implications
We identified eight hypervariable intergenic regions with π > 0.05 that could serve as molecular markers for species identification and population genetics in Pleurothallidinae. Given that traditional morphology-based identification can be challenging in this subtribe, molecular markers are particularly valuable [55,56,57]. The regions identified here (psbK-psbI, atpI-rps2, petN-psbM, psbB-psbT, petD-rpoA, rpoA-rps11, rps3-rpl22, ccsA-ndhD) complement previously reported variable regions and provide additional options for marker development.
Our phylogenetic results confirm the non-monophyly of Lepanthes and Pleurothallis, consistent with earlier molecular studies [2,3]. The placement of L. tachirensis within a Pleurothallis-dominated clade, highlighting a critical limitation of traditional taxonomic frameworks: the floral characters long relied upon to delimit these genera, such as the degree of sepal fusion, labellum morphology, and structural differentiation of floral segments, do not consistently track the evolutionary relationships inferred from molecular data. This discrepancy arises primarily from the high level of homoplasy in floral traits within Pleurothallidinae [2]. These findings further validate the necessity of ongoing taxonomic revisions that integrate molecular phylogenetics with morphological reevaluation [2,19], as they underscore the inadequacy of relying solely on floral characters for generic delimitation in this evolutionarily dynamic subtribe.
The relatively long branch length of Lepanthes in the chloroplast genome coding region-based phylogenetic tree is closely linked to its molecular evolutionary patterns and taxonomic delimitation, and also provides a reference for the screening of molecular markers within this group. Previous studies have shown that the early diversification of the Lepanthes lineage (>1500 species)—dated to around 8 Ma—is associated with the colonization of novel Andean habitats, rapid mountain uplift, and the emergence of specialized pollination systems (e.g., pseudocopulation and food mimicry, reported in closely related taxa) [3,28,58]. The accelerated evolutionary rate in Lepanthes likely drove extensive accumulation of synonymous mutations, thereby amplifying the genetic distance between the Lepanthes clade and related groups and resulting in the observed long-branch phenomenon in phylogenetic reconstructions. Future studies should further verify whether synonymous mutation accumulation is the main cause of branch elongation by performing phylogenetic reconstructions using amino acid sequences from chloroplast coding regions, which can also alleviate the confounding effects of non-synonymous mutations on branch length and taxonomic inference. Meanwhile, integrating nuclear gene sequences and other molecular markers will help clarify the taxonomic boundaries and evolutionary history of Lepanthes. These efforts will provide more reliable molecular evidence for taxonomic revision in this group.
4.5. Limitations and Future Directions
Several limitations should be noted. Our sampling represents a subset of Pleurothallidinae diversity, and patterns observed here may not apply to all lineages. The elevated Ka/Ks values should be interpreted cautiously pending functional validation. The correlation between codon usage and geography requires confirmation with broader sampling. Future studies should expand taxonomic and geographic coverage, validate putative adaptive signatures with functional assays, and integrate nuclear genomic data for comprehensive evolutionary analysis.
5. Conclusions
We analyzed 25 plastomes representing 15 genera of Pleurothallidinae, including 14 newly assembled genomes. All plastomes showed a conserved quadripartite structure with predominantly purifying selection on protein-coding genes. We identified lineage-specific signatures of positive selection, most notably in ndhF of the high-altitude species L. tachirensis (Ka/Ks = 3.65), suggesting potential environmental adaptation. Codon usage in Pleurothallidinae shows a marked departure from the mutation-dominated neutral model, demonstrating that natural selection plays a potential role in driving codon evolution, with distinct patterns diverging across species from different geographic regions. We identified eight hypervariable intergenic regions suitable as molecular markers, and confirmed that Lepanthes and Pleurothallis are non-monophyletic. These findings expand genomic resources for Pleurothallidinae, reveal patterns of lineage-specific plastome evolution, and provide molecular tools for future taxonomic and evolutionary studies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pridgeon A.M. Solano R. Chase M.W. Phylogenetic relationships in Pleurothallidinae (Orchidaceae): Combined evidence from nuclear and plastid DNA sequences Am. J. Bot.2001882286230810.2307/355839021669661 · doi ↗ · pubmed ↗
- 2Karremans A. Genera Pleurothallidinarum: An updated phylogenetic overview of pleurothallidinae Lankesteriana 20161621924110.15517/lank.v 16i 2.26008 · doi ↗
- 3Arias T. Moreno J.S. Reyes S. Almario M.L. Serna-Sánchez A. Iturralde G.A. Valencia J. Baquero L. Zuluaga A. Plastome phylogenomics of the diverse neotropical orchid genus Lepanthes with emphasis on subgenus Marsipanthes (Pleurothallidinae: Orchidaceae)BMC Ecol. Evol.2025257910.1186/s 12862-025-02396-640775299 PMC 12329981 · doi ↗ · pubmed ↗
- 4Song W. Chen Z. Shi W. Han W. Feng Q. Shi C. Engel M.S. Wang S. Comparative Analysis of Complete Chloroplast Genomes of Nine Species of Litsea (Lauraceae): Hypervariable Regions, Positive Selection, and Phylogenetic Relationships Genes 202213155010.3390/genes 1309155036140718 PMC 9498446 · doi ↗ · pubmed ↗
- 5Lu R.S. Li P. Qiu Y.X. The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses Front. Plant Sci.20167205410.3389/fpls.2016.0205428119727 PMC 5222849 · doi ↗ · pubmed ↗
- 6Saina J.K. Gichira A.W. Li Z.Z. Hu G.W. Wang Q.F. Liao K. The complete chloroplast genome sequence of Dodonaea viscosa: Comparative and phylogenetic analyses Genetica 201814610111310.1007/s 10709-017-0003-x 29170851 · doi ↗ · pubmed ↗
- 7Wicke S. Schneeweiss G.M. de Pamphilis C.W. Müller K.F. Quandt D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function Plant Mol. Biol.20117627329710.1007/s 11103-011-9762-421424877 PMC 3104136 · doi ↗ · pubmed ↗
- 8Palmer J. Stein D. Conservation of chloroplast genome structure among vascular plants Curr. Genet.19861082383310.1007/BF 00418529 · doi ↗