Plastome Evolution in Viburnum (Adoxaceae): Comparative Genomics Reveals Hypervariable Markers and Relaxed Selection on Protein Import Genes
Lanruo Mou, Qiang Zhang, Bingyue Zhu, Chao Shi, Jing Yang

TL;DR
This study examines the evolution of Viburnum plant chloroplast genomes, identifying hypervariable markers and relaxed selection on certain genes.
Contribution
The study identifies hypervariable intergenic spacers and reports relaxed selection on genes related to fatty acid biosynthesis and protein import in Viburnum plastomes.
Findings
Plastomes of Viburnum species show conserved structure with variation in intergenic spacers.
Single-copy regions exhibit higher nucleotide diversity compared to inverted repeat regions.
Genes accD, ycf1, and ycf2 show relaxed selection, indicating functional divergence.
Abstract
Background: Viburnum (Adoxaceae) is a species-rich woody genus whose taxonomy is complicated by morphological convergence and hybridization. Methods: We assembled complete plastomes of eight species representing five sections and analyzed their structural variation, sequence divergence, and molecular evolution. Results: All plastomes displayed the conserved quadripartite structure typical of angiosperms, with limited size variation attributable primarily to intergenic spacer-length polymorphisms. Sequence divergence was unevenly distributed, with single-copy regions exhibiting substantially higher nucleotide diversity than inverted repeat regions. We identified multiple hypervariable intergenic spacers such as the region trnK-UUU–rps16, suitable as molecular markers for population genetics and species identification. Selection pressure analysis revealed that while most protein-coding…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
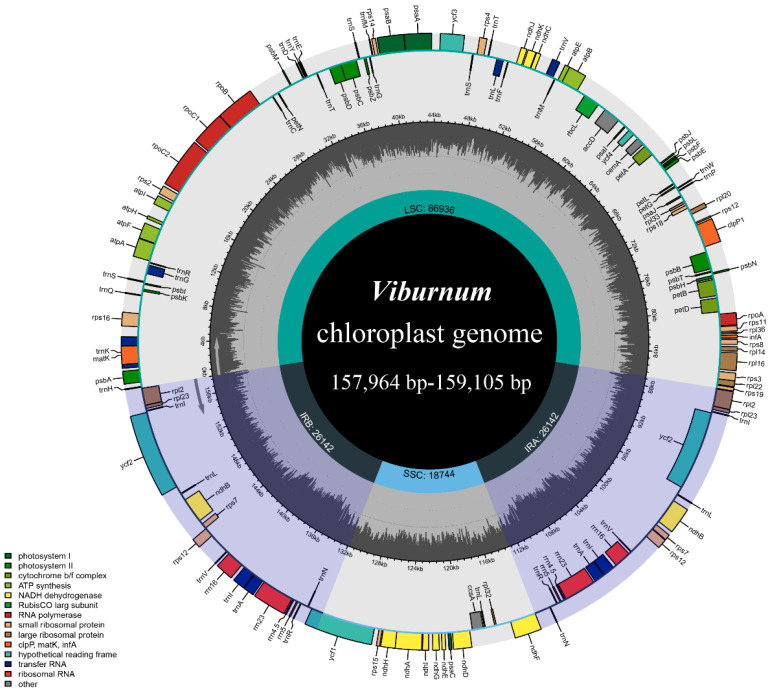 Figure 1
Figure 1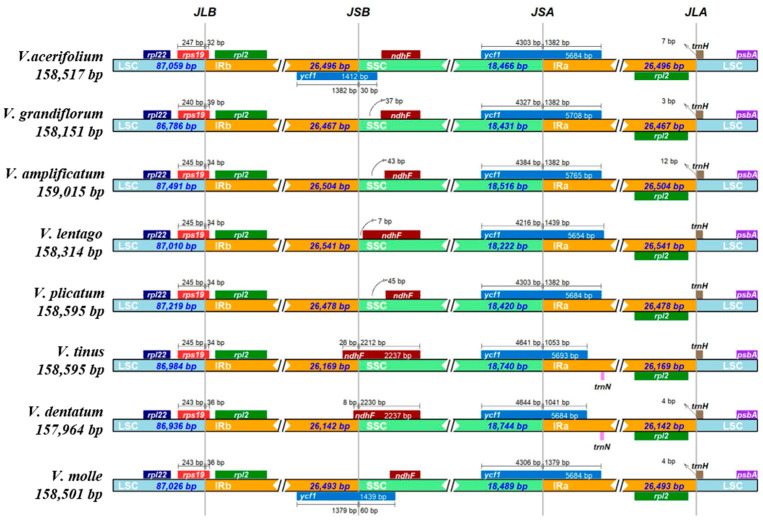 Figure 2
Figure 2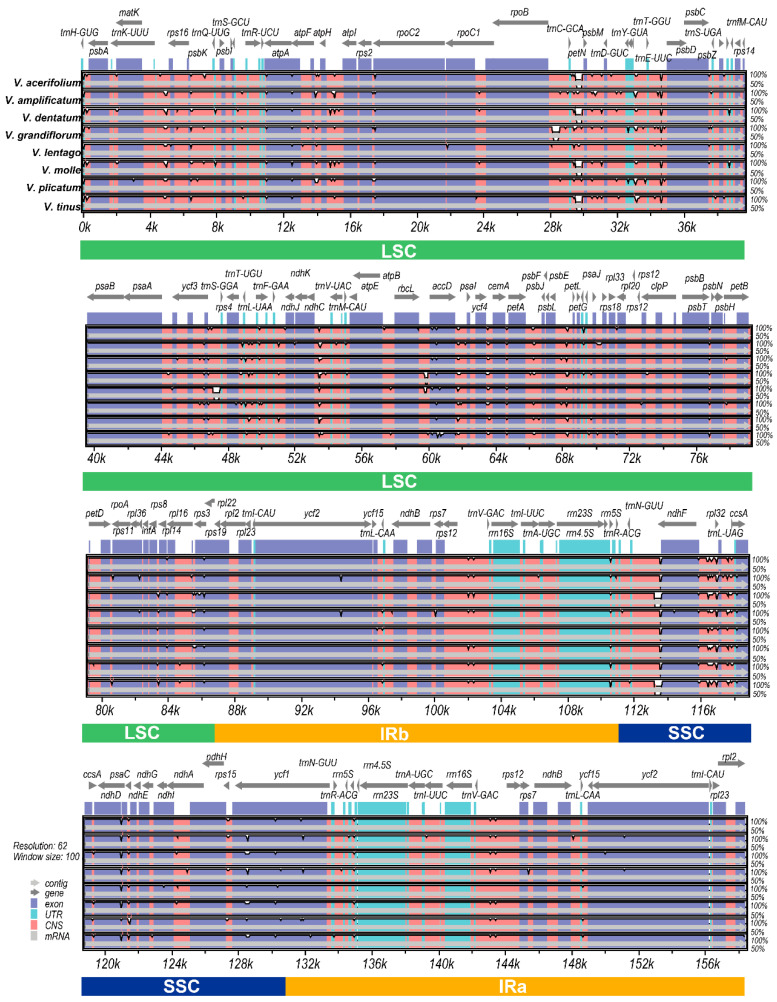 Figure 3
Figure 3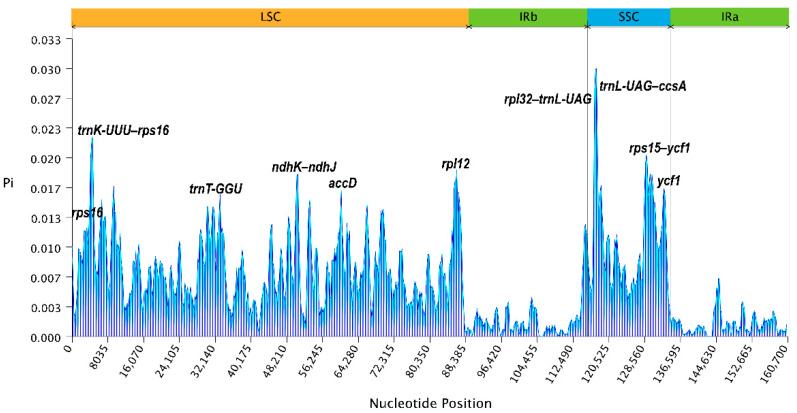 Figure 4
Figure 4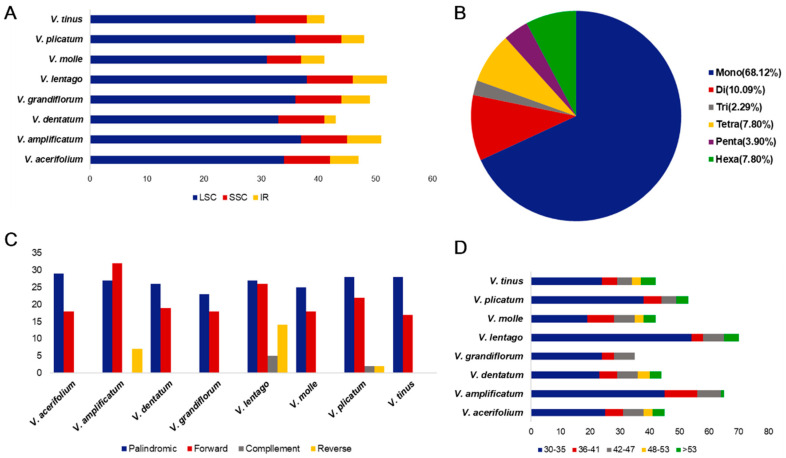 Figure 5
Figure 5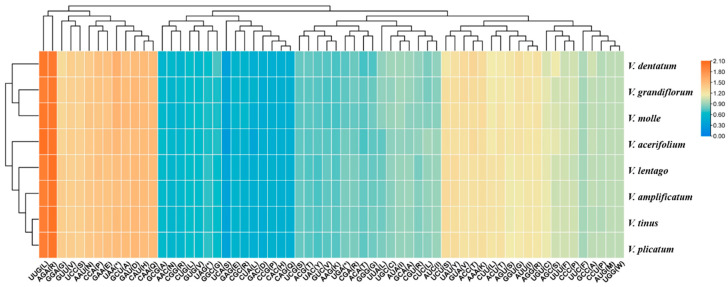 Figure 6
Figure 6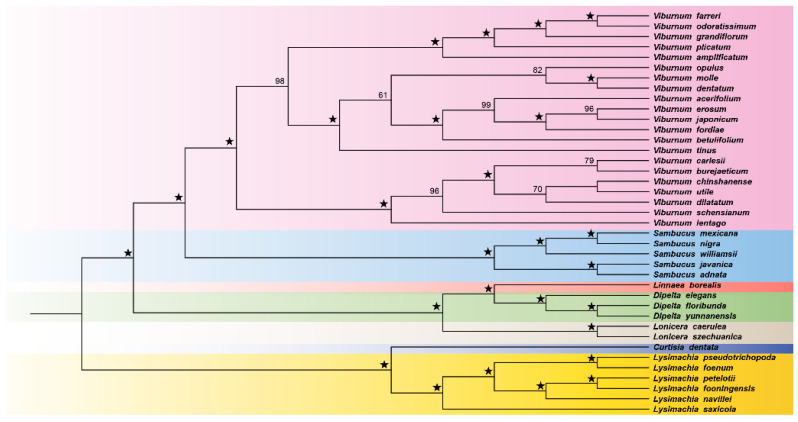 Figure 7
Figure 7- —Shandong Province Natural Science Foundation of China
- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Diversity and Evolution · Genomics and Phylogenetic Studies · Plant Molecular Biology Research
1. Introduction
The genus Viburnum L. (Adoxaceae, Dipsacales) comprises approximately 230 species of shrubs and small trees distributed predominantly across temperate and subtropical regions of the Northern Hemisphere [1]. Asia represents the primary center of diversity, with China alone harboring 96 species—the highest national diversity globally [2]. The genus exhibits remarkable ecological amplitude, occupying habitats ranging from temperate deciduous forests to subtropical cloud forests and Mediterranean shrublands, and many species are cultivated as ornamentals or used in traditional medicine [3,4]. Despite intensive study, species delimitation within Viburnum remains challenging. Morphological characters traditionally used for classification—including endocarp architecture, inflorescence type, and trichome morphology—show extensive homoplasy, and frequent hybridization both in nature and cultivation further obscures species boundaries [5]. Molecular phylogenetic studies have substantially improved understanding of relationships within the genus, revealing that several traditionally recognized sections are not monophyletic and necessitating taxonomic revision [1,6]. Despite these advances, the integration of plastome-scale genomic data with evolutionary and functional interpretations remains limited for Viburnum, particularly at the inter-sectional level.
Chloroplast genomes have proven particularly valuable for resolving relationships within Viburnum. The relatively slow evolutionary rate of plastomes, combined with their uniparental inheritance and lack of recombination, makes them well-suited for phylogenetic reconstruction at various taxonomic levels [7]. Previous studies have sequenced plastomes from numerous Viburnum species, establishing a robust phylogenetic framework and resolving major clades within the genes [6,8]. However, existing studies have largely focused on tree topology, whereas comparative analyses of plastome evolution—especially those integrating structural variation and selection pressures on protein-coding genes—remain scarce in Viburnum. In particular, it remains unclear whether plastome genes exhibit lineage- or section-specific patterns of molecular evolution, and how such patterns may relate to functional divergence or historical biogeography within the genus. Although plastome gene content and organization are broadly conserved across angiosperms, individual genes vary considerably in their evolutionary rates [7]. Genes encoding components of the photosynthetic apparatus typically evolve under strong purifying selection, reflecting their essential metabolic functions. By contrast, genes involved in other processes—including fatty acid synthesis (accD) and protein import (ycf1, ycf2)—often show accelerated evolution, potentially reflecting functional divergence or relaxed constraint [9,10]. Characterizing these patterns across diverse lineages can reveal the evolutionary forces shaping plastome architecture.
In this study, we assembled and analyzed complete plastomes from eight Viburnum species representing five sections and spanning both Old World and New World distributions. By integrating plastome structural comparisons, molecular evolutionary analyses, and phylogenetic inference, we aimed to address three key questions: first, to systematically characterize structural variation in Viburnum plastomes and identify hypervariable regions suitable for molecular marker development; second, to assess selection pressures on protein-coding genes and identify candidates for functional divergence; and third, to evaluate phylogenetic relationships and their congruence with existing classifications. Through these analyses, this work not only supplements the chloroplast genome database of the genus Viburnum and evolutionary research, but also offers new insights into the evolutionary dynamics of plastid genes within a morphologically complex and taxonomically challenging genus.
2. Materials and Methods
2.1. Plastome Assembly and Annotation
Raw sequencing data for eight Viburnum species representing major lineages and geographic regions: Viburnum molle Michx. and Viburnum dentatum L. (section Porphyrotinus, eastern North America), Viburnum acerifolium L. (section Succotinus, eastern North America), Viburnum lentago L. (section Valvatotinus, North America), Viburnum amplificatum Kern (section Crenotinus, Southeast Asia), Viburnum grandiflorum Wall. ex DC. (section Solenotinus, Himalaya), Viburnum plicatum Thunb. (section Tomentosa, East Asia), and Viburnum tinus L. (section Tinus, Mediterranean) were obtained from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/ (accessed on1 February 2026)). Raw reads were processed using Trimmomatic v0.39 [11]. Quality-filtered reads were assembled into complete plastomes using GetOrganelle v1.7.7 [12] with parameters optimized for embryophyte plastomes. This pipeline employs a reference-guided extension algorithm that iteratively maps reads to seed sequences, followed by de novo assembly of mapped reads using SPAdes (v.4.2.0). Assembly graphs were visualized and inspected using Bandage v0.8.1 [13] to verify complete circular assembly and resolve any ambiguities at inverted repeat boundaries. For each species, we confirmed that the assembly graph showed the expected structure with two alternative paths through the inverted repeats. Final assemblies achieved mean sequencing depths ranging from 312× to 587× across species.
Plastome annotation was performed using GeSeq v2.03 [14] with Viburnum japonicum (MH036493) and Viburnum farreri (NC_056112) as references. The annotation pipeline employed BLAT for protein-coding genes, HMMER for rRNAs, and ARAGORN and tRNA scan-SE for tRNAs. All annotations were manually verified and refined in Geneious Prime 2023.1 (Biomatters, Auckland, New Zealand). Start and stop codon positions were checked against amino acid alignments of orthologous genes from related species, and any apparent frameshifts or premature stop codons were examined for possible annotation errors or pseudogenization. Intron-exon boundaries were verified by aligning genomic and CDSs and confirming consensus splice site motifs (GT…AG for group I introns, variable for group II introns). Transfer RNA structures were validated using tRNAscan-SE v2.0 [15] with organellar search mode and a cove score cutoff of 15. Circular genome maps were generated using OGDRAW v1.3.1 [16]. Annotated sequences were deposited in GenBank under accession numbers NC_086516 (V. molle), NC_086692 (V. dentatum), NC_086693 (V. tinus), NC_086694 (V. plicatum), NC_086695 (V. lentago), NC_086696 (V. amplificatum), NC_086697 (V. grandiflorum), and PP101997 (V. acerifolium).
2.2. Comparative Sequence Analysis
Whole-plastome alignments were generated using MAFFT v7.505 [17] with the FFT-NS-i iterative refinement algorithm and default gap penalties. Sequence conservation was visualized using the mVISTA server [18] in Shuffle-LAGAN mode, which performs global alignment with automated detection of rearrangements, using V. molle as the reference sequence. Then, the DNA polymorphism module of DnaSP v6.12.03 [19] was used for nucleotide variability (Pi) analysis with a selected step size of 200 sites and a window length of 800 sites. This window size was selected to capture variation at the scale of individual genes while providing sufficient resolution to identify localized hotspots. Regions with Pi value exceeding 0.02 were flagged as hypervariable and evaluated as candidate molecular markers. Inverted repeat boundaries were compared across species using IRscope [20], which generates graphical representations of the four junction sites (JLB, JSB, JSA, JLA) showing gene positions relative to boundaries. This analysis identifies expansion or contraction events that alter the genes located at IR-SC junctions.
2.3. Repeat Sequence Identification
Simple sequence repeats (microsatellites) were identified using MISA v2.1 [21] with minimum repeat number thresholds of 10 for mononucleotides, 5 for dinucleotides, 4 for trinucleotides, and 3 for tetra-, penta-, and hexanucleotides. These thresholds were selected to balance sensitivity with specificity based on previous plastome SSR studies. Long dispersed repeats were identified using REPuter [22] with a minimum repeat size of 30 bp, maximum size of 300 bp, and Hamming distance of 3 (allowing for up to 3 mismatches per 30 bp). Four repeat types were detected: forward (direct), reverse, complement, and palindromic.
2.4. Codon Usage Analysis
Protein-coding sequences were extracted from each plastome, excluding genes with internal stop codons, frameshift mutations, or incomplete terminal codons. For genes with multiple copies (e.g., those in inverted repeats), a single representative copy was retained. Relative synonymous codon usage (RSCU) was calculated using CodonW v1.4.4 (http://codonw.sourceforge.net/ (accessed on 1 February 2026)). RSCU values were calculated as the observed frequency of a codon divided by the expected frequency under equal usage of synonymous alternatives; values greater than 1.0 indicate preferential usage. RSCU values were visualized as heatmaps using the pheatmap package in R v4.2.0, with hierarchical clustering based on Euclidean distances and complete linkage.
2.5. Selection Pressure Analysis
To ensure accurate codon alignment, protein-coding sequences across Viburnum species were analyzed with MACSE v2 [23]. The assessment of the ratio between non-synonymous (Ka) and synonymous (Ks) substitution rates was conducted using Ka/Ks Calculator v2 [24]. By comparing with V. japonicum, the Ka/Ks values for each Viburnum species were determined. Unrealistic Ka/Ks ratios were excluded to ensure precise screening of conserved and divergent genes. Thus, we adopted a more accurate threshold screening: Ka/Ks < 0.01 treated as qualified conserved genes, Ka/Ks = 1 for neutral selection, Ka/Ks >1 (greater than 1) as positively selected orthologs, and Ka/Ks < 1 (less than 1) for purifying selection.
2.6. Phylogenetic Analysis
For phylogenetic reconstruction, we assembled a dataset comprising the eight newly sequenced Viburnum plastomes, thirteen additional Viburnum species obtained from GenBank (representing major sections not covered by our sampling), and employed a hierarchical set of eighteen outgroup taxa spanning a range of phylogenetic distances to reconstruct the phylogeny. Specifically, closely related Caprifoliaceae taxa (Sambucus and Lonicera) were selected to stabilize internal branching patterns, moderately distant Dipsacales (Dipelta and Linnaea) lineages were used to calibrate key divergence nodes, and distantly related groups (Lysimachia and Curtisia) were included to root the tree reliably. This multi-layered approach ensures the robustness of the inferred phylogenetic topology. Complete plastome sequences were aligned using MAFFT v7.505 with the G-INS-i algorithm, which is optimized for global homology with accurate alignment of conserved regions. Ambiguously aligned regions were removed using trimAl v1.4 [25] with the automated1 heuristic, which selects trimming parameters based on alignment characteristics. The optimal partitioning scheme and substitution models were determined using ModelFinder [26] as implemented in IQ-TREE v2.2.0 [27], testing all models available in IQ-TREE and selecting based on the Bayesian Information Criterion (BIC). Bayesian inference was conducted using MrBayes v3.2.7 [28]. Two independent runs of four Metropolis-coupled MCMC chains (three heated, one cold) were run for 20 million generations, sampling every 1000 generations. The temperature parameter was set to 0.1 to improve chain mixing. Convergence was assessed by examining the average standard deviation of split frequencies (target < 0.01), potential scale reduction factors (target ≈ 1.0), and effective sample sizes (target > 200 for all parameters) using Tracer v1.7.2. The first 25% of samples were discarded as burn-in, and a 50% majority-rule consensus tree was constructed from the remaining samples.
3. Results
3.1. Plastome Structure and Gene Content
Complete plastome assemblies were obtained for all eight Viburnum species, each displaying the canonical quadripartite structure characteristic of most photosynthetic angiosperms. Total plastome sizes ranged from 157,964 bp in V. dentatum to 159,015 bp in V. amplificatum, a difference of 1051 bp representing only 0.66% variation across the sampled species. This size variation was distributed across all four regions but was proportionally greatest in the small single-copy region, which ranged from 18,222 bp (V. lentago) to 18,744 bp (V. dentatum) a span of 522 bp (2.9%). The large single-copy region exhibited slightly less proportional variation (86,936–87,491 bp; 0.64%), while the inverted repeat regions were the most conserved in length (26,142–26,541 bp; 1.5%) (Table 1; Figure 1). These patterns suggest that size variation in Viburnum plastomes is driven primarily by indel accumulation in the single-copy regions rather than by large-scale expansion or contraction of the inverted repeats.
Overall GC content was remarkably uniform across species (38.1–38.2%) but showed the expected asymmetric distribution among regions. The inverted repeats exhibited the highest GC content (43.0–43.2%), attributable to the presence of GC-rich ribosomal RNA genes (rrn16, rrn23, rrn4.5, rrn5). The large single-copy region showed intermediate values (36.3–36.5%), while the small single-copy region had the lowest GC content (32.0–32.2%) (Table 1). This regional variation in nucleotide composition has implications for primer design in regions spanning multiple plastome compartments.
Gene content was highly conserved, with 129–131 genes annotated per plastome: 85–86 protein-coding genes, 36–37 tRNA genes, and 8 rRNA genes (Table 1). The minor variation in gene counts reflected differences in the annotation of boundary genes partially duplicated in inverted repeats and the status of putative pseudogenes. Eighteen genes contained introns: fifteen genes possessed a single intron (atpF, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps16, and six tRNA genes), while clpP, ycf3, and rps12 each contained two introns. Seventeen genes were completely duplicated in the inverted repeats, including all four rRNA species, seven tRNAs, and six protein-coding genes (ndhB, rpl2, rpl23, rps7, ycf2, rps12) (Table S1).
3.2. Inverted Repeat Boundary Variation
Comparison of the four junctions between inverted repeat and single-copy regions revealed both conserved features and lineage-specific variations that illuminate microstructural evolution in Viburnum plastomes. At the JLB junction (LSC/IRb), rpl22 was consistently positioned entirely within the large single-copy region across all species. The adjacent rps19 gene showed partial duplication in all species, with 32–39 bp of its 3′ end extending into IRb (Figure 2).
The JSB junction (IRb/SSC) displayed more substantial variation. In six of the eight species, the ndhF gene was located entirely within the small single-copy region, with its 5′ end positioned 1–89 bp from the junction. However, in V. tinus and V. dentatum, ndhF extended 8–26 bp into IRb, indicating a minor expansion of the inverted repeat into the small single-copy region in these lineages. Notably, these two species also possessed the shortest inverted repeats in our sample (26,142 and 26,169 bp, respectively), suggesting that the ndhF boundary shift was accompanied by contraction elsewhere in the repeat. Additionally, V. tinus and V. dentatum harbored an extra trnN-GUU gene within IRa that was absent from the other six species, further supporting lineage-specific IR restructuring.
The JSA junction (SSC/IRa) was the most conserved boundary, with ycf1 spanning the junction in all species. The larger 5′ portion of ycf1 (4216–4644 bp) resided in SSC while the 3′ portion (1041–1439 bp) extended into IRa. This arrangement creates a truncated ycf1 pseudogene at the JSB junction in IRb, ranging from 1000 to 1200 bp depending on species. The JLA junction (IRa/LSC) showed variable positioning of trnH-GUG: in V. lentago, V. plicatum, and V. tinus, this gene was located entirely within LSC (3–47 bp from the junction), while in the remaining species it extended 3–12 bp into IRa.
3.3. Sequence Divergence Patterns
Global alignment of the eight plastomes revealed high overall sequence conservation punctuated by localized regions of elevated divergence. The mVISTA plot illustrated that sequence identity exceeded 90% across most of the alignment, with the inverted repeats showing particularly high conservation (>99% identity) as expected from their exposure to gene conversion. Divergence was concentrated in intergenic spacers and, to a lesser extent, in certain protein-coding genes, while ribosomal RNA and transfer RNA genes were nearly invariant (Figure 3).
Sliding-window analysis quantified these patterns, revealing a genome-wide mean Pi value of 0.0059 (Figure 4). However, this average masked substantial regional heterogeneity: the small single-copy region showed the highest diversity (Pi value = 0.01), followed by the large single-copy region (Pi value = 0.0074), while the inverted repeats exhibited markedly lower values (Pi value = 0.0012). The 8.3-fold difference in Pi value between the IR and SSC regions reflects both the homogenizing effect of gene conversion on IR sequences and the concentration of rapidly evolving intergenic spacers in the single-copy regions.
The sliding-window analysis identified multiple peaks of elevated diversity distributed across the large single-copy region. The highest Pi value was observed in the trnL-UAG–ccsA intergenic spacer (Pi value = 0.0298). Other notably divergent regions included rpl32–trnL-UAG (Pi value = 0.0275), trnK-UUU–rps16 (Pi value = 0.0222), and rps15–ycf1 (Pi value = 0.0202). Among protein-coding genes, rps16, ndhJ, ndhK, accD, rpl2, rps19, rpl22, ndhD, rps15, ndhH, psaC, and ycf1 showed elevated divergence relative to their functional categories, with Pi values ranging from 0.0160 to 0.0199.
3.4. Repeat Sequence Composition
Simple sequence repeat analysis identified 41–52 microsatellites per plastome, with V. lentago harboring the most and V. molle and V. tinus the fewest (Figure 5A). Mononucleotide repeats predominated, accounting for 68.12% of all SSRs across species and consisting almost exclusively of poly-A or poly-T tracts. This compositional bias reflects the AT-richness of plastome non-coding regions where most SSRs occur. Dinucleotide repeats comprised 10.09% of the total, all with AT/TA motifs, while tri-, tetra-, penta-, and hexanucleotide repeats accounted for the remaining 21.79% in approximately equal proportions. SSRs were non-randomly distributed across the plastome, with the large single-copy region harboring 70–76% of repeats, the small single-copy region containing 15–22%, and the inverted repeats holding only 4–12% (Figure 5B).
Long dispersed repeats (≥30 bp) numbered 41–72 per plastome, varying considerably among species. Forward and palindromic repeats together accounted for more than 73% of long repeats in all species, while reverse and complement repeats were comparatively rare (Figure 5C). The majority of long repeats (45–77%) fell within the 30–35 bp size class, with progressively fewer repeats at larger sizes. Repeats exceeding 50 bp were uncommon (less than 19% of the total) except for the inverted repeats themselves (Figure 5D). The functional significance of these dispersed repeats is unclear, but they may facilitate recombination-mediated rearrangements in some lineages.
3.5. Codon Usage Patterns
Relative synonymous codon usage (RSCU) analysis quantitatively confirmed a strong bias toward A- and T-ending synonymous codons. Of 61 sense codons (excluding the single codons for Met and Trp), 29 showed RSCU values exceeding 1.0, indicating preferential usage (Figure 6). Twenty-five of these twenty-nine preferred codons ended in A or U, while only two G-ending codons (AGG for Arg, UUG for Leu) showed preference. Three C-ending codons (AGC for Ser, CCC for Pro, GCC for Ala) exhibited slight preference. Conversely, 30 codons showed RSCU values below 1.0, predominantly those ending in C or G. The most strongly preferred codons were AGA (Arg; RSCU = 1.98–2.05), UUG (Leu; RSCU = 1.94–2.04), and GAA (Glu; RSCU = 1.44–1.48), while the most avoided codons were UCA (Ser; RSCU = 0.27–0.40), GAC (Asp; RSCU = 0.47–0.51), and CAG (Gln; RSCU = 0.48–0.50).
Hierarchical clustering based on RSCU values grouped the eight species into two clusters. One cluster contained V. molle, V. dentatum, and V. grandiflorum, while the second cluster comprised the remaining five species. The imperfect correspondence between codon usage similarity and phylogeny suggests that factors beyond shared ancestry—potentially including GC content variation, tRNA abundance, and translational selection—influence codon usage evolution in Viburnum plastomes.
3.6. Selection on Protein-Coding Genes
We calculated the Ka/Ks ratios for 78 protein-coding genes in its plastome using V. japonicum as the reference. Due to missing information (Ks = 0) preventing reliable determination for some genes, we chose to exclude these genes from the analysis. The majority of genes exhibited Ka/Ks ratios well below 1, indicating strong purifying selection acting on plastid protein-coding genes. Genes involved in photosynthesis showed particularly low Ka/Ks ratios, with many genes displaying values approaching zero. These included genes encoding photosystem I components (psaA, psaB, psaC, psaJ), photosystem II components (psbA, psbB, psbC, psbD), and ATP synthase subunits (atpA, atpB, atpE, atpH, atpI). In contrast, accD, ycf1, and ycf2 exhibited the highest Ka/Ks ratios among all analyzed genes, although all values remained below 1 (Table S2). This indicates that these genomes have a higher degree of conservation.
3.7. Phylogenetic Relationships
Bayesian analyses produced identical topologies with strong support for most nodes. Within Viburnum, two major clades were recovered, each with maximum support (Bayesian posterior probability = 1.00). The first clade (Clade I) comprised seven species corresponding largely to species traditionally assigned to the sections Valvatotinus and Euviburnum. Within this clade, V. lentago was placed in the Lentago subclade of Valvatotinus, while V. carlesii, V. burejaeticum, V. utile, and V. schensianum formed a well-supported group corresponding to section Euviburnum. The relationships among Euviburnum species were fully resolved, with V. carlesii and V. burejaeticum as sister taxa (Figure 7).
The second major clade (Clade II) contained fourteen species from sections Solenotinus, Tomentosa, Crenotinus, Opulus, Porphyrotinus, Succotinus, and Tinus. This clade was subdivided into two sister groups. The first subgroup united members of Solenotinus, Tomentosa, and Crenotinus, with V. grandiflorum occupying a position at the base of Solenotinus, consistent with its basal morphological features within that section. Within this subgroup, V. amplificatum was determined to be a sister to the Crenotinus clade sensu stricto. The second subgroup comprised sections Opulus, Porphyrotinus, Succotinus, and Tinus. Within Porphyrotinus, V. molle and V. dentatum were placed in distinct subclades, and V. tinus occupied an isolated position within this group.
4. Discussion
4.1. Plastome Structural Conservation in the Context of Viburnum Diversification
The eight Viburnum plastomes characterized in this study exhibit remarkable structural conservation, with size variation of only 0.66% and identical gene content across species representing five sections and divergence times spanning the Oligocene to Miocene. This structural stasis contrasts with the extensive plastome rearrangements documented in some angiosperm lineages, such as Geraniaceae, where inversions, gene losses, and IR expansions have produced highly derived genome architectures [29]. The conservation observed in Viburnum likely reflects both the relatively recent crown age of the genus (~55 Ma) [30] and the strong functional constraints on plastome-encoded genes.
Although overall structure was conserved, we detected subtle variation at IR boundaries that illuminates ongoing microstructural evolution. The finding that V. tinus and V. dentatum share similar IR configurations, including ndhF extension into IRb and an additional trnN-GUU copy, despite belonging to different sections (Tinus and Porphyrotinus) presents an intriguing pattern. One interpretation is that this configuration represents the ancestral state for Viburnum, subsequently modified in other lineages. Alternatively, convergent boundary shifts may have occurred independently in these two species. Resolution of this question will require examination of IR boundaries across a broader sample of species, particularly those branching near V. tinus and V. dentatum in the phylogeny.
The regional heterogeneity in GC content observed in Viburnum plastomes—with IR regions substantially more GC-rich than single-copy regions—is a universal feature of angiosperm plastomes attributable to the presence of GC-rich ribosomal RNA genes in the IRs. However, the functional consequences of this compositional asymmetry remain incompletely understood. Recent studies have suggested that regional GC content may influence mutation rates, recombination frequencies, and gene expression levels, potentially contributing to the observed differences in evolutionary rates among plastome regions [31].
4.2. Hypervariable Regions and Their Utility as Molecular Markers
The identification of eleven intergenic regions with nucleotide diversity exceeding 0.02 provides a robust set of candidate markers for various applications in Viburnum research. Several of these regions, including trnK-UUU–rps16, rpl32–trnL-UAG, and trnL-UAG–ccsA, have been previously identified as phylogenetically informative in Dipsacales and other angiosperm families [32,33], supporting their general utility. The petN–trnC-GCA spacer is particularly promising because it combines high variability with conserved flanking regions that facilitate primer design, and it has been successfully employed for species discrimination in several plant genera.
For practical applications, we suggest a tiered approach to marker selection depending on the taxonomic level of interest. For discrimination among closely related species or infraspecific taxa, the most variable regions (trnK-UUU–rps16, trnL-UAG–ccsA) offer maximum resolution. For genus-wide phylogenetics, moderately variable regions with fewer alignment difficulties are more appropriate. For DNA barcoding applications requiring standardized markers, ycf1 emerges as a strong candidate: it combines high variability with a length suitable for single-amplicon sequencing and has been proposed as a universal barcode for land plants [34].
The predominance of mononucleotide A/T repeats is typical of plastome SSRs and facilitates primer design in flanking regions. However, mononucleotide repeats can be challenging to score accurately due to polymerase slippage during PCR, and di- or trinucleotide repeats may be preferable for population genetic applications where precise allele calling is essential.
4.3. Relaxed Selection on Non-Photosynthetic Genes
The finding that accD, ycf1, and ycf2 evolve under relaxed purifying selection relative to photosynthesis genes is consistent with observations across diverse angiosperm lineages and illuminates the different selective regimes experienced by plastome genes with distinct functions [35]. Photosynthesis genes encode proteins that must function within highly integrated complexes where individual subunits interact with multiple partners; amino acid changes that disrupt these interactions are deleterious and eliminated by selection. In contrast, genes involved in other processes may experience weaker constraint, particularly if their functions can be partially complemented by nuclear-encoded paralogs or if the processes they mediate are themselves subject to adaptive modification.
The elevated Ka/Ks of accD is particularly noteworthy because this gene has been lost from plastomes in multiple angiosperm lineages, including grasses, with its function assumed by nuclear-encoded acetyl-CoA carboxylase [36]. In Viburnum, accD remains intact and functional, but its relaxed constraint may indicate incipient functional transfer or simply reduced selection intensity in a genus where fatty acid metabolism varies among species with different ecological strategies. The observation that accD is also among the most sequence-divergent protein-coding genes suggests that its elevated Ka/Ks translates into substantial sequence variation useful for phylogenetic and barcoding applications.
The relaxed evolution of ycf1 and ycf2 reflects the coevolutionary dynamics inherent in protein import systems. These genes encode components of the TIC translocon, which must recognize and translocate thousands of different nuclear-encoded proteins with diverse transit peptides [37]. Maintaining this broad substrate specificity while preserving transport fidelity may require a degree of sequence flexibility not permitted in the more structurally constrained photosynthesis complexes. Consistent with this interpretation, ycf1 and ycf2 show elevated Ka/Ks across most angiosperm lineages examined, suggesting that relaxed constraint on protein import genes is a general feature of plastome evolution rather than a Viburnum-specific phenomenon.
4.4. Phylogenetic Implications
The phylogenetic relationships recovered in this study are highly congruent with previous molecular systematic analyses of Viburnum [6,8], confirming the robustness of plastome data for resolving infrageneric relationships. The recognition of two major clades—one comprising sections Valvatotinus and Euviburnum, the other containing the remaining sections—appears to represent a fundamental division within the genus that may correspond to an ancient biogeographic or ecological divergence [30]. We inferred that this split occurred in the Eocene, with subsequent diversification in each clade associated with distinct geographic regions and climatic niches.
Two specific results merit discussion in the context of taxonomic revision. First, the placement of V. amplificatum as sister to Crenotinus sensu stricto, rather than within it, suggests that this Southeast Asian species may warrant recognition as a distinct section or subsection. Its intermediate morphological features and isolated phylogenetic position indicate a unique evolutionary trajectory that would be obscured by subsuming it within Crenotinus. Second, the isolated position of V. tinus within Subclade IIb reinforces its distinctiveness as the sole representative of the section Tinus. This Mediterranean endemic possesses a combination of characters—including evergreen habit, coriaceous leaves, and metallic blue drupes—found in no other Viburnum species, consistent with a long period of independent evolution in a geographically isolated region [1].
The relationships among North American species are also clarified by our analysis. The separation of V. molle (Mollotinus) from V. dentatum (Dentata) within section Porphyrotinus, and their collective distinction from V. acerifolium (Succotinus), confirms that these eastern North American species represent at least two independent colonization events from Asian ancestors, as inferred by Spriggs et al. (2015) [38]. This biogeographic pattern of multiple trans-Beringian dispersals is consistent with the genus’s center of diversity in Asia and the relatively recent (Miocene–Pliocene) establishment of Viburnum in the Americas.
5. Conclusions
Comparative analysis of eight Viburnum plastomes has revealed a structurally conserved genome harboring substantial sequence variation suitable for diverse molecular applications. The identification of eleven hypervariable intergenic regions and the characterization of selection pressures across protein-coding genes provide resources for population genetics, phylogeography, and species identification. The finding that genes involved in fatty acid biosynthesis (accD) and protein import (ycf1, ycf2) evolve under relaxed constraints relative to photosynthesis genes suggests that these loci experience distinct selective regimes potentially linked to functional divergence or coevolution with nuclear partners. Phylogenetic analysis confirms established sectional relationships while providing an improved resolution for taxonomically problematic taxa, including V. amplificatum and V. tinus. These results supplement the chloroplast genome database of the genus Viburnum and contribute to understanding plastome evolution in this ecologically important genus.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Winkworth R.C. Donoghue M.J. Viburnum phylogeny based on combined molecular data: Implications for taxonomy and biogeography Am. J. Bot.20059265366610.3732/ajb.92.4.65321652443 · doi ↗ · pubmed ↗
- 2Zhao L. Wang Y. Lyu W. Tang Z. Qiu L. Tang M. A new synonym for Viburnum erosum (Viburnaceae) in East China, based on morphological and molecular evidence P Lo S ONE 202520 e 03129204026714610.1371/journal.pone.0312920 PMC 12017528 · doi ↗ · pubmed ↗
- 3Altun M.L. Yılmaz B.S. Orhan I.E. Citoglu G.S. Assessment of cholinesterase and tyrosinase inhibitory and antioxidant effects of Hypericum perforatum L. (St. John’s wort)Ind. Crops Prod.2013438792
- 4Kajszczak D. Zakłos-Szyda M. Podsędek A. Viburnum opulus L.—A review of phytochemistry and biological effects Nutrients 202012339810.3390/nu 1211339833167421 PMC 7694363 · doi ↗ · pubmed ↗
- 5Donoghue M.J. Baldwin B.G. Li J. Winkworth R.C. Viburnum phylogeny based on chloroplast trn K intron and nuclear ribosomal ITS DNA sequences Syst. Bot.20042918819810.1600/036364404772974095 · doi ↗
- 6Clement W.L. Arakaki M. Sweeney P.W. Edwards E.J. Donoghue M.J. A chloroplast tree for Viburnum (Adoxaceae) and its implications for phylogenetic classification and character evolution Am. J. Bot.20141011029104910.3732/ajb.140001524928633 · doi ↗ · pubmed ↗
- 7Wicke S. Schneeweiss G.M. Depamphilis C.W. Müller K.F. Quandt D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function Plant Mol. Biol.20117627329710.1007/s 11103-011-9762-421424877 PMC 3104136 · doi ↗ · pubmed ↗
- 8Ran H. Liu Y. Wu C. Cao Y. Phylogenetic and comparative analyses of complete chloroplast genomes of Chinese Viburnum and Sambucus (Adoxaceae)Plants 20209114310.3390/plants 909114332899372 PMC 7570041 · doi ↗ · pubmed ↗